



Developmental Malformations
Walker-Warburg syndrome
Apr. 14, 2024
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
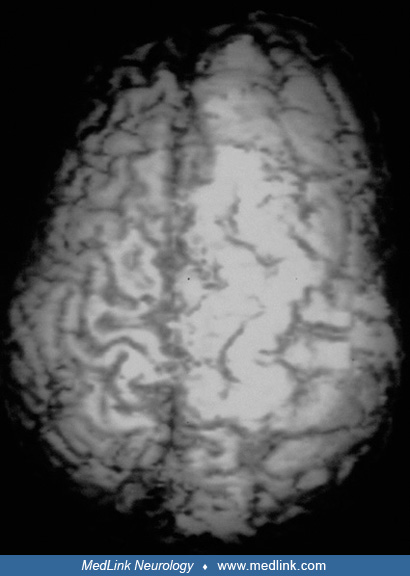
Hemimegalencephaly is a rare central nervous system disorder of neuronal cell lineage, proliferation, maturation, and migration characterized by in utero enlargement of all or most of one cerebral hemisphere. The clinical hallmark is early-onset intractable epilepsy with associated hemiparesis and developmental delays. Hemimegalencephaly may occur in isolation or within the context of a defined syndrome. Occasionally, unilateral cerebral enlargement may also involve the ipsilateral brainstem and cerebellum, as well as rare hypertrophy or overgrowth of half of the face or body. Epilepsy is usually refractory to antiepileptic medications, and cerebral hemispherectomy or disconnection surgery is often a treatment of choice. Outcomes vary depending on the extent of neural dysplasia, association with a neurocutaneous syndrome, and surgical intervention. Currently, pharmacologic, surgical, and early developmental interventions remain at the forefront of long-term treatment strategies. The etiology and pathogenesis have been shown by neuropathological and genetic studies to be a postzygotic somatic mutation involving gain of function in the mammalian target of rapamycin (mTOR) signaling pathway and related genetic and metabolic pathways, such as AKT/PIK3CA. The extent of the cerebral lesion is determined by the timing of the onset of genetic expression in embryonic and fetal life. Hemimegalencephaly is part of a continuum in the spectrum of focal cortical dysplasia type IIb. It is a frequent complication of the two neurologic phenotypes of epidermal nevus syndromes and occasionally of tuberous sclerosis complex.
• Hemimegalencephaly is a disorder characterized by the enlargement of one hemisphere or side of the brain and abnormal cellular structure with megalocytic, dysplastic neurons and balloon cells. | |
• Patients can present from birth to young adulthood but can be diagnosed prenatally or in the neonatal period. | |
• Common presenting symptoms include hemiparesis, hemianopia, intractable epilepsy, cognitive impairment, and global developmental delay. | |
• Hemimegalencephaly may be associated with neurocutaneous syndromes, particularly the keratinocytic and sebaceous types of epidermal nevus syndromes, and occasionally with tuberous sclerosis complex. | |
• Hemimegalencephaly is both neuropathologically and genetically part of the spectrum of focal cortical dysplasia type IIb as a disorder of the mTOR signaling pathway, with onset of the postzygotic somatic mutation earlier in neuroepithelial mitotic cycles than in the smaller, more localized lesions of focal cortical dysplasia type II. | |
• As with other mTOR disorders, phosphorylated tau protein is upregulated in hemimegalencephaly. | |
• Balloon cells are common to focal cortical dysplasia type IIb, hemimegalencephaly, and cortical tubers of tuberous sclerosis complex. | |
• Epilepsy is often refractory to medical control, but early surgical intervention for intractable epilepsy may lead to improved epileptic and developmental prognoses. |
First described by Sims in 1835, hemimegalencephaly is a rare central nervous system disorder of neuronal proliferation and migration characterized by congenital enlargement of all or most of one cerebral hemisphere (142; 43; 86; 17; 151; 44; 98; 93; 06). Clinically, hemiparesis, developmental delays, and intractable seizures are characteristic. In some cases, unilateral cerebral enlargement may also involve brainstem and cerebellum, creating the appearance that two brains of different sizes had been joined in the midline (62). Involvement of the infratentorial neural structures is now known as “total hemimegalencephaly” and is explained by somatic mutations in an early mitotic cycle of the neuroepithelium.
The term “hemimegalencephaly” was coined by Gross and Uiberrak, who performed and published the autopsy of an innocent 7-year-old German girl named Heide who had epilepsy, cognitive impairment, and a facial keratinocytic nevus ipsilateral to the dysplastic cerebral hemisphere (58). However, there is an ethical issue because she did not die of natural causes but was euthanized in a mental institution in Vienna in 1944. She had a form of epidermal nevus syndrome (triad of facial verrucous nevus, ipsilateral hemifacial lipomatosis, and ipsilateral hemimegalencephaly) that is now called “Heidi syndrome” (49; 45; 48). A recounting of the inhumane Nazi medical euthanasia program and other Nazi physicians are well documented by Ronen (122).
Patients with hemimegalencephaly are divided into two groups: one in which hypertrophy is localized to the CNS (isolated) and the other when associated with neurocutaneous syndromes. In one phenotype, hyperplasia may involve the face ipsilaterally (Heide syndrome) or other parts of the body (33; 95; 152; 47; 49). Most early descriptions of hemimegalencephaly were of patients with both brain and somatic hemihypertrophy (161; 63; 125). Thus, comprehensive history and examination remain the foundation of evaluation of any patient with apparent hemimegalencephaly.
Epilepsy, which develops in nearly all children with hemimegalencephaly, is typically refractory to medical management (119; 107) and resective or cerebral disconnection surgery; hemispherectomy remains the treatment of choice in many cases (154; 140; 36; 91; 128; 90; 75; 160; 162). Anatomic hemispherectomy (AH) was first introduced by Dandy in 1928 for the treatment of malignant gliomas and was expanded for the treatment of seizures by McKenzie in 1938 (09). This was initially deemed a last resort for patients with “catastrophic epilepsy,” an uncommon subset of seizure disorders marked by uncontrollable seizures and severe cognitive deficits. Later, Krynauw employed anatomic hemispherectomy in cases of intractable seizures in patients with unilateral CNS abnormalities (80). Although seizure reduction was noted in the first observations following the anatomic hemispherectomy procedure, complications such as hemorrhage, hemosiderosis, and hydrocephalus minimized its utility for the treatment of epilepsy. Today, modern variants, including functional hemispherectomy, first introduced by Rassmusen in 1978, have minimized complications while still achieving comparable seizure outcomes (150; 113; 36). Motor deficits remain unchanged postoperatively, and, in some cases, cognitive functions may improve.
Epilepsy, development and cognitive delays, and contralateral hemiparesis are hallmarks of hemimegalencephaly, with symptoms typically presenting during the neonatal period but can have variability in its clinical presentation (137). Patients with the onset of epilepsy after the neonatal period or in early infancy may exhibit subclinical epileptiform activity on EEG from birth. Seizure onset is often the first manifestation of hemimegalencephaly with earlier seizure presentation classically linked to increased severity of seizures in this patient population, with many having uncontrollable seizures and catastrophic epilepsy (82). Most infants have focal seizures involving the arm and leg contralateral to the side of hemimegalencephaly (107). In some cases, both epilepsia partialis continua (155; 51; 45) and secondary generalization occur over time in children with hemimegalencephaly. Other seizure types, such as infantile spasms and atonic seizures, may occur in addition to focal seizures (18; 148; 107; 156; 118). Hemimegalencephaly may also present as an infantile myoclonic epilepsy (16; 105; 114), Ohtahara syndrome (13), or later evolve to Lennox-Gastaut syndrome (83).
Macrocephaly and cranial asymmetry is apparent during the neonatal period, but commonly missed if the skull is not examined carefully and viewed from the vertex (157). Various degrees of hemiparesis are present, as a result of both malformation of the involved motor cortex and frequent focal motor seizures. Hemianopsia may also be detected in older children. Localized asymmetrical hypertrophy of the extremities as well as hemimegalencephaly are characteristic of Proteus syndrome, a keratinocytic variety of epidermal nevus syndrome caused by somatic mutation of the AKT1 gene affecting mTOR (88; 45). Facial asymmetry is due to infiltrative lipomatosis, which is distinct from and more severe than encapsulated lipoma (49; 45; 129).
Milder symptoms and later onset of seizures may occur presumably due to less severe cytoarchitectural abnormalities (155; 114; 29), especially if hemimegalencephaly occurs in the context of neurofibromatosis or tuberous sclerosis complex (32; 23; 126). Rarely, early developmental motor and cognitive function may be normal (52; 29).
Rarely, an adult with undiagnosed hemimegalencephaly was found by imaging ordered for another reason, such as recurrent migraine headaches (89), though migraine is probably unrelated.
The prognosis of children with hemimegalencephaly is variable given the variable extent of neuronal malformation seen across the spectrum of this disease (152). However, it has been noted that the appearance of seizures prior to 1 month of age foreshadows a poor prognosis (148; 73), whereas seizures beginning after the age of 7 portend a better functional outcome. Additionally, status epilepticus may be a cause of early death (76; 119). Patients with hemimegalencephaly surviving to adulthood without hemispherectomy are uncommon (66). For patients who are not good surgical candidates or decline surgical intervention, relative seizure control may be achieved through medical management, but seizure freedom is rarely achieved. Ikeda and colleagues reported one case of hemimegalencephaly in a 26-year-old-male with severe epilepsy, albeit poorly controlled, who was managed medically; however, these cases are most often associated with a milder seizure disorder and are rarely reported in the literature (66).
A Caucasian girl presented to the neurology clinic at age 2 years with a nonprogressive right hemiparesis. Parents had noted weakness of her right hand greater than leg since shortly after birth. She was developmentally delayed, most notably with delayed walking at 15 months and delayed expressive language, and she had been receiving physical and speech therapies. Exam showed an alert, interactive child without neurocutaneous markers or neurometabolic odors. Head circumference was at the 75th percentile for age, with noted cranial asymmetry, left hemicranium much larger than the right. Right spastic hemiparesis was demonstrated with hemiparetic gait, decreased muscle power (4-/5), increased tone, and increased deep tendon reflexes in the right arm greater than the right leg. Further, family and social histories were negative, and no history of seizures was reported. Brain MRI was consistent with left hemimegalencephaly. Two weeks after MRI, she presented to her local emergency room in focal status epilepticus. AED therapy was begun. She was started on phenytoin but was soon changed to oxcarbazepine. Epilepsy surgery planning was started. Right focal seizures persisted, and she was transitioned to topiramate. Continued seizures persisted despite addition of valproate. Within 2 months of initial seizure, left hemispherectomy was successfully performed. The patient has been postoperatively seizure-free for 5 years and continues to make substantial neurodevelopmental gains.
Hemimegalencephaly is a postzygotic somatic mutation in undifferentiated premigratory neuroepithelial cells, with involvement of the mTOR signaling pathway or related pathways that influence mTOR, such as AKT (87; 134; 101). This pathogenesis has been shown neuropathologically and has been confirmed as etiology in genetic studies (31; 08; 37; 38; 69). Small focal cortical dysplasias exhibit somatic mutations only in neurons; large lesions involve glial cells as well. Dysplastic clones disperse across the cortex at relatively low levels of mosaicism, and small lesions involve neuroblasts only; larger lesions involve glial cells as well (120). Increased levels of nonphosphorylated beta-catenin occur in hemimegalencephaly, which transcriptionally activates cyclin D7 and c-myc genes but reduced levels of Ser33/Ser37/Thr41 phospho-beta-catenin, which is essential for beta-catenin inactivation (169).
Neuropathology. Timing is the essence of determining the size of the lesion because hemimegalencephaly is within the same spectrum as focal cortical dysplasia type IIb (133; 135). Neuropathological confirmation has been made in the midgestation fetus (92). There are approximately 33 mitotic cycles of the human ventricular neuroepithelium that produce all neurons of the cerebral cortex because of logarithmic exponential increase (24). If the somatic mutation, which involves some but not all neuroepithelial cells, is first expressed in a late cycle, a small lesion of focal cortical dysplasia is produced. A somewhat earlier onset yields a wider lesion of several adjacent gyri, and an even earlier onset produces hemimegalencephaly. If the onset is in the first few mitotic cycles, when the neuroepithelium of the supratentorial and infratentorial compartments are still continuous, the result is “total hemimegalencephaly” that not only involves the cerebral hemisphere but also the ipsilateral cerebellar hemisphere and hemi-brainstem (133; 135).
Thus, hemimegalencephaly is characterized by excessive proliferation of both neurons and astrocytes. The principal lesion is histologically classified as a hamartoma because it involves not only disorganization of tissue architecture but abnormal cellular morphology as well.
Involved cortex is thickened, with a flattened inner cortical border, associated with a lack of lamination. There are an increased number of miniature, shallow gyri over the cortical surface, and in some places adjacent microgyri fuse and the cortical surface appears paradoxically smooth (11; 46; 50).
In addition to diffuse enlargement of the cerebral cortex, there is typically dilatation of the ipsilateral occipital horn of the lateral ventricle (46) and expansion of the hemisphere beyond the midline.
Neuropathological findings also include abnormal neuronal growth and cytomorphology in both gray and white matter (50), abnormal neuronal lamination (86; 34; 06), abnormal neuronal orientation (58), and neuronal and leptomeningeal glioneuronal heterotopia (103; 92). Note that “heterotopia” is the plural form of this Greek derivative; the singular form is “heterotopion.”
No abnormality is known in the density of synapses, although there is an increased number in a given cytoarchitectural area because of increased cortical thickness (106). Most cases are also characterized by gliosis (148; 155).
Balloon cells are large globoid cells with eccentric nuclei and coarse, short radiating processes and are of mixed cellular lineage or incomplete differentiation, expressing both neuronal and glial proteins as well as primitive proteins of progenitor cells (168; 131). Balloon cells are immunoreactive for the mTOR substrates phospho-ribosomal S6 protein, similar to cells in tuberous sclerosis (15). Occasionally, however, the cortical dysplasia appears mild (34). Balloon cells are common in focal cortical dysplasia type IIb, hemimegalencephaly, and tuberous sclerosis complex (151; 119; 34; 130; 131).
Immunoreactivity studies have demonstrated that such abnormal neurons in the white matter demonstrate interactions with synaptophysin-reactive axons and are not “isolated islands” of neuronal heterotopia. Such neurons may also contribute to the epileptogenicity associated with clinical hemimegalencephaly (50).
An additional neuropathological lesion demonstrated in both focal cortical dysplasia type II and hemimegalencephaly is a cortical and hippocampal accumulation of an abnormally phosphorylated tau protein similar to that demonstrated in adult Alzheimer disease and certain other adult neurodegenerative diseases with dementia (134; 132). Furthermore, there is a neutral lipid storage in many cortical neurons in at least some cases of hemimegalencephaly, the significance of which remains uncertain (134).
Cortical lesions of hemimegalencephaly resemble the cerebral lesions (ie, tubers) of tuberous sclerosis (ie, tubers). Neuroblast migratory disturbances are accompanied by abnormal gyration (167; 50). Coexistence of hemimegalencephaly and tuberous sclerosis complex has been described (53; 110; 59; 20; 141), but most cases of tuberous sclerosis do not have hemimegalencephaly despite numerous cortical tubers. Tuberous sclerosis is another disorder of the mTOR signaling pathway, and the TSC1 and TSC2 genes are closely related to it, so it is not surprising. Tuberous sclerosis complex genes are both germline mutations as autosomal dominant traits and also postzygotic somatic mutations not of Mendelian inheritance. Immunocytochemical studies show similarities in the megalocytic dysplastic neurons in both, and glial cell involvement also occurs in both. Furthermore, both show similar abnormal tau protein expression (132). A subtle difference is that cortical tubers may exhibit microcalcifications that are rare in hemimegalencephaly and do not occur in focal cortical dysplasia type II. Balloon cells are similar in all. Periventricular nodules and giant cell astrocytomas in tuberous sclerosis, by contrast, contain glial elements but no neurons or balloon cells. Hemimegalencephaly in tuberous sclerosis complex can sometimes be diagnosed by prenatal imaging as early as midgestation (20).
Neoplastic transformation of the hamartoma of hemimegalencephaly into glioblastoma multiforme is rare but has been described in isolated patients (28). The authors have also seen one such case, which has not yet been published.
Genetics. The fundamental genetic defect in hemimegalencephaly is a disorder of the mTOR signaling pathway, including any of the various associated pathways that affect mTOR, such as PIK3CA, AKT, DEPDC5, and GATOR (87; 72; 54). This genetic basis occurs in both isolated hemimegalencephaly and when this malformation is associated with epidermal nevus syndromes, tuberous sclerosis complex, and also in focal cortical dysplasia type IIb (101).
Neuronal heteroploidy has been suggested by quantitative histochemical studies demonstrating enlarged nucleoli, nuclei, and increased DNA and RNA content in neurons of the involved hemispheres of two patients (17; 93). The first cultured cell line developed from cerebral cortex came from a patient with hemimegalencephaly (123), suggesting that hyperdiploidy may confer a proliferative enhancement to neurons. In addition to the primary genetic etiology of postzygotic somatic mutation due to an mTOR defect, DNA from a case of isolated hemimegalencephaly showed a heterozygous deletion of 15q11.2-15q13.1. The region of the deletion includes at least 30 known genes and could present a susceptibility locus for hemimegalencephaly (15). Studies also suggest a possible correlation with platelet activating factor and cell adhesion molecule L1, two substances thought to be involved in neuronal migration, but no precise mechanism for this brain malformation has been elucidated (153; 64). One case study identified somatic uniparental disomy of the ZNF597 gene of chromosome 16 in patients with hemimegalencephaly, indicating that overexpression of maternally expressed ZNF597 may play a role in aberrant hemispheric development (57).
In a female neonate with refractory seizures since birth and hemimegalencephaly, whole genome sequencing revealed a novel, maternally inherited 3Kb deletion encompassing exon 5 of the NPRL3 gene of chromosome 16, which encodes a subunit of the GATOR complex (26). GATOR is a regulator of the mTOR signaling pathway, and, thus, this variant is consistent with a mutation of one of several pathways affecting mTOR.
Neuroimaging. Advanced MRI technology with diffusion tensor imaging has shown asymmetry in interhemispheric fiber tracts and white matter of not only the enlarged hemisphere but also the “normal” hemisphere in patients with hemimegalencephaly (70). Fiber tracts passing through the corpus callosum showed aberrant pathways from the unaffected side to incorrect areas of the corresponding lobe and to noncorresponding lobes. Abnormal projections of the cortico-ponto-cerebellar pathway are also demonstrated by diffusion tensor imaging (41). These images also demonstrated abnormal fiber tract volumes on the affected side when compared to the unaffected side.
Dysplasia of the corpus callosum was also confirmed neuropathologically (50). Takahashi and colleagues hypothesize that the abnormal fiber tracts could be due to immature neuronal cells connecting with abnormal axonal projections during development (146). Transcranial magnetic stimulation has shown abnormal axon orientation and excess spread of corticospinal excitation associated with multiple defects of cortical inhibition in the affected hemisphere in a patient with hemimegalencephaly (29).
In addition to the more clinically obvious enlarged hemisphere, abnormal findings outside the involved hemisphere have been reported. MRI and neuropathological abnormalities reported include enlargement of ipsilateral olfactory bulb (119) and optic nerves, ipsilateral cerebral vascular dilations, ipsilateral cerebellar and brainstem enlargements, and abnormal cerebellar folia (both ipsilateral and contralateral). These findings confirm the great importance of high-quality performance and interpretation of diagnostic neuroimaging studies (138).
The diagnosis of hemimegalencephaly can sometimes be demonstrated prenatally by fetal ultrasound confirmed by fetal MRI (163) and as observed in the authors’ practice (49).
Ictal and interictal single photon emission computed tomography (SPECT) and positron imaging tomography (PET) often detect hypometabolism in the overgrown cerebral hemisphere. Another special MRI sequence, arterial spin labelling, is an additional noninvasive functional imaging biomarker of cerebral perfusion using magnetically labelled blood as an endogenous contrast agent; it shows reduced cerebral blood flow that correlates with the hypometabolism of PET and SPECT, except for ictal increased blood flow during peri-ictal phases in hemimegalencephaly (147).
Hemimegalencephaly remains an infrequent malformation that is reported worldwide and in all ethnic groups; it is sometimes associated with syndromes, especially in the neurologic phenotypes of epidermal nevus syndromes, thereby making comprehensive, unbiased, and detailed epidemiological studies difficult (149; 47; 45). Thus, longitudinal assessment throughout the childhoods of all patients with hemimegalencephaly is essential.
Because hemimegalencephaly has a developmental pathogenesis related to altered gene expression, no current methods of prevention are available to the developing infant (169). However, prenatal diagnosis by ultrasound and fetal MRI are possible before midgestation.
Megalencephaly (but not hemimegalencephaly) may also occur with a variety of metabolic disorders, including leukodystrophies such as Canavan disease and Alexander disease, lipidoses such as Tay-Sachs disease, or mucopolysaccharidoses. In addition, general disturbances of growth, such as Sotos syndrome (cerebral gigantism), Beckwith syndrome, or achondroplasia, may cause megalencephaly, but these typically have other distinguishing features (97; 158). Megalencephaly occurring with neurocutaneous syndromes, such as neurofibromatosis (124; 32) or tuberous sclerosis (23; 126; 149), present a more difficult diagnostic challenge, particularly if there is asymmetrical hypertrophy, as may be seen with intracerebral hemangiomatosis of Sturge-Weber syndrome, Klippel-Trenaunay-Weber syndrome (95; 21; 104; 159), or “cutis marmorata telangiectatica congenita” (56), which is now known as “megalencephaly capillary malformation or MCAP.” In a Japanese nationwide survey, 16 of 44 patients with hemimegalencephaly had underlying neurocutaneous syndromes (137); this has been confirmed by others (45).
Neurocutaneous syndromes and hemimegalencephaly. The most frequent neurocutaneous syndromes associated with hemimegalencephaly are the neurologic phenotypes of epidermal nevus syndromes, both the linear nevus sebaceous and keratinocytic types, in approximately one half of cases (112; 67; 111; 45). They are distinguished by a midline facial nevus, occasional facial hemihypertrophy caused by hamartomatous-lipomatous lesions, and venous sinus dysplasias (25; 12; 19; 170; 30; 136; 61). Hemimegalencephaly has also been reported with both keratinocytic and sebaceous types of epidermal nevus syndrome associated with severe epilepsies (39; 127; 40; 60; 90; 45).
Hypomelanosis of Ito is another neurocutaneous syndrome that is inconstantly associated with hemimegalencephaly and epilepsy (115; 14; 143; 27; 117) and Proteus syndrome (96; 05; 13; 88; 49). Still another neurocutaneous syndrome that may present with hemimegalencephaly is neurocutaneous melanocytosis (81).
The diagnosis of hemimegalencephaly is often suspected because of an enlarged hemicalvarium. Though hemimegalencephaly may be detected prenatally by ultrasound or magnetic resonance imaging, post-natal high-resolution imaging remains the current standard in the diagnostic evaluation (65; 02).
MRI is the most sensitive study for diagnosing hemimegalencephaly (01), which occurs in association with pachygyria, polymicrogyria, nodular heterotopia, and a small hippocampus in some patients (100; 99).
Unilateral lissencephaly or agyria may also occur in some cases (84). Ipsilateral ventricular enlargement occurs in most cases, often with a straightened and uplifted frontal horn.
Greater enlargement of the involved cerebral hemisphere is seen in patients with polymicrogyria and normal subcortical white matter, whereas patients with significant subcortical gliosis have less enlargement (84). Because hemicalvarial enlargement does not occur in all patients with hemimegalencephaly, CT or MRI studies may be required to demonstrate enlargement of at least one lobe of one hemisphere.
This is particularly true for normocephalic infants being evaluated for refractory partial seizures. In addition, the abnormally enlarged hemisphere over time may show impairment of growth, causing a later relative micrencephaly. MR imaging, especially with gadolinium enhancement, is useful in distinguishing hemimegalencephaly from disorders characterized by leptomeningeal angiomatosis or intraparenchymal vascular malformations.
Evaluation of the contralateral nonmegalencephalic hemisphere is also important, not only to assess improvement following hemispherectomy of the abnormal cortex, but also to identify possible dysgenesis of the nonmegalencephalic hemisphere. SPECT (79) and PET (118) may be used to identify possible hypometabolism of "normal" cortex. Neuropathological examination of the “normal” hemisphere may disclose mild histopathological abnormalities (134).
Electroencephalography is essential for localizing zones of epileptic activity. This is important because seizures may rarely originate from cortex other than that which is considered abnormal on the basis of neuroimaging studies (155). EEG findings over the involved hemisphere typically demonstrate depression of background voltage with bursts of numerous spikes during wakefulness, and unilateral suppression-burst during sleep (154). This has been labeled hemihypsarrhythmia (148; 83; 30), though a good response to ACTH is not assured (114). In neonates and infants, interictal fast oscillations on the affected hemisphere may represent an epileptic tendency (166). There is no consensus concerning the prognostic value of EEG (78; 79; 152). Nevertheless, video EEG is considered important to verify the stereotypic pattern of seizures and to demonstrate the onset of seizures from the malformed cerebral hemisphere (154).
Cerebral angiography, although not typically required for diagnosis, may demonstrate arteriovenous abnormalities in areas of aberrant neuronal migration, as normal vessels that coalesce from venules and venous channels within fully developed cortex do not form (10). Ultrasonographic studies have also provided descriptive information (85; 42); however, these are not likely to provide as much specificity and prognostic information as MRI (108).
Literature has documented the superiority of MRI over neurosonography in accurate diagnosis and avoidance of misdiagnosis of hemimegalencephaly, particularly prenatally (102). In a case report by Romero and colleagues, MRI was done as early as 22 weeks gestational age, resulting in a definitive diagnosis of hemimegalencephaly (121). In older patients who do not present with hemiparesis, cortical mapping studies can be performed prior to epilepsy surgery for counseling on prognosis. Transcranial magnetic stimulation performed on a patient with hemimegalencephaly showed enlargement of the cortical motor map (29).
Antiseizure medication is typically required for infants or children with hemimegalencephaly, although a few patients with milder cytoarchitectural abnormalities may not develop early seizures until later in childhood, or not at all. Antiseizure medications used in this age population seldom result in seizure freedom, even with the newer agents available since 1993. Seizures may improve with ketogenic diets, including a modified Atkins diet (27).
Though medical control of seizures is desirable (152), surgery is most likely to be the best option for infants and children with refractory seizures (76; 150; 154; 55; 07; 144; 35; 03; 71; 116; 137; 140; 36; 91; 20; 68) and should not be considered a last resort. The surgical strategy for these patients depends in large part on the extent of neuroimaging abnormalities as well as motor impairment (109; 140; 36; 91). For infants and young children with significant hemiparesis, cerebral hemispherectomy is recommended (76; 154; 137; 140; 36; 91). In a large multicenter study of 1267 hemispherectomies for intractable epilepsy in 32 centers of 12 countries, 85% were seizure-free at 12 months postoperatively, though not all patients in this cohort had hemimegalencephaly (162).
With mild hemiparesis, resection of more clearly demarcated structural or electrical abnormalities is appropriate (108); however, postoperative seizure control may be less satisfactory than that achieved following hemispherectomy (04; 71; 140; 36). Hemispherotomy has also been described and attempted for patients with hemimegalencephaly (94). Results were not favorable, possibly due to incomplete disconnection of the affected hemisphere. Early surgical intervention is thought advisable to permit brain "plasticity" maximum benefit, but it is not likely to be helpful in patients with cortical dysplasia or hypometabolism of the nonmegalencephalic hemisphere (79; 118; 145; 137). Kawai and colleagues reported a modification of the vertical hemispherectomy for refractory epilepsy in seven patients (74). Four patients were children with hemimegalencephaly and the other three were adults with ulegyric hemisphere. Surgical procedure was completed without complication in all cases, and there was no case that required CSF shunting. Seizure outcome was Engel's class I in six and class IV in one. For patients that are poor surgical candidates in which the seizures arise from the contralateral hemisphere or if the risk of a hemispherectomy is unacceptably high, implantation with vagus nerve stimulation may also be considered. Vagal nerve stimulation in this population can lead to significant reduction in partial and generalized seizures (greater than 60%) as well as psychological improvement in the form of calmer effect and improved communication in patients with severe cognitive impairments (28).
A promising treatment that has not yet been officially approved but has been successful in some isolated cases is the use of mTOR inhibitors, such rapamycin (165).
Early hemispherectomy, at a few months of age, has been associated with a significantly improved outcome, including seizure freedom or more than 75% seizure reduction, as well as developmental and cognitive improvements (154; 155; 118; 35; 90; 162). Even infants just beyond the neonatal period have undergone hemispherectomy for retractable seizures in hemimegalencephaly, either isolated (134) or complicating tuberous sclerosis complex (139).
Improvement of cognitive function of the remaining hemisphere, following surgery for intractable epilepsy, is a function of preoperative glucose metabolism as determined by PET (22; 118). Some patients with hemimegalencephaly seem to experience poorer outcomes following hemispherectomy presumably because of greater involvement of the non-megalencephalic hemisphere. An explanation for this is that there are bilateral cerebral hemispheric abnormalities, though significantly less in the “non-megalencephalic” hemisphere (128). Postnatal evolution of cortical malformation in the previously unaffected hemisphere has been reported following hemispherectomy for intractable hemitonic and asymmetrical epileptic spasms (77).
As the child with hemimegalencephaly grows, the involved hemisphere may fail to grow normally as does the nonmegalencephalic hemisphere. This results in an eventual hemiatrophy of the previous megalencephalic hemisphere (164), and may ultimately lead to macrocephaly, though the pathogenesis for such aberrant growth remains unknown (126).
Pregnancy is not altered by a fetus with hemimegalencephaly, with the exception of macrocephaly, which may preclude safe vaginal delivery and require Caesarean section. The diagnosis of hemimegalencephaly can be established prenatally by fetal ultrasonography or MRI (20) and in the authors’ experience (49).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Harvey B Sarnat MD FRCPC MS
Dr. Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See Profile
Laura Flores-Sarnat MD
Dr. Flores-Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See Profile
Bernard L Maria MD
Dr. Maria of Thomas Jefferson University has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Developmental Malformations
Apr. 14, 2024
Developmental Malformations
Apr. 09, 2024
Developmental Malformations
Mar. 22, 2024
Developmental Malformations
Mar. 22, 2024
Developmental Malformations
Mar. 22, 2024
Developmental Malformations
Mar. 22, 2024
Developmental Malformations
Mar. 13, 2024
Developmental Malformations
Mar. 11, 2024