















Abnormalities of tetrahydrobiopterin metabolism
Apr. 07, 2024
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
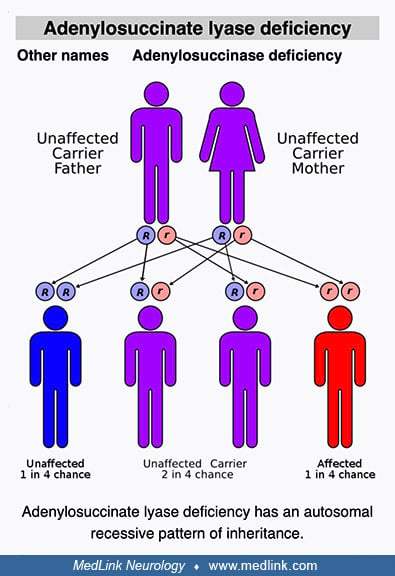
Adenylosuccinate lyase deficiency is a rare, autosomal recessive defect of purine metabolism affecting purinosome assembly and reducing metabolite fluxes through both de novo purine synthesis (DNPS) and purine nucleotide recycling pathways. The purinosome is a multienzyme complex of DNPS enzymes (including adenylosuccinate lyase) that cells transiently assemble in their cytosol to address both depletion of and increased demand for purines. Clinically, adenylosuccinate lyase deficiency is generally categorized into three phenotypes: a fatal neonatal form, a severe form (type I), and a milder form (type II). Neurologic symptoms are the most common and prominent clinical problems associated with adenylosuccinate lyase deficiency. In the severe form of the disease, neurologic symptoms and signs are typically evident soon after birth. Common neurologic presentations include acute encephalopathy, chronic encephalopathy, and behavioral abnormalities in various nonspecific combinations with seizures and developmental delay or regression. Diagnosis of adenylosuccinate lyase deficiency may be delayed or missed because patients can present with nonspecific features, such as developmental delay, autism spectrum disorder, or epilepsy. Adenylosuccinate lyase deficiency can be diagnosed by detection of elevated metabolites along the DNPS pathway (succinylpurines) in body fluids. No specific FDA-approved treatment is available for adenylosuccinate lyase deficiency.
• The clinical presentation of adenylosuccinate lyase deficiency varies greatly with respect to age of onset, clinical manifestations, and rate of disease progression. | |
• Patients with adenylosuccinate lyase deficiency can present with nonspecific symptoms, such as developmental delay, autism spectrum disorder, or epilepsy, including infantile spasms. | |
• Due to lack of specific features and later onset of symptoms, diagnosis is difficult. | |
• Selective screening for adenylosuccinate lyase deficiency should be performed in infants who have neurologic disease without clear etiology, especially if supportive MRI findings are also present (eg, delayed or lack of myelination, abnormal white matter signal, and atrophy of the cerebrum or cerebellum). | |
• Detection of succinylpurines in body fluids by high-performance liquid chromatography or liquid chromatography-tandem mass spectrometry is the preferred biochemical test for adenylosuccinate lyase deficiency. | |
• No specific FDA-approved treatment is available for adenylosuccinate lyase deficiency. |
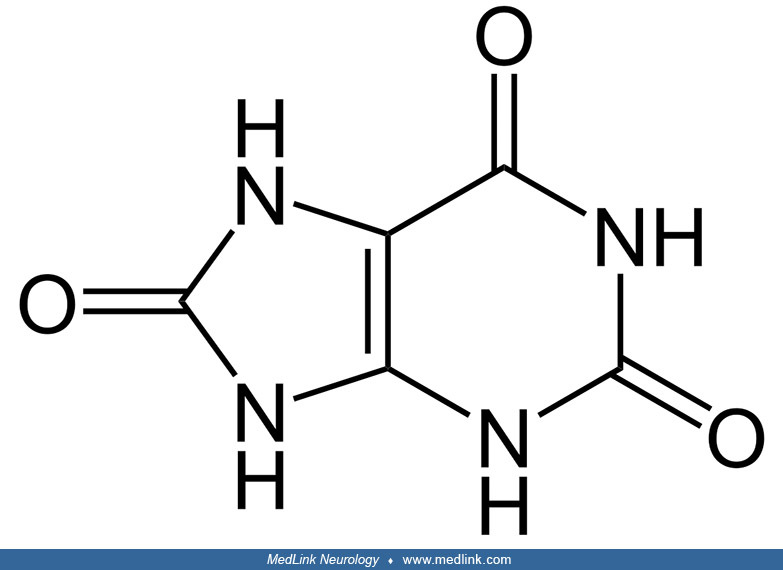
Origins of purine biochemistry. The word purine was coined by the German chemist Emil Fischer (1852-1919), who derived the German chemical term purin from the Latin purum uricum, for pure + uric acid; this was subsequently modified to an -ine ending in English, giving purine.
Fischer, working at the University of Würzburg, synthesized purine for the first time in 1898. Purine is a specific, water-soluble, aromatic organic compound that consists of two heterocyclic rings (ie, a 6-member pyrimidine ring and a 5-member imidazole ring) fused together. Given the name that Fischer chose, it is perhaps not surprising that he employed uric acid as the starting material for the reaction sequence that led to purine.
Uric acid had been known for a century, having been isolated from kidney stones in 1776 by Swedish-German pharmaceutical chemist Carl Wilhelm Scheele (1742-1786).
In 1891, 7 years before his synthesis of purine, Fischer, in conjunction with German chemist Oscar Piloty (1866-1915), had synthesized the pentose L-ribose.
Piloty started studying chemistry under Adolf von Baeyer (1835-1917) at the University of Munich in 1888 but transferred to the University of Würzburg in 1889 to work with Fischer on the chemistry of sugars. Piloty transferred after Baeyer awarded him failing marks on an exam, but the low exam score did not reflect Piloty's knowledge or abilities and was apparently instead Bayer's adverse reaction when he learned that Piloty had fallen in love with his daughter. Piloty earned his PhD under Fischer in 1890, helped Fischer synthesize L-ribose from L-arabonic acid in 1891, then married Baeyer's daughter in 1892, and that same year followed Fischer to the University of Berlin. A family reconciliation ultimately occurred, and Piloty and his wife returned to Munich in 1900 when his father-in-law offered Piloty a position at the University of Munich (even though Fischer had made him a better offer).
Fischer received the 1902 Nobel Prize in chemistry (only the second ever awarded to this point) "in recognition of the extraordinary services he has rendered by his work on sugar and purine syntheses" (20). Baeyer subsequently received the 1905 Nobel Prize in chemistry "in recognition of his services in the advancement of organic chemistry and the chemical industry, through his work on organic dyes and hydroaromatic compounds."
After Fischer's pioneering work, purine biochemistry advanced markedly over the first half of the 20th century, in part propelled by his students. In 1909, Lithuanian-American biochemist Phoebus Levene (1869-1940) and his assistant (later associate), American Walter Jacobs (1883-1967), recognized that D-ribose was a natural product, the enantiomer of Fischer and Piloty's L-ribose, and an essential component of nucleic acids.
Levene had spent a summer with Fischer at the University of Berlin in 1902, and Jacobs had only recently earned his PhD in chemistry under Fischer in Berlin in 1907. When Jacobs returned to New York following his doctoral work in Germany, he was appointed as a postdoctoral fellow in Levene's laboratory at the newly established Rockefeller Institute for Medical Research. Not only did Levene ultimately discover the carbohydrate components of both RNA and DNA (ie, ribose and deoxyribose, respectively), but he also correctly inferred that DNA is composed of a series of nucleotides and discovered the order of the three major components of a single nucleotide (ie, phosphate-sugar-base). Unfortunately, although widely accepted for decades, Levene had, in fact, stumbled with his overly simplistic tetranucleotide hypothesis (1909), which proposed that DNA is composed of equal parts adenine, guanine, cytosine, and thymine, resulting from a repeating linear sequence of the four nucleotides (ie, tetranucleotide repeats).
The genetic role of DNA was then unknown, and it was instead believed that Levene's proposed repeating structure excluded the possibility that DNA could store genetic information; instead, the protein component of chromosomes was then erroneously assumed to be the basis of heredity.
Several key steps beginning in the mid-1940s and extending to the early 1950s allowed a correct formulation of the structure of DNA: (1) demonstration that genes are composed of DNA, not protein; (2) discovery of "base pairing" through formulation of the Chargaff rules, which effectively disproved Levene's tetranucleotide hypothesis; (3) x-ray crystallography findings that indicated DNA had a helical structure. Work that began with Fischer ultimately culminated in the double helix model of deoxyribonucleic acid (DNA), by James Watson (b. 1928) and Francis Crick (1916-2004) in 1953.
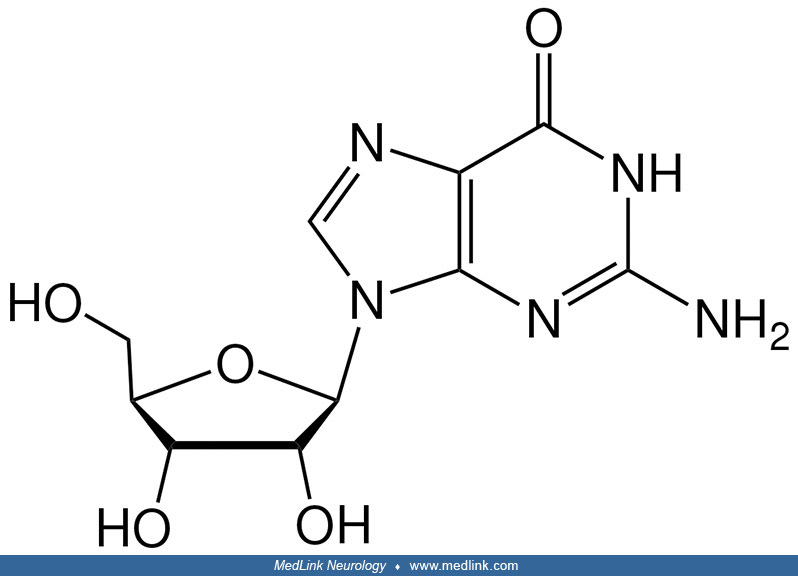
Purine nucleosides and nucleotides. The purines as a class of molecules include the purine bases adenine (A, 6-amino-purine) and guanine (G, 2-amino-6-oxypurine), both of which can be synthesized in vivo from inosine monophosphate or can be obtained either from the diet or from breaking down and recycling existing compounds and derivatives.
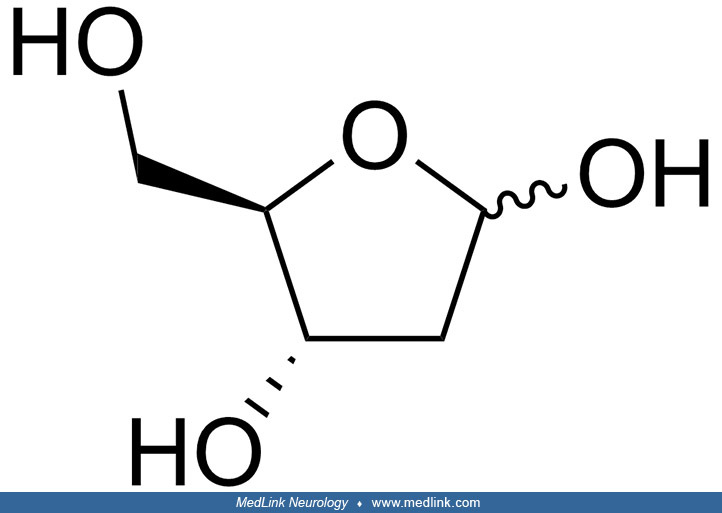
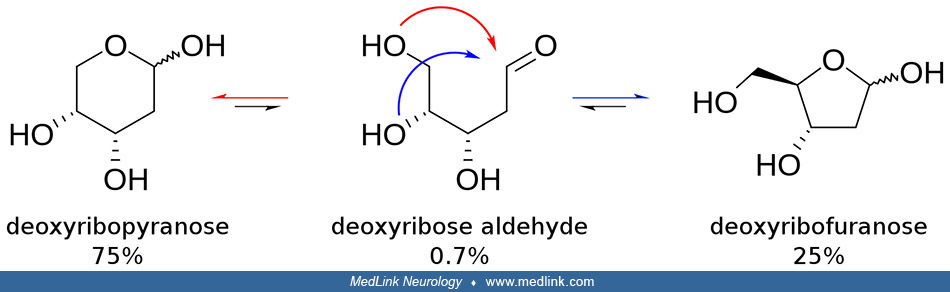
The purine bases may be attached to the pentose sugar ribose to form the ribonucleosides adenosine and guanosine, respectively. Ribose exists in equilibrium in multiple forms: an open-chain form and cyclic forms with 5- or 6-member rings.
Several of the forms of D-ribose: the open-chain form (top), and Haworth projections of two of the four cyclic forms, alpha-D-ribopyranose (middle), and beta-D-ribofuranose (bottom). Ribose exists as a mixture of cyclic forms i...
The purine bases may also be attached to the pentose sugar deoxyribose to form the deoxyribonucleosides 2'-deoxyadenosine and 2-deoxyguanosine, respectively. Ribose also exists in equilibrium in multiple forms: an open-chain form and cyclic forms with 5- or 6-member rings.
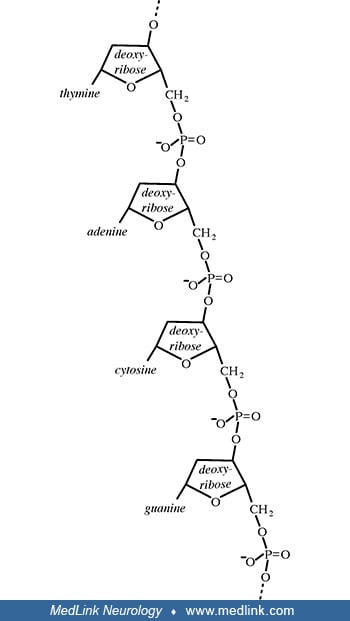
Addition of a phosphate group to the 5' carbon of ribose in the ribonucleosides adenosine and guanosine results in the formation of the ribonucleotides adenosine-5-monophosphate and guanosine-5-monophosphate, which are important metabolic and signaling molecules and building blocks of ribonucleic acid.
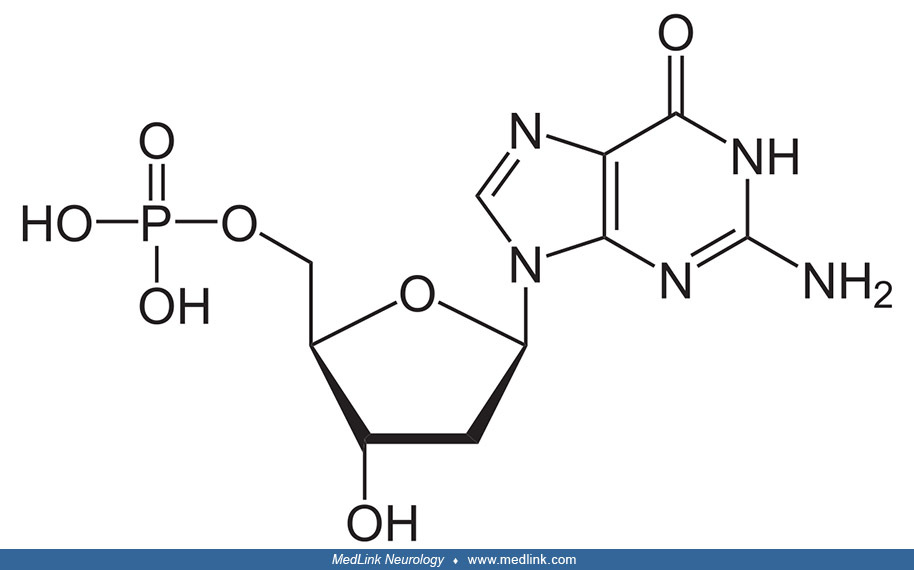
Addition of a phosphate group to the 5' carbon of deoxyribose in the deoxyribonucleosides 2-deoxyadenosine and 2'-deoxyadenoguanosine results in the formation of the deoxyribonucleotides deoxyadenosine monophosphate (dAMP; also known as deoxyadenylic acid or deoxyadenylate) and deoxyguanosine monophosphate (dGMP; also known as deoxyguanylic acid or deoxyguanylate), which are building blocks of deoxyribonucleic acid.
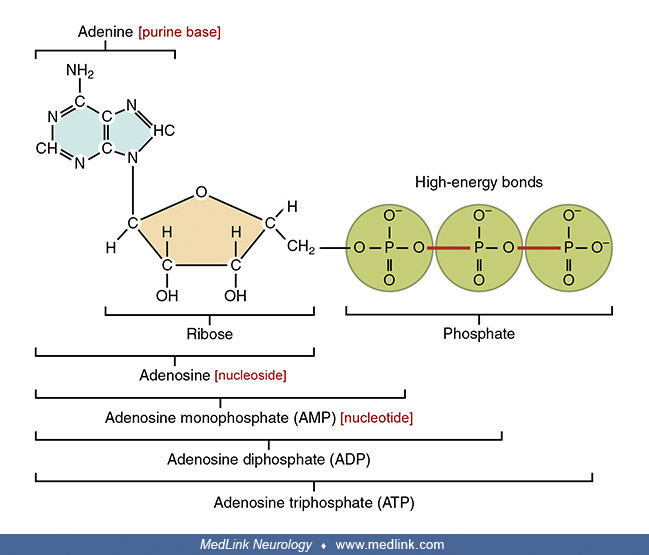
Examination of the structural components of adenosine triphosphate, the energy-carrying molecule that provides energy to drive many processes in living cells, will illustrate the components of nucleotides and nucleosides and how they are linked.
Adenylosuccinate lyase deficiency. Adenylosuccinate lyase deficiency, the first enzyme deficiency reported in the DNPS pathway in man, was discovered during a systematic study of amino acids in cerebrospinal fluid before and after acid hydrolysis (30). In three children with severe psychomotor retardation and autistic features, this procedure released abnormally large, equimolar amounts of aspartate and glycine. The additional identification by gas chromatography of an equimolar amount of ribose led to a search for purine compounds. Anion-exchange high-pressure liquid chromatography of deproteinized but not hydrolyzed cerebrospinal fluid, plasma, and urine revealed the presence of two UV-absorbing compounds that were undetectable in control samples: succinyl aminoimidazole carboxamide riboside (SAICA-riboside) and succinyl-adenosine (S-Ado). These succinylpurines are the products of the dephosphorylation by 5'-nucleotidases of succinyl aminoimidazole carboxamide ribotide (SAICAR) and succinyladenosine monophosphate (S-AMP, sometimes abbreviated SAMP), respectively.
Nearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125