


Epilepsy & Seizures
Hippocampal and parahippocampal seizures
Apr. 22, 2024
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Benign adult familial myoclonic epilepsy (or familial adult myoclonic epilepsy) is an inherited epileptic syndrome characterized by cortical hand tremors, myoclonic jerks, and rare convulsive seizures. In most affected individuals, the disease takes a benign course; however, at an advanced age, worsening of the tremor and myoclonus is common, and slight intellectual disability is present in a subset of patients. Prevalence is unknown but is estimated to be less than 1 in 35,000. It is transmitted autosomal dominantly with high penetrance, and anticipation has been noted in some families. This is a well-delineated disease with remarkable features that clearly distinguish it from other forms of myoclonic epilepsies. Genetic studies of the families show heterogeneity, and different susceptible chromosomal loci have been identified. Diagnosis is based on the clinical and electrophysiological findings. It must be differentiated from epilepsy syndromes with prominent myoclonus features. Valproate, levetiracetam, and benzodiazepines are the most beneficial treatments.
|
• Benign adult familial myoclonic epilepsy usually presents in the second decade of life with a mild hand tremor that is myoclonus-exacerbated by fatigue or emotional stress. Myoclonus usually appears around the same age and consists of erratic, arrhythmic, segmental jerks of the upper limbs heightened by posture and action. Rare or sporadic convulsive seizures are also a manifestation and are often precipitated by photic stimulation, emotional stress, and sleep deprivation. | |
|
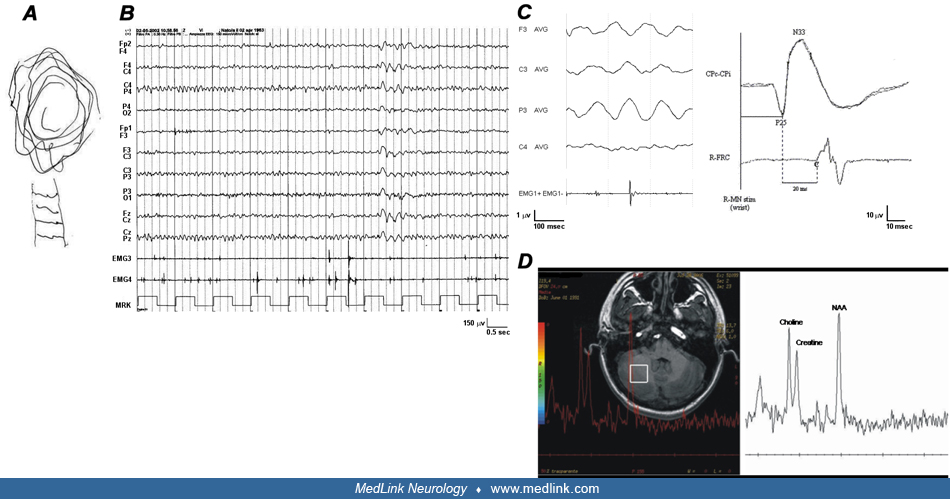
• Diagnosis is based on clinical and electrophysiological findings. EEG findings include a photomyoclonic response along with abnormality of polyspikes and waves. Patients also display extremely enlarged cortical components of somatosensory-evoked potentials and an enhanced C-reflex. Jerk-locked average analysis reveals positive-negative, biphasic spikes preceding myoclonus. | |
|
• Benign adult familial myoclonic epilepsy is transmitted autosomal dominantly, and it must be differentiated from idiopathic epilepsy syndromes with prominent myoclonus features (eg, juvenile myoclonic epilepsy syndrome) and from progressive myoclonus epilepsies. | |
|
• Valproate, levetiracetam, and benzodiazepines are the most beneficial treatments. Convulsive seizures are rare and usually responsive to therapy. |
The term “cortical tremor” was introduced by Ikeda and colleagues to indicate a postural and action-induced shivering movement of the hands mimicking essential tremor but showing the electrophysiological findings of cortical reflex myoclonus, that is: (1) brief EMG burst of about 50 msec duration; (2) no definite synchronization or reciprocity in antagonist’s muscles; (3) positive spikes preceding EMG bursts at the jerk-locked averaging; (4) enlarged cortical components of somatosensory-evoked potentials; (5) enhanced long-loop reflex I (or C-reflex) (21). Initially, it was likely conflated with other familial myoclonus epilepsies, especially progressive myoclonus epilepsies. As progressive myoclonus epilepsies became better understood clinically and genetically, this group began to stand out and was first recognized as such in Japan (02).
Uyama and colleagues first reported a patient with adult-onset fine finger tremulous movement, myoclonic jerks, and two generalized seizures coming from a family that was affected with the same condition through three generations with high penetrance (51). None of the patients showed other neurologic signs nor abnormal neuroradiological findings; the electrophysiological study indicated cortical reflex myoclonus. Subsequently, the same group reported four unrelated families, including 27 affected members spanning three generations, with high penetrance (50). In 1991, Yasuda used the term "benign adult familial myoclonic epilepsy" (BAFME) to describe two pedigrees in which affected members showed autosomal dominant hand tremor, myoclonus, and epileptic seizures with a nonprogressive course (58). Also, in these patients, electrophysiological studies showed evidence of cortical reflex myoclonus. In 1999, Plaster and colleagues and Mikami and colleagues mapped the disease on chromosome 8q24 (34; 37).
Although this condition was exclusively reported from Japan until the 1990s, several reports on pedigrees with similar clinical features but with different genetic identifiers appeared from different European countries and worldwide over the past decade with different names, such as autosomal dominant cortical myoclonus and epilepsy, familial adult myoclonic epilepsy, familial cortical myoclonic tremor, familial essential myoclonus and epilepsy, familial benign myoclonus epilepsy of adult-onset, and familial tremor and epilepsy. Despite phenotypic and genetic differences, the clinical and electrophysiological data point toward one syndrome (46; 47).
Genetic studies of families have revealed a large genetic heterogeneity with different identified loci, including 8q24 (FCMTE1), 2p11.1-q12.2 (FCMTE2), 5p15.31-p15.1 (FCMTE3), and 3q26.32-3q28 (FCMTE4), but a unique underlying causative mechanism, ie, pentameric intronic expansions (see section 4).
Nearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125