








Abnormalities of tetrahydrobiopterin metabolism
Apr. 07, 2024
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Complex II deficiency results from reduction in activity of the mitochondrial enzyme succinate-ubiquinone oxido-reductase. This enzyme complex participates in the mitochondrial electron transport chain to generate ATP and serves in the Krebs cycle. Abnormalities of complex II activity are associated with a wide range of phenotypes. Most affected individuals have an encephalomyelopathy, with various degrees of CNS, skeletal muscle, and cardiac muscle involvement. CNS involvement includes microcephaly, seizures, ataxia, spasticity, developmental delay, psychiatric abnormalities, optic atrophy, oculomotor abnormalities, and radiographic abnormalities compatible with Leigh disease or leukodystrophy. Initial workup includes blood pH, lactate, pyruvate, creatine phosphokinase, and liver enzymes. Final diagnosis depends on muscle biopsy.
|
• Complex II deficiency is caused by a reduction in activity of the mitochondrial enzyme succinate-ubiquinone oxido-reductase. | |
|
• This enzyme complex participates in the mitochondrial electron transport chain to transfer reducing equivalents from succinate to coenzyme Q (as part of the process of generating ATP) and serves a dual role in the Krebs cycle (to convert succinate to fumarate). | |
|
• Abnormalities of complex II activity are associated with a wide range of phenotypes. | |
|
• Most affected individuals have had an encephalomyelopathy, with various degrees of central nervous system, skeletal muscle, and cardiac muscle involvement. | |
|
• Central nervous system involvement has included microcephaly, seizure disorder, ataxia, spasticity, developmental delay, psychiatric abnormalities, optic atrophy, oculomotor abnormalities, and radiographic abnormalities compatible with Leigh disease or leukodystrophy. | |
|
• Initial diagnostic workup of persons with complex II deficiency should include an examination of the blood pH, lactate, pyruvate, creatine phosphokinase, and liver enzymes. Final diagnosis is dependent on muscle biopsy. |
The electron transport chain, located in the inner mitochondrial membrane, is the site of oxidative phosphorylation, the metabolic pathway in which cells use enzymes to oxidize nutrients, thereby releasing energy that is used to reform adenosine triphosphate (ATP).
In this pathway, the NADH and succinate generated in the Krebs citric acid cycle are oxidized, releasing energy to power ATP synthase.
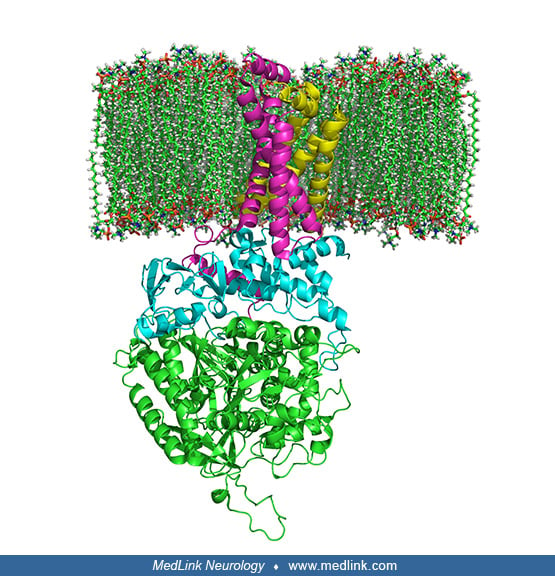
Within mitochondria, complex II links the two key cellular energy-conversion pathways: complex II serves as succinate dehydrogenase (SDH) in the Krebs cycle, and as succinate ubiquinone oxidoreductase, one of five complexes of the mitochondrial oxidative phosphorylation system. In particular, complex II couples the oxidation of succinate to fumarate in the citric acid cycle with the reduction of ubiquinone (coenzyme Q) to ubiquinol in the electron transport chain.
Complex II is a heterotetramer (ie, is protein containing four noncovalently bound subunits, wherein the subunits are not all identical) with four dissimilar subunits: SdhA, SdhB, SdhC, and SdhD.
Two of these subunits are hydrophilic (SdhA and SdhB) and two are hydrophobic membrane anchor subunits (SdhC and SdhD). SdhA contains a covalently attached flavin adenine dinucleotide (FAD) cofactor and the succinate binding site. Human mitochondria contain two distinct isoforms of SdhA (Fp subunits type I and type II). SdhB contains three iron-sulfur clusters: [2Fe-2S], [4Fe-4S], and [3Fe-4S].
The subunits form a membrane-bound cytochrome b complex with six transmembrane helices containing one heme b group and a ubiquinone-binding site.
Two phospholipid molecules, one cardiolipin and one phosphatidylethanolamine, are also found in the SdhC and SdhD subunits in the hydrophobic space below the heme b.
Although all the elements of the respiratory chain function in the mitochondria, many of the subunits are encoded genetically in the nucleus and imported into the mitochondria. Complex II is made of four subunits (A, B, C, D), all of which are autosomally encoded (ie, encoded in the nucleus) by the succinate dehydrogenase (SDH) genes (SDHA, SDHB, SDHC, and SDHD). Other factors and cofactors needed for complex II activity are riboflavin, iron, cytochrome b560, and ubiquinone (coenzyme Q). Complex II activity is often measured together with complex III activity as succinate cytochrome C reductase activity.
Isolated complex II deficiency was first inferred in two siblings with mental retardation, seizures, myoclonus, and ataxia (48). Since that report a variety of other phenotypes have been reported, including Leigh disease (40; 13), leukodystrophy (03; 45), encephalomyopathy (29), Kearns-Sayre syndrome and other mitochondrial myopathies (49; 52), rhabdomyolysis and hemolytic uremic syndrome (42), cardiomyopathy with variable associated neurologic manifestations (psychiatric abnormalities, optic atrophy, oculomotor abnormalities, progressive polyneuropathy) (04; 52; 02; 14), and late-onset progressive neurodegeneration (54). In persons of northern Swedish descent, a myopathic phenotype due to a combination of complex II, complex III, and aconitase deficiencies has been described (24). Complex II (alone or in combination with other elements of the mitochondrial oxidative phosphorylation system) has also been reported to be abnormal in many neurodegenerative disorders, but these biochemical findings are most likely secondary events.
Nearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125