



Behavioral & Cognitive Disorders
Academic underachievement
Apr. 18, 2024
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Choroid plexus tumors of childhood are a relatively rare form of childhood brain cancer, comprising only 1% to 2% of all types of tumors that occur in patients younger than 18 years of age. Both choroid plexus papillomas and carcinomas can occur. Treatment of choice for both lesions is gross total resection. Patients with choroid plexus papillomas who have undergone complete resection rarely need alternative forms of adjuvant treatment. For patients with choroid plexus carcinomas, the need for adjuvant therapy following a gross total resection is unclear. Both radiotherapy and chemotherapy have shown questionable benefits for patients with subtotally resected choroid carcinomas. In this article, the author discusses some of the controversies in management and the genetic implications of a diagnosis of choroid plexus tumors.
• Choroid plexus tumors, although relatively rare, do make up a significant component of central nervous system tumors occurring in the first year of life. | |
• Separation of choroid plexus papillomas from atypical choroid plexus papillomas and atypical lesions from carcinomas is subjective, and the clinical course of atypical choroid plexus papillomas is less well characterized compared to other choroid tumor types. | |
• Degree of surgical resection is likely the single most important factor in determining outcome for all types of choroid plexus tumors. |
Choroid plexus tumors of childhood are a rare form of childhood neoplasm, constituting no more than 2% of all childhood primary central nervous system tumors (48). Although other types of central nervous system tumors may arise in the choroid plexus, the vast majority of childhood choroid plexus tumors are either papillomas or carcinomas (26). In Kernohan and Fletcher-Kernohan's 1937 study of 109 cases of ependymoma, choroid plexus papillomas were classified as a subvariety of ependymoma (31). This type of classification was widely used as late as the 1970s. In the most recent World Health Organization classification of tumors of the central nervous system, choroid plexus tumors are considered a distinct variety of neuroepithelial tumor and are subclassified as choroid plexus papillomas (Gr I), atypical choroid plexus papillomas (Gr II), or choroid plexus carcinomas (Gr III). Papillomas that are well-differentiated are relatively easy to categorize; however, some papillomas have increased cellularity, nuclear pleomorphism, and increased mitotic activity and may be classified as atypical choroid plexus papillomas (41). The updated WHO classification of tumors of the central nervous system continues the same classification (42; 43).
Choroid plexus carcinomas vary significantly in size at the time of diagnosis. Usually, children with choroid plexus papillomas present with signs and symptoms of increased intracranial pressure, such as vomiting, headaches, and unsteadiness (27). Because up to 70% of choroid plexus tumors occur in children less than 2 years of age, with a median onset of 9 months, macrocephaly is often present, especially in congenital tumors (30; 29). In a series of 18 intracranial tumors diagnosed in the fetal stage, 5 were either choroid plexus carcinomas (2) or papillomas (3). Three had associated hydrocephalus (07). Given the chronicity of the tumor, it is somewhat surprising that patients usually have a relatively abrupt onset of symptoms before diagnosis, most commonly ranging from 1 week to 2 months. The hydrocephalus in patients with choroid plexus papillomas is believed to be secondary to the overproduction of cerebrospinal fluid (37; 18; 66). There may also be secondary hemorrhage within the cerebrospinal fluid and increased ventricular dilatation due to subarachnoid reabsorption block.
The clinical symptomatology of children with choroid plexus carcinomas differs somewhat from that of children with choroid plexus papillomas (40). Carcinomas tend to be more locally invasive. Children with choroid plexus carcinomas frequently have focal neurologic deficits without hydrocephalus early in the illness. Children younger than 3 years of age commonly present with vomiting and lethargy, although there is often associated hemiparesis. Older children are more likely to have focal neurologic deficits, which are later associated with headaches or vomiting, but no frank evidence of increased intracranial pressure until later in the illness.
Metastatic spread occurs more commonly in choroid plexus carcinomas than in papillomas. Although such dissemination may be symptomatic, in the majority of cases of disseminated choroid plexus carcinoma, dissemination is asymptomatic and is found on staging studies.
A 9-month-old child was brought to his pediatrician because of a 1-month history of vomiting. On examination, the child looked somewhat pale, was lethargic, and was mildly dehydrated. The child had a head circumference of 48 cm, which is well above the 98th percentile. Both the anterior and posterior fontanelles were open and the anterior fontanelle was somewhat full. On neurologic examination, the child was arousable but irritable. Pupils were equal and reactive and the discs were slightly pale. Extraocular movements were full, except for some limitation in upgaze. The remainder of the child’s cranial nerve examination was normal. On motor and coordination testing, there was mild to moderate hypotonia. The child, who was sitting by 6 months of age, could no longer sit. The child was supporting weight poorly. Reflexes were diffusely increased. On CT scan, a lobulated mass was found in the right lateral ventricle. The mass was hyperdense in comparison to the normal surrounding brain and enhanced homogeneously. There was marked associated hydrocephalus, with dilatation of both the lateral and third ventricle. On MRI, the mass appears papillary with relatively well-defined margins. At surgery a well-circumscribed choroid plexus papilloma was resected. The child had an intraventricular bleed following surgery and required 7 days of extraventricular drainage. The drain was removed and the hydrocephalus resolved without need for a ventricular peritoneal shunt.
Carcinomas may occur as part of the familial Li-Fraumeni syndrome, which is an autosomal dominant disorder caused by a germ-line mutation in the TP53 gene on chromosome 17p13 (45). Li-Fraumeni syndrome is a cancer predisposition syndrome manifest by germline mutations in the TP53 cancer suppressor gene. In 1 series of 42 children, 6 (16.7%) had phenotypic or genotypic characteristics consistent with Li-Fraumeni syndrome. Four of 11 with choroid plexus carcinomas were positive for TP53 germline mutations (21). Another series also found an association with TP53 germline mutations and choroid plexus carcinomas (19).
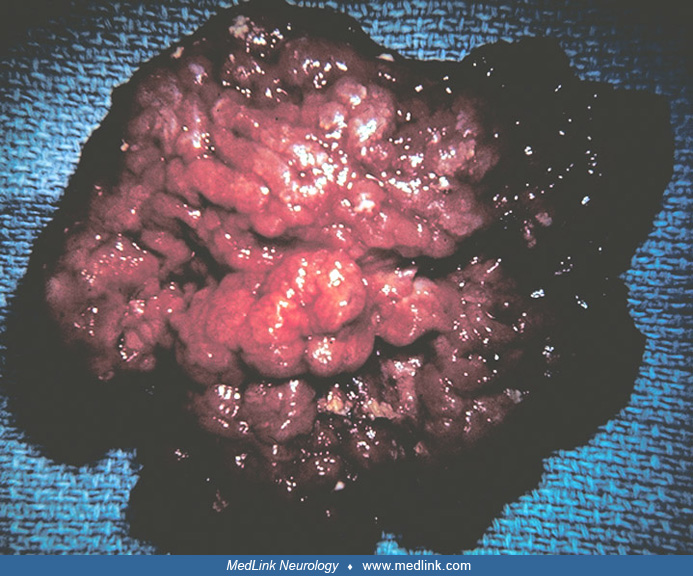
The choroid plexus papilloma is an epithelial tumor arising in the choroid plexus of the cerebral ventricles, composed of simple or pseudostratified layers of cuboidal or columnar cells, resting on a basement membrane, overlying papillary, vascularized connective tissue cores (58).
It may be difficult to distinguish a papilloma from the more orderly normal choroid plexus, or from a papillary ependymoma on a small biopsy. Cytological atypia, occasional mitoses, and less commonly, foci of necrosis may be present. Demonstration of immunoreactivity to transthyretin may aid in the differential diagnosis. The World Health Organization classification recognizes an oncocytic variant (32). The classification makes no comment on malignant choroid plexus papillomas and uses the term "choroid plexus carcinoma" to designate a more malignant form of tumor. Choroid plexus papillomas are believed to cause ventricular dilatation due to overproduction of cerebrospinal fluid, although a partial block in cerebrospinal fluid reabsorption may also be present.
The choroid plexus carcinoma is a choroid plexus tumor with histological evidence of anaplasia, including increased mitotic activity, nuclear atypia, loss of papillary differentiation with transition to pattern-less cellular sheets, and necrosis (32).
In adults, a rare variant may be difficult to distinguish from metastatic carcinoma to choroid plexus. Metastatic spread of nonprimary central nervous system tumors to the brain is rare in childhood.
The molecular biology of choroid plexus tumors is being elucidated. Transgenic mouse models have increased the understanding of choroid plexus carcinomas. The tumors were developed by activating the MYC oncogene and deleting the tumor TP53 suppressor gene in murine neural stem cells. These tumors resembled human choroid plexus carcinomas and exhibited multiple whole chromosomal losses (68).
Unique molecular signatures distinguish choroid plexus carcinomas from papillomas, including atypical choroid plexus papillomas; there are no clear molecular differences between papillomas and atypical papillomas (51). The majority of choroid plexus carcinomas harbor TP53 mutations, and those tumors that have 2 mutant copies are the most likely to be aggressive and result in poorer survival (51). Notch 3 signaling, the transcription factor TWIST1, platelet-derived growth factor receptor, and the tumor necrosis factor-related apoptosis-inducing ligand pathway have also been implicated in papilloma tumorigenesis (59). Molecular analysis has also shown that some choroid plexus carcinoma tumors, especially tumors of infancy, have been misdiagnosed as embryonal tumors (61).
Immunoreactivity for cytokeratin, vimentin, S-100 protein, transthyretin, and carboxyanhydrase, but not for epithelial membrane antigen, typifies choroid plexus carcinomas (16; 09; 20). The use of transthyretin as an immunohistochemical marker has been a significant advance in the diagnosis of choroid plexus tumors, especially in the distinction of primary choroid plexus tumors from other tumors that may invade the choroid plexus. Transthyretin is a transport protein for thyroxin and retinol. Transthyretin is believed, in theory, to be synthesized by the choroid plexus epithelium, and is, thus, useful as a marker for a primary choroid plexus neoplasm. In 1 review of 52 patients, the absence of transthyretin in tumor cells of patients with choroid plexus tumors was associated with a poorer prognosis (20). Other factors associated with a poorer prognosis included decreased immunoreactivity with the S100 protein, brain invasion by neoplastic cells, absence of stromal edema, and necrotic areas (04). The prognostic significance of atypical features of choroid plexus papillomas remains uncertain. In a series of 164 patients, elevated mitotic activity (more than 2 mitosis per 20 high-power field) was the sole feature independently associated with a higher likelihood of recurrence (28).
Methylation profiling has demonstrated 3 distinct subgroups: cluster 1 consisting of low risk tumors that contained primarily choroid plexus papillomas and atypical papillomas occurring in young children, primarily in the supratentorial space and having an excellent prognosis; cluster 2 arising in adulthood with predominant infratentorial origin and intermediate to excellent prognosis; and cluster 3, also found most commonly in the supratentorial space of young children, consisting of choroid plexus carcinomas, papillomas, and atypical papillomas. Cluster 3 may demonstrate a p53 abnormality, and those with p53 abnormalities have the poorest prognosis (63).
Furthermore, choroid plexus carcinomas clustered on DNA methylation profiles with homozygous TP53 mutations separately from those with heterozygous TP53 mutation or wild type TP53 and had the worst prognosis (56; 64).
In a proteogenomic characterization, choroid plexus tumors clustered in an “epithelial” cluster and demonstrated upregulation of immune-related pathways, as well as CTLA4 and PD-1 molecules (55).
Choroid plexus tumors make up less than 2% of all childhood brain tumors. The choroid plexus papilloma outnumbers the choroid plexus carcinoma by a ratio of 4 to 1. Seventy percent or more of childhood choroid plexus tumors occur in children less than 2 years of age, with a median age of onset of diagnosis ranging from 7 to 17.5 months (14; 50; 35; 13; 10). Carcinomas are more likely to be diagnosed before 5 years of age than papillomas (10).
Choroid plexus papillomas in childhood primarily arise in the lateral ventricles. Fourth ventricular choroid plexus papillomas are more common in adults. Choroid plexus carcinomas arise primarily in the lateral ventricles, but may also arise in the fourth ventricle, or in the third ventricular region. In 1 series, the majority of patients with carcinomas were less than 3 years of age at the time of diagnosis, although 3 of 11 children were greater than 7.5 years at the time of detection.
The risk factors for development of choroid plexus papillomas and carcinoma are unknown.
The primary differential diagnosis in the evaluation of a choroid plexus tumor is primary separation of a carcinoma from a papilloma, although embryonal tumors can be confused with carcinomas (61). As stated previously, papillomas more commonly present with hydrocephalus without localizing neurologic signs. Carcinomas are more likely to present with localizing neurologic deficits, due to invasion into brain parenchyma. Carcinomas may or may not cause hydrocephalus, depending on their location within the central nervous system. Symptoms of increased intracranial pressure are more commonly due to the mass effect caused by the lesion and surrounding edema.
In adults, metastatic lesions, such as breast and lung cancer, may spread to the choroid plexus and cause clinical confusion. However, such metastatic deposits in childhood are rare. Differentiation of choroid plexus papillomas from ependymomas may be difficult, especially on biopsy. However, with the use of immunohistochemical markers, such as transthyretin, such a distinction is usually possible.
CT and MRI, which often may be complementary, are the primary components of the diagnostic workup of a child with a presumed choroid plexus carcinoma or papilloma. Neuroimaging of the entire craniospinal axis is indicated after detection for both tumors, although, the incidence of dissemination is much higher in children with choroid plexus carcinomas than in patients with papillomas (53).
CT and MRI are essentially the only tests needed to diagnose a choroid plexus tumor. Plain skull x-rays, although often abnormal, are nonspecific. Plain skull x-rays usually show spreading of the cranial sutures and an enlargement of the cranial vault. Presence of mottled calcifications within the ventricular pathway is highly suggestive of a papilloma, but is only seen in a minority of patients. CT studies of choroid plexus papillomas usually demonstrate well-marginated, smooth or lobulated masses filling the lateral ventricles (65). The mass is usually isodense or hyperdense, does not seem to invade the surrounding brain parenchyma, and tends to enhance homogeneously.
There is usually significant hydrocephalus and there may or may not be peritumoral edema. On MRI, choroid plexus papillomas appear as papillary or cauliflower-like tumors with well-defined margins. On T1-weighted images, the tumor may be isointense, in comparison to brain parenchyma. The lesion tends to show marked contrast enhancement and there may be associated foci of necrosis or hemorrhage. Calcification is usually better appreciated on CT than on MRI.
Invasion of choroid plexus carcinomas into the surrounding brain is usually demonstrated by both CT and MRI. Peritumoral vasogenic edema is usually present. The typical appearance of a choroid plexus carcinoma on CT is that of an isodense to hyperdense heterogeneous mass. After contrast injection, the solid portion of the tumor usually shows intense homogeneous enhancement. Calcifications and cysts may be present. On MRI, choroid plexus carcinomas are typically hypointense on T1-weighted images. The solid portion of the tumor tends to enhance intensely and homogeneously with contrast. On T2-weighted images, the solid portion of the tumor may have associated cystic areas or necrotic regions. Although not essential for diagnosis, arteriography may be valuable as part of an intravascular embolization procedure to reduce blood flow to the tumor, prior to surgical excision.
Because of the presence of hydrocephalus in patients with papillomas and increased intracranial pressure, due to local tumor invasion and associated vasogenic edema in patients with carcinomas, cerebrospinal fluid usually cannot be sampled prior to surgical intervention. In patients with both papillomas and carcinomas, cerebrospinal fluid protein will be elevated. Children with papillomas also frequently have hemorrhagic cerebrospinal fluid due to the vascular nature of the tumor. Cerebrospinal fluid is usually obtained following surgery in patients with carcinomas for evaluation of presymptomatic tumor spread.
The management of choroid plexus papillomas is gross total surgical resection, when possible (22; 24; 23; 08; 52; 44). The complications of such resection have already been reviewed, including the possibility of death following surgery, due to tumor hemorrhage, hydrocephalus, or the development of postoperative subdural hematomas. Brain shift, secondary to acute CSF drainage, has been recognized a significant factor in morbidity (34). There is no evidence that postoperative radiation improves survival of children after gross total resection. Radiotherapy has been used in children with subtotally resected choroid plexus papillomas, and some have used radiation following initial attempts at surgery, to shrink the tumor so that a second surgical resection could be attempted. The benefits of radiotherapy in treating subtotally resected tumors, or in shrinking disease prior to an attempt at surgical resection are poorly documented. The reported experience with chemotherapy is essentially nil.
Although choroid plexus carcinomas are malignant invasive tumors, surgery remains the primary modality of treatment (14; 54; 25). In various reviews, children with total surgical resections have fared much better than those with partial resections. In 1 review, 11 of 14 children had prolonged disease-free survival after gross total resections, only 2 of who received adjuvant therapy. By contrast, only 2 of 20 children with choroid plexus carcinomas were reported to be alive and free of progressive disease with less than total resections, independent of the type of adjuvant therapy received.
Although radiation therapy has been recommended for children with choroid plexus carcinomas after gross total resection, there is little proof of its benefit. In children with partially resected tumors, the benefits of radiation therapy are also questionable (03). There is some evidence that patients treated with radiotherapy after subtotal resections experience longer disease remissions, but there is no evidence that radiation therapy is curative (69). Stereotactic radiosurgery has also been used with possible success (15).
The role of chemotherapy for choroid plexus carcinomas is far from settled (47; 01; 11; 69; 35; 03). A variety of different chemotherapeutic agents have been used, including a 4-drug regimen of cyclophosphamide, vincristine, cisplatinum, and VP-16. A subgroup of infants treated with this regimen did well; however, the majority who fared well had gross total resection prior to the initiation of treatment (12; 08). A meta-analysis of case reports suggests that chemotherapy may be of some benefit (71); however, there is no consensus over what chemotherapy regimen is best. In a registry study of 35 children, retrospective analysis of nonrandomized patients suggested a carboplatin/etoposide regimen was superior to cyclophosphamide and etoposide dual treatment (70). High-dose chemotherapy with autologous hematopoietic stem cell rescue has also been used with some benefit (72). Loss of TP53 expression may result in increased resistance to anticancer agents (33).
Because of the relatively high incidence of TP53 mutations and association of Li-Fraumeni syndrome, some have suggested that all patients with choroid plexus carcinomas be tested for germline mutations. In those testing positive, a possible approach is to screen all siblings, especially early in life, for development of intracranial tumors and to also perform more frequent screening in adult relatives for development of related malignancies such as breast, colon, and lung tumors (21). It has yet to be proven that earlier detection will improve overall outcome.
The overall 5-year prognosis for children with choroid plexus papillomas has improved. In early series, evaluating patients treated primarily before 1970, a surgical mortality rate as high as 30% was reported (67; 57; 02). Surgical mortality has fallen in series reviewing patients treated in the 1970s and 1980s (38). In a French series of 54 children with choroid plexus papillomas, 5-year survival was 100%; for 26 children with atypical papillomas, it was 96.2% (60). Because the majority of children with choroid plexus papillomas have significant hydrocephalus at the time of diagnosis, part of the surgical morbidity is associated with the development of subdural hematomas, secondary to collapse of the ventricular system after cerebrospinal fluid drainage or tumor removal. In addition, choroid plexus papillomas are highly vascular, and secondary hemorrhage within the brain after tumor removal may occur. In patients with gross total resections of choroid plexus papillomas who survive the postoperative period, overall survival is excellent. The vast majority of children who survive surgery and have a total removal of the tumor are cured of their lesion (44). A variety of surgical approaches have been used, including an open transcortical approach (36; 49). In a SEER registry review, with current treatment, over 95% of children with papillomas were found to be alive (10). The proliferative index of choroids plexus tumors may be somewhat predictive of outcome (04). In addition, methylation of the telomerase reverse transcriptase (TERT) promoter may portend poorer prognosis or transformation from a papilloma to a carcinoma (06). Stromal invasion does not signify a poorer prognosis (39). The quality of life of children after gross total resection is less than optimal, especially in extremely young children with choroid plexus papillomas. Complications include hydrocephalus with subdural collections, mental retardation, and motor impairments.
The prognosis of children with choroid plexus carcinomas is poorer than those of children with papillomas (05), with overall survival at 5 years of 65% in a series of 22 children (60). However, in this series, relapses were frequent, so event-free survival was only 25% at 5 years. The majority of reported children who have undergone partial resections have died of disease progression (54; 08; 69). In contradistinction, over two-thirds of children treated with gross total resections are alive and free of disease 5 years following treatment (14; 54; 17; 62; 10; 46; 25). Children with choroid plexus carcinomas may have significant residual neurologic compromise, especially with tumors that have invaded eloquent areas of the brain. These sequelae include hemiparesis, seizure disorders, and cognitive dysfunction.
Because the vast majority of children with choroid plexus papillomas and carcinomas have increased intracranial pressure at the time of diagnosis, anesthesia techniques for patients with increased intracranial pressure must be used.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Roger J Packer MD
Dr. Packer of Children’s National Medical Center and George Washington University has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Behavioral & Cognitive Disorders
Apr. 18, 2024
Behavioral & Cognitive Disorders
Apr. 17, 2024
Developmental Malformations
Apr. 14, 2024
Developmental Malformations
Apr. 09, 2024
General Child Neurology
Apr. 05, 2024
General Child Neurology
Apr. 01, 2024
General Child Neurology
Mar. 29, 2024
General Child Neurology
Mar. 29, 2024