










Epilepsy & Seizures
Ambiguous paroxysmal events
Mar. 22, 2024
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Among the focal epilepsies, mesial temporal lobe epilepsy associated with hippocampal sclerosis (MTLE-HS) and characterized by seizures originating in the hippocampal formation is the most common. Data accumulated from surgical series strongly suggest that MTLE-HS represents a distinctive epilepsy presentation. Early diagnosis of this epilepsy syndrome is important because disabling seizures and their consequences can be prevented by surgical treatment, either by resection or ablation. Resection has a well-established seizure-free rate of 70% to 90% of patients and low complication rates at epilepsy surgery centers. Ablation is an alternative technique that does not produce as high a seizure-freedom rate but is less invasive. The correct identification of hippocampal and parahippocampal seizures is often noninvasive, but intracranial EEG recording is sometimes required. However, the diagnosis of MTLE-HS or associated with other mesial temporal lesions can be achieved without invasive methods based on seizure history, clinical presentation, progressive nature, medication resistance, and special features in electrophysiological, neuropsychological, structural, and functional imaging. Differentiation from other temporal lobe epilepsy types and evaluation of the memory is important if resection of the hippocampal formation is considered.
• Hippocampal and parahippocampal seizures are common manifestations of mesial temporal lobe epilepsy. | |
• Hippocampal and parahippocampal seizures have characteristic features, including auras with paramnestic features and impaired awareness without convulsive motor features. | |
• Hippocampal and parahippocampal seizures are associated with localized structural and functional abnormalities. |
In 1880, John Hughlings Jackson described the symptomatology of “dreamy states,” and in 1889, he described other variants of “uncinate fits” as “a particular variety of epilepsy,” which subsequently were called “psychomotor seizures” by Gibbs and colleagues and “temporal lobe seizures” by Jasper and Kershman (100; 147). Although Anderson and then Jackson and Beevor had noted the association of temporal lobe tumors with olfactory hallucinations and dreamy states (04; 141), it was the postmortem finding of a small cystic lesion restricted to the uncinate gyrus in a patient who had suffered from seizures with dreamy states, elaborated automatisms, and amnesia (142) that led Jackson and Stewart to the concept of “uncinate fits” with “origin of the discharge... in a region of which this gyrus [ie, the gyrus uncinatus] is part...” (143).
Jackson and Stewart described masterly the most characteristic symptomatology of the mesial temporal lobe seizures (143). Insights into temporal lobe function with description of the now classical aura-types, automatisms (mainly of the oroalimentary type and including deambulation), and amnesia were fostered by results of stimulation and ablation in monkey experiments reported by Ferrier (92). However, the verification of the tight relation in humans of certain temporal lobe structures, specifically the anterior mesial ones, with the observed and described variety of symptoms and signs did not happen until the era of epilepsy surgery, with reproduction of these symptoms by electrical brain stimulation as well as electroencephalographic, pathological, anatomical, and physiological studies (245; 234; 247; 90; 188; 242; 243; 244; 30; Feindel and 243; 243; 250; 13; 65; 306; 01; 104; 146; 176; 88; 62; 106; 112; 14; 260; 192; 230; 273; 350; 193; 327).
Based on a stereoelectroencephalographic study on the electroclinical features of psychomotor seizures, Wieser proposed five localization-related subtypes: the temporobasal limbic type, the temporal pole type, the frontobasal-cingulate type, the opercular (insular) type, and the posterior neocortical temporal type (327).
Bouchet and Cazauvieilh described the pathology of mesial temporal lobe epilepsy as early as 1825 and presented autopsy data with abnormalities in the Ammon’s horn in seven of 18 patients who suffered from epilepsy and psychiatric symptoms (“mental alienation seizures”) (37). At that time, the abnormalities were believed to be an effect, rather than a cause, of epilepsy. Jackson recognized limbic-type seizures and associated them with lesions in mesial temporal structures but not with Ammon’s horn sclerosis (Jackson 1931-1932). Sommer and Bratz both suggested that Ammon’s horn sclerosis might be an epileptogenic lesion (286; 38), and subsequent evidence has accumulated suggesting that distinctive structural damage, ie, hippocampal sclerosis, is typical for temporal lobe epilepsy. This pathology was accurately described and illustrated by Bratz, with destruction of the pyramidal cells in Sommer’s sector (currently termed, sector CA1), preservation of the cells in the neighboring subiculum, and cell loss in the hilus of the dentate gyrus and adjacent sector CA3 but preservation of neurons in sector CA2 and of the dentate granule cells (38). Granule cell dispersion is a common finding in hippocampal sclerosis in patients with intractable focal epilepsy. It is considered to be an acquired, post-developmental rather than a pre-existing abnormality, involving dispersion of either mature or newborn neurons, but the precise factors regulating it and its relationship to seizures are unknown. Thom and colleagues presented two cases of granule cell dispersion with associated CD34-immunopositive balloon cells, a cell phenotype associated with focal cortical dysplasia type IIB, considered to be a developmental cortical lesion promoting epilepsy (312).
The International League Against Epilepsy (ILAE) Commission of Classification and Terminology 2005-2009 revised concepts, terminology, and approaches for classifying seizures and forms of epilepsy (23) without making changes in epilepsy syndromes already recognized and updated in the 2006 classification (80). The Commission’s work was aided by the Montreal workshop (45). In the new classification, the concept of generalized and focal with regard to seizures was redefined. Generalized seizures were defined as seizures occurring in and rapidly engaging bilaterally distributed networks, whereas focal seizures emerge within either discretely localized or more widely distributed networks that are limited to one hemisphere. Function MRI connectivity studies have provided evidence for this more distributed abnormality in mesial temporal lobe epilepsy with hippocampal seizures (214; 119). The new scheme includes lists of generally agreed-upon epileptic seizure types and epilepsy syndromes. The description of focal seizures in the new classification adopts the main principals of the diagnostic scheme of the Task Force approved in Buenos Aires in 2001 and is based on the ictal phenomena, which has been drawn from the glossary of ictal semiology (35) and is revised from the original 1981 classification. The Cleveland group proposed a somewhat similar seizure classification based exclusively on ictal semiology and conceptually concentrated on the so-called symptomatogenic zone (187). The symptomatogenic zone is one of five zones that must be determined to define the “epileptic focus.” The epileptogenic zone, on the other hand, is more closely related to the epileptic syndrome. None of the classification revisions so far have negated the 1981 classification of epileptic seizures.
The focal onset (partial) seizures as before were classified according to the degree of impairment of the consciousness during seizures:
Without impairment of consciousness or awareness.
With observable motor or autonomic components. This roughly corresponds to the concept of “simple partial seizure.” “Focal motor” and “autonomic” are terms that may adequately convey this concept depending on the seizure manifestations.
Involving subjective sensory or psychic phenomena only. This corresponds to the concept of an aura, a term endorsed in the 2001 Glossary.
With impairment of consciousness or awareness. This roughly corresponds to the concept of complex partial seizure. ‘‘Dyscognitive’’ is a term that has been proposed for this concept (35).
Evolving to a bilateral, convulsive seizure (involving tonic, clonic, or tonic and clonic components). This expression replaces the term “secondarily generalized seizure.”
In the new classification, the distinction of complex and simple partial seizures is eliminated. Nevertheless, the Commission recognized that such distinction based on dyscognitive features can be further used for specific purposes in individual patients. Dyscognitive seizures with or without automatisms were not exactly synonymous with the term “complex partial seizures,” which were defined on the basis of impaired consciousness only and do not necessarily involve limbic areas. Dyscognitive seizures with or without automatisms, as well as the term “psychomotor,” conforms more to the original intent of the term “complex partial seizures” in the 1970 ILAE Classification of Epileptic Seizures. It was implied that mesial temporal limbic areas and their immediate connections are involved in the clinical manifestations, although seizures may have been initiated elsewhere.
The ILAE Commission on Classification and Terminology uses the terms “structural,” “genetic,” “infectious,” “metabolic,” “immune,” and “unknown” as the categories of etiological factors of the disease (269). For other than well described distinct electroclinical syndromes with characteristic clinical, electrographic, developmental, and genetic features, the Commission proposed the method of defining other clinical entities based on constellations of the clinical, electrographic, and specific cerebral lesions. The category of these types of clinical entities includes mesial temporal lobe epilepsy, hypothalamic hamartoma with gelastic seizures, epilepsy with hemiconvulsion and hemiplegia, and Rasmussen syndrome (23).
Hippocampal and parahippocampal seizures as seen in association with mesial temporal lobe epilepsy are typically characterized by auras evolving into focal seizures with impaired awareness with initial behavioral arrest (motionless stare), followed by oroalimentary and manual automatisms. However, MTLE-HS probably consists of more than one syndrome, and it is not certain whether features of patients with hippocampal sclerosis clearly differentiate them from those with other mesial temporal lesions. Nevertheless, observation by Heuser and colleagues showed distinct phenotypic tendency in MTLE-HS patients: higher incidence of simple partial seizures and seizures with ictal psychiatric and autonomic manifestations as well as higher incidence of childhood febrile seizures and lower age of onset than temporal lobe epilepsies without hippocampal sclerosis (128). In the current classification, this syndrome is listed under the category “epilepsy syndromes by age of onset and related conditions, with less specific age relationship” (80).
Hippocampal and parahippocampal seizures show unique characteristics. In comparison with some frontal lobe seizures that cause brief and clustered seizures with little or no postictal disturbances and nocturnal predilection, hippocampal and parahippocampal seizures are less-frequent events with profound postictal disturbances and have slower propagation. Hippocampal and parahippocampal seizures almost always require local spread for clinical manifestation, which typically occurs over many seconds and may involve the insula, amygdala, hypothalamus, and other limbic structures. Contralateral propagation is slower for hippocampal seizures when compared to neocortical seizures (328).
Hippocampal and parahippocampal seizures as seen in association with mesial temporal lobe epilepsy typically are characterized by auras evolving into seizures with impaired awareness and initial behavioral arrest (motionless stare), followed by oroalimentary and manual automatisms with the occurrence of manual automatisms, suggesting an onset in the posterior hippocampus and includes the parahippocampal gyrus (363). Vegetative-autonomic signs and symptoms are prominent and may involve cardiovascular, pupillary, gastrointestinal, sudomotor, vasomotor, and thermo-regulatory functions. Auras occur frequently in isolation and typically consist of autonomic features. Epigastric auras are common and include abdominal discomfort, nausea, emptiness, tightness, churning, butterflies, malaise, pain, and hunger. Such sensations may rise to the chest or throat and may have high specificity. Henkel and colleagues found that all patients with temporal lobe epilepsy had at least one seizure with abdominal aura evolving into oral and manual automatism, and most of these patients had mesial temporal lobe epilepsy (125).
Experiential phenomena include affective, mnemonic, or composite perceptual phenomena and illusory or composite hallucinatory events. Hallucinatory perceptions have no corresponding external stimuli, whereas illusions are alterations of actual percepts and involve visual, auditory, somatosensory, olfactory, or gustatory phenomena. These phenomena have subjective qualities similar to those experienced in life but are recognized by the subject as occurring outside of the actual context. When elementary, the visual hallucinations are typically contralateral, whereas experiential (complex) visual hallucinations are typically nonlateralized. The experiential visual hallucinations may be spatial distortions and either familiar or unfamiliar visual scenes, and this may relate to the role of the hippocampal-parahippocampal system in encoding visual experiences with memory (258). Overall, the auras may appear alone or in combination and include feelings of depersonalization.
The term dyscognitive describes events in which disturbance of cognition is the predominant or most apparent feature. Components of cognition are perception (symbolic conception of sensory information), attention (appropriate selection of a principal perception or task), emotion (appropriate affective significance of a perception), memory (ability to store and retrieve percepts or concepts), and executive function (anticipation, selection, monitoring of consequences, and initiation of motor activity including praxis, speech).
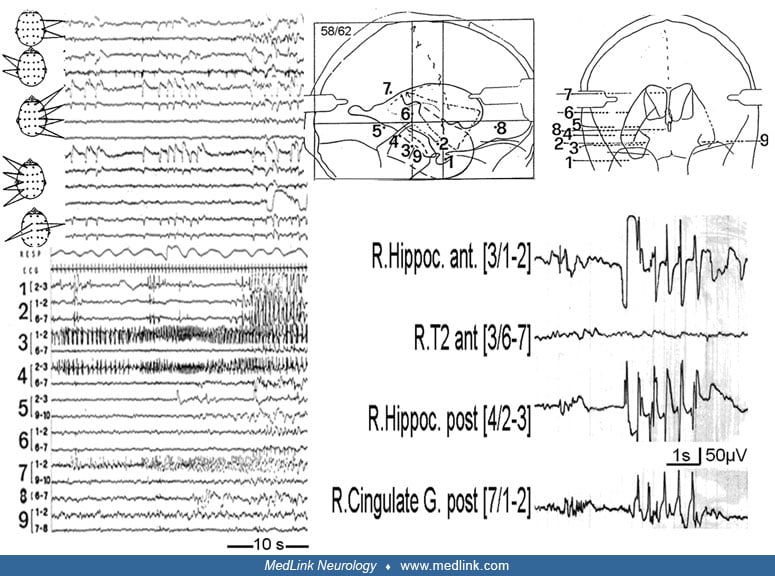
Emotional experiences such as fear, dysmnesias (dreamy states, déjà-vu, déjà-vecu, déjà entendu, and other kinds of recollections), alterations of self-perception (in time and space), focal sensory phenomena with olfactory or gustatory symptoms, and vague bilateral sensory phenomena such as tingling are relatively common in hippocampal and parahippocampal seizures with ipsilateral spread to the amygdala and anterior neocortical temporal regions. Affective components include fear, depression, joy, and (rarely) anger and suggest an onset in the anterior hippocampus and amygdala (363). Mnemonic components reflect ictal dysmnesia, such as false feelings of familiarity (déjà-vu) and unfamiliarity (jamais-vu). Vignal and colleagues restudied dreamy states (318). They had observed a total of 15 sensations of déjà vecu, 35 visual hallucinations consisting of the image of a scene and five “feelings of strangeness.” These were recorded during 40 stimulations in 16 subjects and 15 spontaneous seizures in five subjects; 45% of dreamy states were evoked by stimulation of the amygdala, 37.5% by the hippocampus, and 17.5% by the parahippocampal gyrus. During both spontaneous and provoked dreamy states, the electrical discharge was localized within mesial temporal lobe structures without involvement of the temporal neocortex.
Automatisms consist of apparently purposeless, coordinated, repetitive motor activity usually occurring when awareness and memory are impaired. Automatism without cognitive impairment is a relatively rare phenomenon observed predominantly in seizures arising from the nondominant temporal lobe (75). They often resemble a voluntary movement that is out of context or socially unexpected and may consist of an inappropriate continuation of ongoing preictal motor activity. Automatisms may be ictal or postictal, de-novo, or reactive. Ictal automatisms associated with temporal lobe seizures frequently consist of oroalimentary symptoms, such as lip-smacking, lip-pursing, chewing, licking, tooth-grinding, or repetitive swallowing. Manual automatisms are also frequently observed and are often unilateral, ie, fumbling, picking, or exploratory movements with the hand, directed toward self or the environment. They may resemble movements intended to lend further emotional tone to speech. Mimetic automatisms are facial expressions suggesting an emotional state, often fear. Ictal and postictal automatic movements have localizing value and are seen more commonly in mesial temporal seizures than in neocortical temporal seizures. Ictal unilateral motor automatism is predominantly ipsilateral to the epileptic focus in mesial temporal lobe seizures (often because of contralateral posturing) and contralateral nonautomatism motor abnormality in neocortical temporal seizures. Concomitant ipsilateral distal motor automatism and contralateral dystonic posturing (flexion of the wrist and elbow and extension of the fingers) is exclusively seen in mesial temporal lobe seizures (71). Unlike distal automatism, the proximal upper arm and shoulder automatic nonmanipulative stereotyped circular movements are related to the contralateral mesial temporal lobe seizures (157). Postictal facial- and nose-wiping is another reliable lateralizing sign with high predictive value for ipsilateral mesial temporal seizures (129).
Hyperkinetic automatisms are a hallmark of frontal lobe seizures. They involve predominantly proximal limb or axial muscles and produce irregular sequential ballistic movements, such as pedaling, pelvic thrusting, thrashing, or rocking movements. The emergence of an intense affective and behavioral state during a temporal lobe seizure could be related to the involvement of a network of structures, including the anterior temporal lobe, orbitofrontal cortex (15), and thalamus.
Clonic or tonic-clonic motor phenomena indicate supra-Sylvian seizure spread. An exception is dystonic posturing, which might indicate ictal involvement of the striatopallidal system. Kotagal and colleagues have pointed out that posturing of one extremity is a valid lateralizing sign pointing to contralateral ictal onset (167). Secondary generalization can occur, particularly in children, but is relatively infrequent in adults receiving standard antiseizure medications.
The ictal semiology of seizures involving the limbic system is particularly rich and includes aspects of consciousness. When impairment of aspects of consciousness is used as a major criterion for classification, operationally, it refers to the degree of awareness or responsiveness of the patient to externally applied stimuli. Typically, awareness is gradually lost or at least impaired in hippocampal and parahippocampal seizures with spread to the opposite hemisphere. Animal studies suggest that impaired awareness in mesial temporal seizures could be related to the frontal and cingulated cortical deactivation through the activation of the septal and mediodorsal thalamic connections (83).
Typical for the semiology of seizures in mesial temporal lobe epilepsy are varying degrees of postictal confusion with amnesia for the ictal event as well as persisting postictal memory deficit. Postictal aphasia with left temporal lobe seizures also is typical.
The clinical overall gestalt of mesiobasal limbic seizures can be best described within the following groups. First, there are those seizures that, from the viewpoint of an observer, appear absence-like (“fausses absences”) (233); a second group is characterized by “psychomotor symptoms” and automatisms. Third is a group of seizures with predominant psychosensory or pure psychic intellectual, cognitive, and emotional symptoms. It is tempting to correlate these seizure types with the localization-related subtypes classification (327). However, at present, only some relatively vague correlations have been obtained. Many symptoms seem to depend on a characteristic seizure discharge constellation; that is, they depend on the type of propagation along preferential pathways and appear within a characteristic “march of symptoms.”
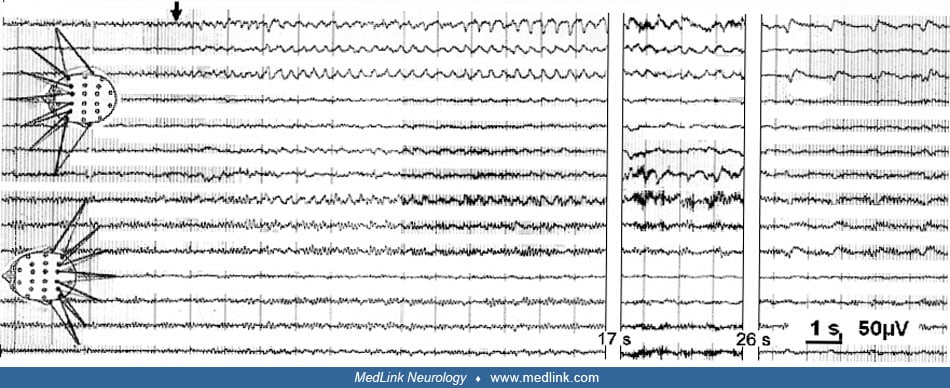
Absence-like focal seizures express themselves with an initial motionless stare. Clinically, one observes clouded consciousness associated with a stare of 10 to 20 seconds, often with the expression of fearful astonishment followed by abrupt and restless looking around and discrete tongue movements. Without the EEG, the distinction from true 3 Hz spike-wave absences may sometimes be difficult. However, focal seizures with the arrest reaction usually lack the palpebral cloni and the sursum-vergens of the bulbi, seen in 3 Hz spike-wave absences. Moreover, during a true 3 Hz spike-wave (petit mal) absence, the patient usually has a vacant and dull but not fearful and tensed look and has very brief or no postictal confusion.
The predominance of automatisms characterizes a second seizure manifestation, which usually starts with a short arrest reaction but then evolves rapidly into the “automatic” phase. The impaired awareness is usually more pronounced, and recovery is slower. Automatisms have been classified into eupraxic (well-adapted) and dyspraxic (mal-adapted), but practically, this is not helpful. Gastaut and Gastaut differentiated them into (1) automatisms that represent a continuation of the previous activity and (2) de-novo automatisms (97). Within the de-novo automatism, one can distinguish (a) reactions of the already confused patient to environmental stimuli; these stimuli are inadequately interpreted, and, therefore, the reactions are usually mal-adapted. Another type of de-novo automatisms might be (b) reactions of the patients to ictal experiences (“reactive automatisms”). Finally, (c) archaic motor patterns can be released. Although extremely rare, antisocial and aggressive behavior might be witnessed (327), and eupraxic urination or defecation and spitting automatisms are described (121). Along with the psychopathological classification, a symptomatic one can be employed and is preferred by clinicians differentiating between oroalimentary, mimic, gestural, verbal, and ambulatory automatisms. In mesial temporal lobe epilepsy, the oroalimentary automatisms are most frequent and consist of degustation and deglutination, lip-smacking, pursing lips, chewing, and swallowing. As a rule, marked autonomic symptoms are associated with oroalimentary automatisms, and both correlate fairly well with epileptic discharges of the amygdala and periamygdalar region. It is important to note that automatisms are not bound to temporal lobe seizures. They can also be observed with frontal and parietal onset seizures, although with different characteristics (98; 99; 324).
Phenomenologically, a third seizure manifestation might be differentiated with marked psychosensorial symptoms. Psychosensorial symptoms are illusions and hallucinations that affect one sense only (unimodal) or more than one (polymodal), and this in a simultaneous or successive fashion. As psychosensorial and pure psychic symptoms require identification and memorization, in a global sense the consciousness must be intact; therefore, these symptoms usually are reported very early in the course of a seizure as an “aura” or a “primictal [signal]” symptom.
Psychosensorial symptoms are classified according to their nature into (1) elementary (simple) and (2) structured (ie, complex, elaborate, or formed) and according to their sensorial quality (visual, auditory, somesthetic, vertiginous, olfactory, or gustatory). Moreover, “positive” and “negative” symptoms can be differentiated. Generally, elementary hallucinations do not occur in mesial temporal lobe epilepsy, and well-structured hallucinations occur only rarely. Occurring more often, however, are delusions.
Olfactory illusions are typically confined to hyperosmias and parosmias. Ictal olfactory hallucinations in patients with temporal lobe epilepsy are often combined with gustatory hallucinations. They are usually unpleasant and often reported as a “metallic” or “burning” character. Gustatory sensations can be classified as hypergeusia or parageusia.
As a general rule, elementary illusions and hallucinations point to an ictal onset in or near the primary sensory areas, whereas complex ictal hallucinations are typical for ictal onset in or ictal propagation to associated areas. As Penfield as well as Gloor and colleagues have pointed out, most complex ictal hallucinations, produced either by electrical stimulation or occurring spontaneously with epileptic discharges, are experiential phenomena in the sense of being recollections of past experiences (242; 107).
Seizures characterized by the predominance of psychic symptoms constitute a third category of hippocampal and parahippocampal seizures. Following Jackson’s classical description, one can differentiate between intellectual and affective-emotional phenomena. Jackson’s description of the “dreamy state” (état de rêve) includes (1) recollections in the sense of déjà vu, déjà-entendu, déjà-vécu; (2) unfamiliarity or unreality (jamais vu, jamais entendu, or jamais vécu); (3) forced thinking, including what the French called “pensée parasite”; and (4) the rapid recollection of the past, the so-called “panoramic vision.” Emotional auras are relatively rarely reported; Gowers stated that he found them in 15 of 25 patients with auras (Gowers 1885). The most common is fear, often associated with restlessness and irritability. Sadness (“aura de tristesse”) (226), pleasure, elation, exhilaration, satisfaction, and the “eureka-feeling” are well documented. According to Lennox, about 0.9% of auras have a pleasurable quality (176).
Some aura symptoms may not have counterparts in human experience and cannot be described; a “strange ineffable feeling” is very often reported. Moreover, some patients experienced auras in the past but no longer have them. For example, during recordings of seizures, some patients may consistently press the alarm button at the beginning of a seizure yet deny any warning afterward. This could be due to seizure-related retrograde amnesia (77; 82).
Ictal autonomic phenomena can be divided into “visceromotor” symptoms and “viscerosensitive” sensations. Measurable autonomic visceromotor changes occur during the course of most temporal lobe seizures. When they characterize the clinical picture, the terms “visceral” or “autonomic seizures” may be used (243; 96). According to the effector systems, one can distinguish between (1) cardiovascular, (2) respiratory, (3) pupillary, (4) sudo-and pilomotor, (5) salivation, (6) gastrointestinal, and (7) genitourinary.
Cardiovascular symptoms are frequent and include tachy- and bradycardia as well as arrhythmias, hypertension, flush, or pallor. A vast amount of experimental work has shown the importance of the amygdala, particularly its central nuclear group, and the functionally connected perifornical so-called HACEAR (hypothalamic area controlling emotional responses) (21; 283). The direction and type of heart rate and blood pressure changes during seizures, electrostimulation, and induced after-discharges vary considerably, and ictal bradycardia and tachycardia have been reported with inconsistent lateralization value (39). However, reasonable evidence supports bradycardia as more often left hemispheric and less consistent evidence that tachycardia is more often right hemispheric when seizures remain focal (213; 76).
A short respiratory arrest or a deep inspiration is common in the initial seizure phase and can be reproduced by mesiobasal limbic electrical stimulation (220; 327). In the later course of complex focal seizures, hyperpnea as well as hypopnea may occur.
Pupillary dilatation (ie, mydriasis) is a common symptom associated with the arrest reaction. Mydriasis is sometimes asymmetrical (329). Miosis and hippus pupillae have been observed. A feeling of “shivering cold” is sometimes associated with piloerection (342). Salivation is a common symptom, but lacrimation or nasal secretion is more rarely encountered. According to the study of Shah and colleagues, hypersalivation as a prominent ictal finding in complex partial seizures of temporal lobe origin is more likely to be of nondominant temporal lobe origin (279).
Gastrointestinal symptoms, such as vomiting, borborygmus, and eructation, often represent major seizure symptoms (144). Ictal vomiting and other symptoms of gastrointestinal alteration, such as ictal defecation (168) and ictal flatulence (302), were predominantly seen in nondominant temporal lobe seizures.
Bladder dysfunction, such as ictal urinary urgency, is a relatively infrequent symptom seen in nondominant temporal lobe seizures (183). There are occasional reports of penile erection or even ejaculation during focal seizures (300).
It was calculated that 6% of patients presenting with a single seizure would eventually develop MTLE-HS. Some patients with MTLE-HS have seizures that are readily controlled by medication, but those with medication-resistant forms are more often characterized because they present at tertiary epilepsy centers. Today, it is not clear whether the benign and severe forms of mesial temporal lobe epilepsy represent two different pathophysiological conditions or a spectrum of a single pathophysiological condition. Patients with dual pathology may have hippocampal sclerosis that is a nonspecific result of the primary epileptogenic lesion and not in itself epileptogenic; they may have hippocampal sclerosis that is secondary to the primary epileptogenic lesion but also epileptogenic. Chassoux and colleagues analyzed the distribution of the epileptogenic zone according to stereo-EEG with intralesional recordings in four patients evaluated for intractable partial epilepsy associated with focal unilateral polymicrogyria, involving the posterior temporal region in two, the perisylvian area in one, and the temporoparietal junction in the other (52). Although intralesional recordings demonstrated intrinsic epileptogenicity in polymicrogyria, these authors concluded that unilateral focal polymicrogyria belongs to a large epileptogenic network extending beyond the MRI lesion.
Several studies have concluded that patients with mesial temporal lobe epilepsy with MRI-identified hippocampal sclerosis are more likely to have intractable seizures than patients with other MRI-identified lesions. A large study from a tertiary center in Paris found that only 11% of patients with hippocampal sclerosis and only 3% with dual pathology had been seizure-free in the past year (277), whereas another study from a primary center in Glasgow, also found hippocampal sclerosis to be associated with the most intractable seizures but reported that 46% of these patients had been seizure-free in the past year (298). The difference between these observations in the tertiary and primary centers confirm a view that there is a relatively high incidence of benign MTLE-HS, although it is unclear how many of these patients appearing at the primary center were in their “silent” period and would eventually develop refractory epileptic seizures. A characteristic interictal behavioral feature of MTLE-HS is material-specific memory deficit, but this can also be seen with mesial temporal lobe epilepsy due to other mesial temporal lesions. Many other psychiatric and psychological problems, especially depression, have been reported to be more prevalent in MTLE-HS.
Latent and silent periods between the initial precipitating incidents and onset of habitual seizures have been described as characteristic features of MTLE-HS. But there are patients without identifiable initial precipitating incidents, and some have habitual seizures that begin immediately after the initial precipitating incidents. A silent period occurring between the first habitual seizure and the onset of intractability exists in many patients (24). The silent period, indicating that seizures are initially easily controlled for some time before they become medically refractory, strongly suggests that the pathological substrate is progressive. Progressive behavioral changes, particularly increasing memory deficit, and an increased appearance of contralateral EEG spikes over time are other observations in the same direction.
Treatment options.
Antiseizure medication therapy. The response is usually satisfactory in the beginning regardless of the antiseizure medication used. Later, however, a substantial percentage of patients have seizures that become resistant to medications used in monotherapy or in combination treatment. About 40% of patients with epilepsy have seizures that are resistant to medical treatment and can be considered for epilepsy surgery (173). The definition of drug-resistant epilepsy has evolved over the years and is based on the consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies; drug-resistant epilepsy is currently defined as failure of adequate trials of two tolerated, antiseizure medications in monotherapy or in combination to achieve sustained seizure freedom (172).
Surgery. Surgical therapy in medically refractory patients with mesial temporal lobe epilepsy is highly effective and renders about 80% of patients seizure-free (79; 346; 57). Most centers have modified temporal lobe surgery in mesial temporal lobe epilepsy to resect mesial temporal lobe structures more radically and to minimize lateral temporal lobe resection. Selective amygdalohippocampectomy with the trans-Sylvian approach (349; 355; 356; 358) or the subtemporal approach (131) and the so-called Spencer operation (resection of mesial temporal structures, of temporal pole, and of only a small amount of anterior lateral temporal cortex) have been strongly advocated in mesial temporal lobe epilepsy (61). This inclusion of the hippocampus with amygdala-piriform cortex complex, and the parahippocampal gyrus has been identified as indicative of a greater seizure freedom rate even when controlling for the total volume of the resection (102). There is evidence that sparing of the lateral temporal lobe cortex has advantages in terms of neuropsychological outcome and that originally hypometabolic lateral temporal lobe structures show a trend for normalization of their metabolism. In well-chosen candidates for amygdalohippocampectomy who already have unilateral material-specific memory and learning deficits, no additional clinically relevant deficits occur postoperatively, and the contralateral material-specific memory performance usually increases (333). Paglioli and colleagues reported data suggesting that postoperative verbal memory scores may even improve in patients who undergo selective resection of a sclerotic hippocampus in the dominant temporal lobe (232). Similar findings were reported with classical anterior temporal lobe resections (255). Patients without preexisting memory deficits, particularly those not becoming seizure-free following left temporal lobe resections, usually worsen in their verbal memory. To assist the prediction of postoperative memory and learning in patients considered to be at risk, intra-carotid amobarbital tests are sometimes used, but the use of this test has declined in recent years.
Stereotactic laser ablation has emerged as a surgical option that can selectively ablate the hippocampus with a less invasive procedure than either the standard resection or the selective amygdalohippocampectomy. Its comparative efficacy and safety are now established with efficacy that is slightly less than open resection but recovery that is usually faster (154; 69; 359). The selectivity varies according to the extent of the ablation volume, and a higher seizure-freedom rate has been associated with the percentage of the parahippocampal gyrus that is ablated (265). This may relate to the observation that parahippocampal gyrus connectivity abnormality is a leading predictor of seizure freedom after surgical treatment for temporal lobe epilepsy (103). As other surgical options, radiofrequency thermoablation and chronic electrical stimulation of the hippocampus are continuing to be developed (64; 175).
Without surgery, the prognosis of medically refractory patients with mesial temporal lobe epilepsy is relatively poor. Both severity and frequency of seizures may increase, and memory may decline, possibly resulting in severe psychosocial disturbances. Early surgical intervention (ie, relief of disabling seizures before the negative consequences of mesial temporal lobe epilepsy interfere critically with vocational and social development) results in the best psychosocial outcome (Khan and 333) and should be envisaged in this prototype of a surgically remediable epileptic syndrome. Several groups have also reported good surgical results in children with temporal lobe epilepsy (206; 216). Surgery-related major complication rates are low, with reports of below 1% from most centers (346).
The response to epilepsy surgery during the first follow-up year is a reliable indicator of the long-term postoperative outcome, which was found to be better in patients with a desecrate preoperative lesion, such as hippocampal sclerosis, and significantly worse in patients with the presence of preoperative secondary generalized tonic-clonic seizures (201). Discontinuation of antiseizure medications after successful epilepsy surgery is possible in a high percentage (270; 341; 336; 161). Seizure recurrence after planned discontinuation of antiseizure medications in seizure-free patients after epilepsy surgery can occur and does not correlate with any antiseizure medications of different mechanisms of action (07). The recurrence rate in adults in four studies was 33%, with maximum follow-up ranging from 1 to 5 years. In one study of children with temporal lobe epilepsy, the recurrence rate was 20%; more than 90% of adult patients with seizure recurrence regained seizure control with reinstitution of previous therapy (272). Seizure freedom without aura at 1 year or more is a reasonable indication for the attempt at medication discontinuation. Younger age at the time of surgery and a shorter disease duration seem to affect successful medication discontinuation for a long-term period.
A 45-year-old man had a history of unprovoked epileptic seizures for more than 20 years. His seizures started with queasy abdominal sensations rising to the throat, followed by loss of awareness. He was told that he stared into space, had lip-smacking mouth movements, and sometimes postured his right hand. The seizures lasted 20 to 30 seconds; thereafter, he remained confused for an additional 1 or 2 minutes before returning to baseline. His habitual seizures happened on average four to five times a month. In his lifetime, he had about three bilateral tonic-clinic seizures, and all happened after missing a few doses of medications. It had been 3 years since his last bilateral tonic-clonic seizure. At the time of presentation for this vignette, he was taking levetiracetam 1000 mg three times daily and oxcarbazepine 750 mg twice daily. He had tried four other antiseizure medications alone and in combination and had never achieved lasting remission except for a few months of improvement following the initiation of new medications.
The patient’s interictal and ictal EEGs showed left anterior temporal spikes. MRI showed left hippocampal atrophy. FDG-PET scan of the brain showed left mesial temporal hypometabolism.
Diagnosis. The patient was diagnosed with left temporal mesial epilepsy.
Classification of seizure. Seizure classification was focal onset seizures with impairment of consciousness.
The patient had left anterior temporal lobectomy and became seizure-free. (Grade 0, class I of Engel’s surgical outcome criteria at 2-year postsurgical visit.)
The oldest phylogenetic parts of the cortex differ from the 6-layered iso- or neocortex and are termed allocortex (321).
The allocortex consists of the archicortex (hippocampus and subiculum), the paleocortex (in essence, olfactory cortex), and the peri-archicortex (in essence, area entorhinalis, regio retrosplenialis, and cingularis). At the base of the temporal lobe, the rhinal fissure represents the border between allo- and isocortex.
Functionally, the most important difference between allo- and neocortex is that the allocortex does not have direct thalamic afferents, whereas the neocortex does (63), though there is an indirect pathway connecting hippocampal formation to thalamus through the mammillary nuclear tracts (138). The efferent pyramidal cells of the allocortex receive their inputs directly from the afferent fibers; that is, the output neurons are simultaneously the input neurons.
Phylogenetically, in parallel with the increasing neocorticalization, the percentage of allocortex in relation to the total cortex steadily decreases. In humans, the allocortex comprises only 4% of the total cortex. Broca coined the term “grand lobe limbique” for that region but without giving cytoarchitectonic references (40).
A part of the allocortex and its pathways has close anatomical and functional relations to the bulbus olfactorius and is, therefore, named rhinencephalon. In the older literature, the term rhinencephalon has been used in a broader connotation, including the totality of the allocortex and its subcortical connections. In the narrow sense, however, the rhinencephalon comprises only the bulbus olfactorius to the regio prepiriforms, the amygdala, the regio septalis, and the periseptalis.
The term limbic system is more comprehensive and embraces, according to the original definition of Papez, the allocortex and the connections of the hippocampus, ie, the fornix, the corpus mamillare, and further on from here, over the Vicq d’Azyr bundle (tractus mamillothalamicus), the anterior thalamic nucleus (239). From the thalamus, there are projections to the anterior cingulate cortex, and from the cingulum back to the hippocampus. Thus, this limbic circuit, proposed as a kind of reverberating circuit, includes the cingulate gyrus, which receives thalamic inputs and, thus, represents neo- or “transitional” cortex. Within the concept of the limbic system, the hippocampal-cingulate connections are considered to be of less importance, and several authors emphasize the connections of the limbic core structures to the hypothalamic regions, as well as (via nucleus accumbens) to the striatopallidal system that controls motor functions.
Initially, Papez proposed the limbic circuit as a kind of “mediator of emotions” (239); later he coined the term “visceral brain” (240). Nauta enriched the limbic system concept with the limbic midbrain area (219).
Hippocampal formation. In 1587, Arantius described the hippocampus, comparing the protrusion on the floor of the temporal horn to a sea-horse, a “hippocampus” (05). Winslow in 1732 suggested “ram’s horn” (352). In 1742, Garengeot probably introduced the term “cornu ammonis” by analogy with the horned Egyptian god Ammon (Amun) (72).
In current, most frequent terminology, the terms cornu ammonis and pes hippocampus are used synonymously. Although the terminology varies, most authors use “hippocampal formation” to refer to the “hippocampus proper” with its four cornu ammonis fields, plus dentate gyrus and subiculum. The name hippocampus applies to the entire ventricular protrusion and consists of two cortical laminae, rolled up one inside the other: the cornu ammonis and gyrus dentatus. The subiculum and entorhinal area represent a transitional cortex between the cornu ammonis and the rest of the temporal lobe and are sometimes joined to the hippocampus, constituting a functional unit--the hippocampal formation (55).
The hippocampus contains multiple fields. The innermost part is the dentate gyrus, which wraps around the end of the cornu ammonis and consists of fascia dentata and hilus. The dentate gyrus underlies by CA4 filed of the cornu ammonis followed by CA3, CA2, and CA1, which then extends to the subiculum
From its deepest level to the surface, ie, from the ventricle towards the hippocampal sulcus, the cornu ammonis is divided into seven layers: alveus, stratum oriens, stratum pyramidale, stratum lucidum, stratum radiatum, stratum lacunosum, and stratum moleculare. The dentate gyrus, on the other hand, is composed of layers: polymorphic layer, stratum granulosum, and stratum moleculare.
A striking feature of the connectivity of the hippocampal formation is that it has its cortical connections only through the entorhinal cortex, which itself, however, is a link in numerous multisensory corticocortical networks.
The intrinsic wiring of the hippocampal formation is mainly unidirectional. The predominantly superficial layers of the entorhinal cortex give rise to a powerful excitatory projection called the perforant path (which “perforates” the subiculum to reach the hippocampus).
The perforant path comprises glutaminergic fibers to the dentate gyrus and CA3. The axons of the granule cells of the dentate gyrus, the mossy fibers, have a large zinc content (202). Mossy fibers project to CA3 and CA4. Axons of CA3-CA4 enter the alveus and then the fimbria, but they first emit the Schaffer collaterals, which reach the apical dendrites of CA1 in the strata radiatum and lacunosum. The axons of CA1, by entering the alveus, produce collaterals that reach the subiculum. The subiculum emits by glutaminergic fibers the principal efferent pathways, which follow the fimbria, the crus, the body of the fornix, and then the postcommissural fornix (behind the anterior commissure) to reach the anterior thalamic nuclei, either directly or via the mamillary bodies.
The entorhinal cortex receives inputs from the sensory association areas of the temporal lobe, conveying visual, auditory, somatosensory, gustatory, nociceptive, and olfactory signals (316; 136; 137). These inputs are distributed along the rostrocaudal extent of the entorhinal cortex, with the olfactory inputs occupying the most rostral part.
Fibers from layers II and III that form the origin of the perforant path exhibit a further differentiation. Whereas fibers from layer II distribute almost exclusively to the dentate gyrus and CA3, fibers from layer III project exclusively to CA1 and the subiculum.
Thus, the perforant path is the main afferent to the hippocampus. A second path, the alvear path, was described by Cajal (44). Although the core elements of this neuronal chain (ie, entorhinal area, gyrus dentatus, Cornu Ammonis, and subiculum) are of disparate anatomy, they constitute a single functional unit and are, therefore, rightly grouped into the “hippocampal formation” (253; 291).
Nuclei amygdalae. The amygdalae are located directly above the temporobasal cortex and ventrorostral to the tip of the temporal horn of the lateral ventricle. Each is usually differentiated into three main nuclear groups: the phylogenetically older corticomedial, the younger basolateral, and a central group. In essence, the corticomedial group receives afferences from various areas of the olfactory cortex, in particular from the periamygdalar cortex, whereas the basolateral group receives its afferences from the inferotemporal neocortex (Brodman area 20).
In the rhesus monkey, nearly all cortical areas of the temporal lobe, major parts of the frontal lobe, and the insular cortex project to the amygdala (21). These projections have a differential distribution within the amygdala. Some parts of some amygdaloid nuclei receive several cortical sensory projections. The lateral part of the central nucleus, the laterobasal nucleus, and the dorsomedial part of the lateral nucleus are areas where substantial convergence of cortical input occurs. It is well-documented that visual, auditory, olfactory, and, to some extent, taste information reaches the amygdala. Somatic sensory input is less clear, but there is reason to believe that all five modalities have some convergence in the dorsomedial part of the lateral nucleus. For example, the dorsomedial part of the lateral nucleus receives projections from the orbitofrontal area, which responds to olfactory stimulation, and this part is also the major amygdaloid projection zone of the cortical taste area. In addition, posterior insular cortex projections to this area carry visceral and probably other somatic information. Moreover, auditory input from the temporal polar cortex projects powerfully to this region. Visual projections are directed primarily to the dorsolateral part of the lateral nucleus.
Efferent fibers from the amygdala are the stria terminalis and, to a lesser degree, the ventrofugal bundle with overlapping targeting areas in the medial and rostral hypothalamus (nuclei ventromedialis and dorsomedialis hypothalami, regio praeoptica), regio septalis (nuclei lateralis septi, “bed nucleus” of the stria terminalis, diagonal band), as well as the posterior part of the magnocellular nuclei dorsomedialis thalami. The latter connects the amygdala with the orbitofrontal cortex and constitutes a part of the second, ie, “basolateral limbic circuit,” formulated by Yakovlev and reemphasized by Livingston and Escobar (354; 182).
Most of the amygdala nuclei, except the medial subdivision of the central nucleus, have low afterdischarge threshold (280) and are sensitive to kindling due to its high concentration of glutamate (266), which likely determines the high propensity for epileptogenicity of the amygdala. With its abundant connections to other medial temporal structures, the amygdala plays a significant role in the pathophysiology of mesial temporal lobe epilepsy. Animal studies show that medial division of central nuclei may suppress propagation to the contralateral hemisphere. Neuroanatomical connections of amygdala with claustrum-insula-perirhinal cortex may also play a role during transition between oral automatism to tonic-clonic convulsion (280).
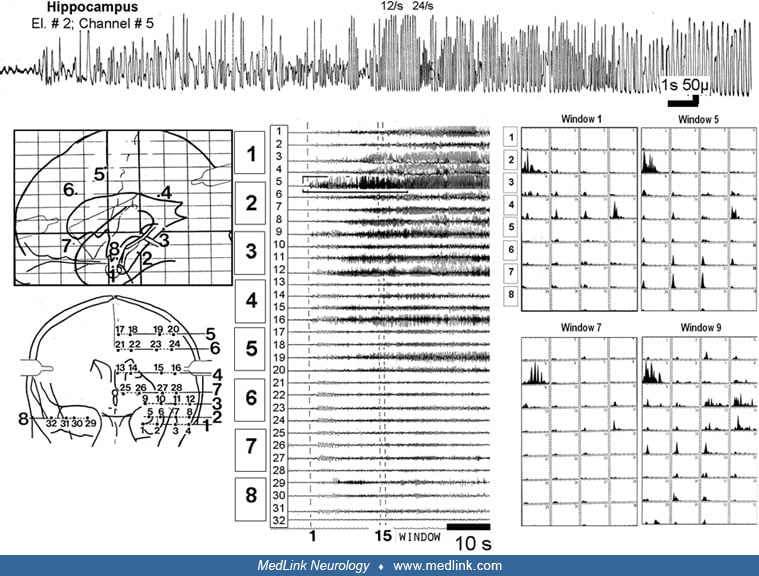
Functional anatomy of the hippocampal formation. In both animals and humans, hippocampal slice experiments have been a cornerstone of the investigation of the basic mechanism of epileptogenesis and have clarified the functional anatomy of the hippocampal formation. The trisynaptic pathway is tightly restricted in the rostrocaudal dimension so that separate lamellae can be identified. According to this plan, the hippocampal formation is organized in a series of functional lamellae arranged perpendicularly to the longitudinal axis of the hippocampal formation and operating independently. Because of this lamellar organization, thin slices can be cut that preserve at least some important hippocampus functions during maintenance in vitro. Hippocampal slice studies have demonstrated that hippocampal CA3, but not CA1, pyramidal cells become pacemakers for interictal spiking. This disparity can be accounted for by certain intrinsic membrane conductances in CA3 pyramidal cells along with recurrent excitatory connections between CA3 pyramidal cells, both lacking in CA1.
On the other hand, ictal events develop in CA1 but not in CA3 (185). The dentate gyrus serves as a critical control point for regulating epileptogenesis. Biochemical operations and morphology of dentate granule cells are altered as a function of epilepsy. The granule cells at this location undergo an enduring decrease of calbindin (a calcium-binding protein) in kindled animals (12). Several studies showed altered neurogenesis in the epileptic dentate gyrus. Evidence suggests that seizure-induced neural proliferation (22) and altered synaptic integration of dentate gyrus (364) play a significant role in an early phase of epileptogenesis. However, during the chronic phase, there is a decline of dentate gyrus neurogenesis, which is implicated in the development of spontaneous recurrent motor seizures, depression, and impairment of memory and learning (120).
Functional loops between the basal ganglia and the temporal lobe. With a view towards some of the well-known symptoms of temporal lobe seizures, such as déjà vu, jamai vu, macropsia, micropsia, delusions of familiarity, depersonalization, and the rediscovered contralateral tonic or dystonic posturing (190; 03; 167; 221), the connections of the temporal lobes with the basal ganglia are important.
The basal ganglia (ie, the caudate, the putamen, and the ventral striatum) are known to receive inputs from the frontal, parietal, and temporal lobes. The output from the basal ganglia via the thalamus addresses the frontal motor and the prefrontal cortex. Basal ganglia loops with the cortex have been thought to “funnel” or collect information from diverse cortical areas to direct motor output and the executive functions of the frontal lobe. Evidence has been provided that the output nuclei of the basal ganglia, the substantia nigra pars reticulata, projects via the thalamus to the inferotemporal cortex (area TE), ie, targets at least one visual area in the inferotemporal cortex (207), which is critically involved in recognition and discrimination of visual objects.
This might explain why dysfunction of the basal ganglia loop with the inferotemporal cortex leads to alterations in visual perception, including visual hallucinations. Mishkin and colleagues have proposed that visuomotor associations, including “habit memories,” involve the “visual striatum,” (ie, the tail of the caudate) and caudal or ventral portions of the putamen (11; 41; 191). These findings explain that damage to basal ganglia impairs visual perception. They also explain visual hallucinations produced by lesions in substantia nigra pars reticularis or brainstem compression (“peduncular hallucinosis,” seeing fully-formed images of people or animals), and possibly also visual hallucinations seen as a major side effect of the use of L-dopa or other dopaminergic agents.
Moreover, these findings may account for the dystonic posturing, an important lateralizing sign, and occur in 15% to 70% of patients with mesial temporal lobe seizures (167; 86). Indeed, ictal SPECT studies have equated this dystonic posturing with increased blood flow in the basal ganglia ipsilateral to the seizure onset (221).
The pathology, which is related to the etiological factors of mesial temporal lobe epilepsy, differs in adults and children. The most common pathologic substrate of mesial temporal lobe epilepsy in adults and older children is hippocampal sclerosis (38; 10; 256); in children below the age of 6 years old, the most common pathologic substrates are prenatal developmental, neurogenetic, and low-grade neoplastic abnormalities (256). The two most common pathologic terms that have been used synonymously with hippocampal sclerosis are mesial temporal sclerosis and Ammon’s horn sclerosis, although these terms imply different degrees of anatomical involvement (87; 192; 06). Other specific types of hippocampal pathology associated with mesial temporal lobe seizures include tumors (eg, gangliogliomas, dysembryoplastic neuroepithelial tumors, astrocytomas, oligodendrogliomas, pleomorphic xanthoastrocytomas or other low or high-grade gliomas), dysgenetic malformations, and vascular malformations (56) as well as postinfectious and autoimmune encephalitis (114; 31; 181). The term mesial temporal sclerosis has advantages because it more appropriately accounts for the amygdala often having equally severe pathology of neuronal loss and gliosis as the hippocampus does (105).
Hippocampal sclerosis. The term hippocampal sclerosis should only be used for the specific type of hippocampal cell loss with marked loss of neurons in the CA1 and hilar region and some loss in endfolium (CA3/CA4) but relative sparing of the CA2 region. The subicular complex, entorhinal cortex, and other transitional cortex and temporal gyri are relatively resistant to cell loss. Hippocampal sclerosis is associated with other characteristic features, such as mossy fiber sprouting (303) and selective loss of somatostatin and neuropeptide Y-containing neurons (68). It is found in up to 70% of patients with medically refractory temporal lobe epilepsy who undergo surgery (10). In epilepsy specimens, the granule cell layer is often wider and somehow disorganized (132). Dispersed granule cells may extend into the surrounding molecular layer and sometimes show a bilaminar pattern.
However, the pathology of mesial temporal lobe epilepsy is not completely uniform; other cerebral diseases can also cause hippocampal damage but usually show a different pattern with variable accentuation of special subfields, mostly involving the CA2 region. Gliosis might involve anterior mesial temporal lobe structures in addition to the hippocampus and might consist of “dual pathology.” Moreover, in autopsy specimens from patients with intractable seizures, hippocampal sclerosis is very often a bilateral condition, although frequently with a unilateral preponderance (192). Blümcke and colleagues have proposed a new clinicopathological classification system for mesial temporal sclerosis (34). Chronic, ambulatory hippocampal recordings have characterized this; however, patient selection was based on the high likelihood of bilateral hippocampal seizures (281; 162). Nevertheless, considerable variability in the lateralization ratio has been observed.
The events that initiate the process of hippocampal sclerosis are not entirely known in detail, but there is no doubt that the epileptogenicity of this disorder results from the loss of specific neurons in the hippocampus and synaptic reorganization of surviving cellular elements with resulting hypersynchronization and hyperexcitability (82). Mathern and colleagues showed that patients with initial precipitating injuries had hippocampal sclerosis, whereas those with idiopathic temporal lobe epilepsy showed fewer neuron losses and worse post-resection seizure relief (195; 197). Patients with nonseizure initial precipitating injuries were on average older at injury, had a longer latent period, and showed fewer neuron losses in Ammon’s horn, CA1, and prosubiculum than those with seizure-associated initial precipitating injuries. Initial precipitating injury patients with repetitive, nonprolonged seizures showed the shortest latent period, earliest age of temporal lobe epilepsy onset, and less CA2 damage than the other initial precipitating injury groups. CA1 and prosubiculum neuron losses were greater in patients with temporal lobe epilepsy for longer than 22 years. Initial precipitating injuries after age 4 years were associated with latent periods shorter than 10 years compared with variable and longer latent periods of initial precipitating injuries before 4 years of age. Similarly, O’Brien and colleges observed the high rate of early precipitating events in patients with mesial temporal sclerosis. In their series of 31 patients with mesial temporal sclerosis and 14 patients with temporal neocortical lesions, they found that more patients with mesial temporal sclerosis (58%) had a history of febrile convulsions than patients with temporal neocortical lesions (26%). Also, more patients with mesial temporal sclerosis had an early, before age 4, cerebral event than the patients with temporal neocortical seizures (225). The results of Mathern and colleagues indicate that in surgically treated temporal lobe epilepsy, hippocampal sclerosis and good seizure outcomes are associated with initial precipitating injuries (195; 197). Most of the hippocampal damage found at surgery and the clinical time course of the habitual temporal lobe epilepsy are influenced by the pathogenic initial precipitating injury mechanism. The same group also studied the correlation of the type of epileptic seizures and the duration of epilepsy with the development of mesial sclerosis by reviewing different pathological specimens of pediatric patients with hippocampal, temporal neocortical, and extratemporal seizures (194). They concluded that childhood hippocampal seizure as an initial precipitating event can damage the hippocampal granular cells, causing an early neuronal loss and development of the aberrant neuronal circuits, which subsequently results in development of chronic hippocampal seizures. They also showed that childhood extrahippocampal generalized seizures do not impact the hippocampus and cause hippocampal sclerosis.
Besides a proven role of an early precipitating event, such as febrile seizures, in the development of mesial temporal sclerosis, studies show a link between HHV6B virus and mesial temporal sclerosis (156). Theodore and colleagues demonstrated that two thirds of patients with mesial temporal sclerosis had findings for active HHV6B virus, likely causing noninflammatory excitotoxicity leading to the development of mesial sclerosis (309). Also, studies show association of HHV6, a pathogen for common childhood infection roseola, with prolonged febrile convulsion (84), which by itself is a major risk for the development of mesial sclerosis.
Mody and Heinemann found that dentate granule cells in the kindled animal are more sensitive to agonists that activate the N-methyl-D-aspartate (NMDA) type of glutamate receptors compared to nonkindled animals (212). Binding studies indicate that dentate granule cells have abundant receptors for NMDA (20) and that their expression and operation are altered after kindling (357). These findings are very interesting in light of experimental results showing that maximal dentate activation and changes in maximal dentate activation can be opposed by NMDA antagonists (301).
Morphological changes of dentate granule cells as a consequence of epilepsy were initially reported by Tauck and Nadler, who demonstrated sprouting of the axons (mossy fiber sprouting) in animals (307). Several laboratories have documented that it also occurs in the human epileptic brain (304; 303; 196; 254). It is reasonable to assume that mossy-fiber sprouting contributes significantly to epileptogenesis by providing recurrent excitation of adjacent cells.
In a (1)H magnetic resonance spectroscopy study of temporal lobe epilepsy patients, Hetherington and colleagues found that the ratio of N-acetyl aspartate to creatine (NAA/Cr), a measure of neuronal injury and loss, was significantly reduced in both the ipsilateral and contralateral hippocampi and thalami (127). NAA/Cr in the ipsilateral hippocampus was significantly correlated with the ipsilateral and contralateral anterior and posterior thalami, putamen, and contralateral hippocampus. Müller and colleagues found clear differences in T2 relaxation patterns and rate in temporal lobe epilepsy with versus without mesial temporal sclerosis (215).
Besides the morphological changes primarily affecting the hippocampus, patients with mesial temporal lobe sclerosis also have findings supporting the involvement of the neocortical temporal lobe. Thom and colleagues studied the resected temporal lobe specimens of 272 patients with mesial temporal lobe sclerosis (311). Thirty of 272 patients had neocortical temporal lobe sclerosis findings, more severely affecting the temporal pole, in addition to the typical hippocampal sclerosis. Temporal lobe changes showed a reduction of neurons in layer II/III and laminar gliosis.
Parent and Lowenstein as well as Scharfman reviewed seizure-induced neurogenesis (241; 268). Several lines of evidence implicate newly generated neurons in structural and functional network abnormalities in the epileptic hippocampal formation of adult rodents. These abnormalities include aberrant mossy-fiber reorganization, the persistence of immature dentate granule cell structure (eg, basal dendrites), and the abnormal migration of newborn neurons to ectopic sites in the dentate gyrus. These findings suggest a pro-epileptogenic role of seizure- or injury-induced neurogenesis in the epileptic hippocampal formation. Conversely, neurogenesis also might contribute to network homeostasis; Jakubs and colleagues found that new neurons born into the pathological environment differed regarding synaptic drive and short-term plasticity of excitatory and inhibitory afferents (145). The new granule cells formed in the epileptic brain exhibited functional connectivity consistent with reduced excitability. By this means, adult neurogenesis might contribute to network homeostasis in the epileptic temporal lobe (158).
Recurrent seizures in infancy may interrupt synapse maturation and produce persistent decreases in molecular markers for glutamatergic synapses—particularly components of the NMDA receptor complex implicated in learning and memory (305).
“Lesional” temporal lobe epilepsy. This is defined by lesions in the temporal lobe other than hippocampal sclerosis. In the Montreal series (1928-1973) of surgically excised epileptogenic temporal lobes, Mathieson studied 857 of 878 patients and found in 202 patients the following discrete focal lesions: gliomas and gangliogliomas (105), meningocerebral cicatrix and remote contusion (39), vascular malformations of brain or pia (19), hamartomas (14), residuum of cerebral abscess (10), tumors other than gliomas (10), tuberous sclerosis and formes frustes (4), and residuum of old infarct (1) (198). Even higher percentages of lesions were found in a neuropathological study enrolling 247 patients with mesial temporal lobe resections (252). Neoplasms and microscopic clusters of oligodendroglia-like foci of 30 to 100 cells (interpreted as precursor lesions for neuroepithelial tumors) were found in 126 patients. Similar results of lesional mesial temporal lobe epilepsy for other etiologies than mesial sclerosis were found by Clusmann and colleagues (56). In their series of 78 patients with partially resected mesial temporal lobe for lesions other than mesial sclerosis, they found ganglioglioma and dysembryoplastic neuroepithelial tumors to be the most prevalent pathologies, followed by low-grade tumors and dysplasias. Thus, neuroectodermal tumors, predominantly low-grade gliomas and gangliogliomas, are frequently encountered. Although focal dysplasia was already precisely described as early as 1971 (308), dysembryoplastic neuroepithelial tumors (67; 66) and several types of cortical dysgenesis have only been increasingly recognized in more recent years, mainly as a result of improved in vivo diagnosis due to the advent of high-resolution MRI. However, microdysgenesis is still not visible on neuroimaging and remains a pathological diagnosis. Thus, it is likely that several subtle forms of migrational disorders are underdiagnosed in most series (236; 237; 238).
A useful classification scheme for cortical dysgenesis presenting with epilepsy in general was proposed by Raymond and colleagues (257). These authors also list “dysgenesis of the archicortex (duplication or dispersion of the dentate fascia, ie, most probably some forms of hippocampal sclerosis).” The latter, ie, the disorganization of the dentate gyrus in temporal lobe epilepsy, has increasingly attracted researchers (132; 133; 205). Of 34 temporal lobe specimens from patients with temporal lobe epilepsy, Mello and colleagues found the granule cells normally arranged in 44%, generally dispersed in 38%, and displayed in a bilaminar arrangement in 18% (205). Varying degrees of hippocampal sclerosis were observed in all cases. The pathogenesis of these abnormalities is controversial. Arguments for the pre-and postnatal mechanisms have been put forward as well as evidence for being both the cause and the effect of epilepsy. Some arguments are in favor of a migrational defect: the occurrence of granule cells in the molecular layer suggests that they have migrated beyond their target position; the presence of granule cells in the hilus of the dentate gyrus suggests that some cells have not completed their normal migration; their elongated form is reminiscent of developing forms; and their alignment in vertically-oriented rows is similar to the radial arrangement of migrating cells (133). Mathern and colleagues quantified hippocampal mossy-fiber synaptic reorganization and neuron losses to determine the pathological features associated with epileptogenic fascia dentata, differentiating patients with temporal lobe epilepsy and mesial temporal sclerosis with seizure genesis in the hippocampus, or temporal mass lesions with seizures that were probably extrahippocampal (196). They found that inner molecular layer mossy-fiber puncta densities and neuron losses are greater in patients with mesial temporal sclerosis than in those with lesions and concluded that mossy-fiber sprouting contributes to the pathophysiology of hippocampal seizures. However, some patients with extrahippocampal lesions had mossy-fiber sprouting similar to mesial temporal sclerosis patients, suggesting that hippocampi in lesion patients may also be capable of epileptogenesis from synaptic reorganization.
Several subtypes of MTLE-HS might exist, including at least a primary form, possibly a secondary form, and a familial form (165; 91).
Neuronal loss and gliosis. Neuronal loss and gliosis are typical pathological findings in mesial temporal lobe epilepsy but do not, per se, prove an active seizure disorder. Studies show autopsy specimens of hippocampal pathology without antemortem seizures (322; 59; 204). In other words, not every damaged hippocampus is epileptogenic, and not every severe seizure history causes hippocampal sclerosis.
Dual pathology. If hippocampal sclerosis is associated with other temporal lobe pathology, we speak of a dual pathology (177). Cortical microdysgenesis and cortical dysplasia (ie, alterations of neuronal migration), as well as hamartomas, small tumors, and cavernomas, may be found in addition to hippocampal sclerosis. Diagnoses such as microdysgenesis, heterotopia, and hamartomas depend heavily on the pathological criteria to define an area as abnormal. It is also not clear what pathology is responsible for seizure genesis. Numerous studies have retrospectively analyzed the neuropathological findings in patients with temporal lobe epilepsy, including such with proven seizure onset in the hippocampal formation (290; 296; 198; 60; 70; 203; 42; 85; 252; 353; 195; 34). Salanova and colleagues studied the occurrence of dual pathology in their series of 240 patients with temporal lobe epilepsy who underwent temporal resections following a comprehensive pre-surgical evaluation (263). Thirty-seven (15.4%) of these had hippocampal sclerosis or temporal lobe gliosis in association with another lesion (dual pathology). Eighteen of 37 patients with dual pathology had heterotopia of the temporal lobe; nine had cortical dysplasia; four had cavernous angiomas or arteriovenous malformations; one had a dysembryoplastic neuroepithelial tumor; one had a contusion; and four patients had cerebral infarctions in childhood. Abnormal head MRI was found in 68.5% of the patients, abnormal positron emission tomography scans in 91.3%, and abnormal ictal SPECT in 96%. The intracarotid amobarbital procedure showed impaired memory of the epileptogenic side in 72% of the patients. Twenty patients had left-sided and 17 had right-sided en bloc temporal resections, including the lesion and mesial temporal structures. Twenty-six (70.2%) became seizure-free; eight (21.6%) had rare seizures; two (5.4%) had worthwhile seizure reduction, and one (2.7%) had no improvement (range of follow-up 1 to 16 years, mean = 7.4 years). The dual pathology was almost exclusively seen in patients whose lesions were congenital or occurred early in life.
Besides pathological findings, some clinical features help to define MTLE-HS, such as early insults, severe febrile convulsions, age and modes of onset, higher incidence of seizures within a family, clinical course, development of drug-resistance, and material-specific memory deficits (284; 285; 48; 50; 93; 134; 351; 276; 197).
Some patients with familial mesial temporal lobe epilepsy are documented to have intractable seizures with hippocampal sclerosis; in these patients, the genetic defect may cause mesial temporal lobe epilepsy, which then leads to hippocampal sclerosis with or without febrile seizures (51; 166). There is no evidence to suggest that familial partial epilepsies with variable foci, partial epilepsies with auditory features, or temporal lobe variants of benign childhood epilepsy with centrotemporal spikes ever evolve into MTLE-HS.
Both benign childhood epilepsy with centrotemporal spikes and mesial temporal lobe epilepsy can begin in childhood with generalized seizures. However, the partial seizures of benign childhood epilepsy with centrotemporal spikes usually have sensory- or motor-lateralized symptoms localized around the mouth or the upper extremities. Interictal EEG spikes are also different in these two syndromes: The broad centrotemporal EEG spike is located more posteriorly and superiorly and has a characteristic transverse dipole, whereas in mesial temporal lobe epilepsy, the spike or spike-wave discharges are located more anteriorly and basally with a characteristic oblique dipole direction. Differentiation from temporal lobe epilepsy due to other lesions in or near the mesial temporal lobe is usually easy by MRI. Clinical signs and symptoms might be similar, although in mesial temporal lobe epilepsy, age of seizure onset is usually earlier, and there is often a history of complicated febrile seizures and an increased incidence of family members with seizures.
The “unknown” temporal lobe epilepsy denotes temporal lobe epilepsies without a pathological substrate. It is difficult to estimate the incidence of this epilepsy category. Available figures from surgical series are heavily biased, as are the figures of the familial temporal lobe epilepsy syndrome with a benign course (26). Although in Mathieson’s surgical series no histopathological abnormality was found in 173 (20%) of 857 resected temporal lobes (198), Plate and colleagues’ Zurich study found no pathology in only 2% of 224 available mesial temporal lobe tissue specimens (252). Because the surgical outcome of patients without pathology is usually worse than those with histological abnormalities, it must be considered that in patients without histopathological abnormalities and those who did not become seizure-free following temporal lobe resection, the temporal lobe was not the origin of the seizures. Therefore, this category might soon disappear in surgical series with more sophisticated diagnoses.
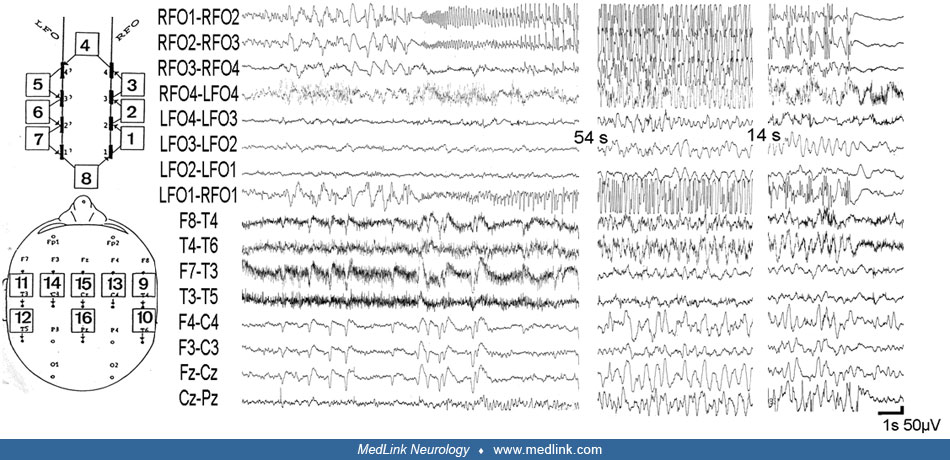
Lateral neocortical temporal lobe seizures are much rarer, and they usually show a lesion invading the lateral temporal cortex alone or in combination with the insula. However, seizures of lateral temporal origin and without a gross morphological lesion do exist and have been documented by stereoelectroencephalography (SEEG) (329). Such seizures, as a rule, spread to the ipsilateral mesial temporal structures, which may act as a kind of “amplifier” sustaining and prolonging the seizure discharges. Clinical signs and symptoms supportive of seizures arising from the lateral temporal neocortex commonly derive from epileptic discharges involving the cortex of more than one lobe. No absolute specific symptoms and signs are indicative of lateral temporal seizure onset, but the following symptoms are more frequently encountered in lateral neocortical temporal seizures: ictal aphasia if the dominant hemisphere is involved; auditory hallucinations if the posterior insula (Heschl gyrus) and the superior temporal gyrus are involved (326; 348); and vestibular hallucinations, which have been documented with posterior-temporal-parietal discharges (329). Visual hallucinations, in particular macropsia and micropsia, as well as teleopsia, plagiopsia, dysplatopsia, achromatopsia, or erythropsia and xanthopsia, loss or enhancement of 3-dimensional apperception, polyopia, palinopsia, and quick-motion as well as slow-motion, can occur with discharges in the temporo-parieto-occipital junction and inferotemporal cortex.
Motor symptoms with contralateral tonic or clonic manifestations and head or eye deviation are more frequently seen in neocortical lateral than in mesiobasal seizures, whereas dystonic posturing occurs more frequently (in about 40% of patients) with documented mesial temporal lobe onset seizures.
Insular seizure onset has been found in six of 50 consecutively SEEG-explored patients by Isnard and colleagues (139). This group reported a relatively typical ictal semiology beginning with a sensation of laryngeal constriction and paresthesiae, often unpleasant, affecting large cutaneous territories, most often at the onset of a complex partial seizure. It was eventually followed by dysarthric speech and focal motor convulsive symptoms. The insular origin of these symptoms was supported by the data from functional cortical mapping of the insula by using direct cortical stimulations.
Seizures of frontal lobe origin usually have features (eg, brief and clustered, no postictal disturbances, nocturnal predilection, hyperkinetic automatisms) that help to distinguish them from temporal onset seizures. However, the differential diagnosis might be difficult without invasive recordings if frontal lobe onset seizures invade one or both temporal lobes (148).
Hippocampal and parahippocampal seizures are the hallmarks of mesial temporal lobe epilepsy. At the Istanbul ILAE Meeting, the Commission on Neurosurgery discussed in depth whether mesial temporal lobe epilepsy with hippocampal sclerosis is a syndrome. More participants were in favor of considering MTLE-HS a subtype of a greater syndrome of mesial temporal lobe epilepsy rather than a distinct syndrome, implying that mesial temporal lobe epilepsy, regardless of the cause, represents a sufficient cluster of signs and symptoms to make up a syndromic diagnostic entity. There was no consensus, however, and arguments against this position depended on the actual or potential features that make MTLE-HS unique—such as its progressive nature, genetic predisposition, and initial precipitating incidents—whereas the other subtypes due to specific static or neoplastic etiologies could be considered diseases. It was generally agreed that there were several subtypes of MTLE-HS, including at least a primary form, possibly a secondary form, and a familial form with the further differentiation in functional abnormalities (33; 58). A major point is that MTLE-HS appears to involve areas of structural and functional disturbances that are much more extensive than the hippocampus and maybe even the mesial temporal area (336). Seizures originating in the amygdala are relatively rare and usually invade the hippocampus (335; 335).
Diagnosis of MTLE-HS requires a constellation of signs and symptoms and cannot be made based on a single criterion. Today, hippocampal atrophy and hippocampal sclerosis can be reliably diagnosed in vivo with MRI (25; 351; 171). MRI distinguishes MTLE-HS from MTLE due to other lesions. However, diagnosing hippocampal and parahippocampal seizures relies on typical ictal onset location and pattern (259; 74). It strongly depends on semi-invasive and invasive EEG, although there are relatively typical scalp EEG findings in mesial temporal lobe epilepsy.
EEG. Interictal regional slowing in patients with temporal lobe epilepsy not associated with a mass lesion is topographically related to the epileptogenic area and, therefore, has a reliable lateralizing, and possibly localizing, value. Its presence is irrelevant to the severity or chronicity of the epilepsy and to lateral deactivation secondary to neuronal loss in the mesial temporal structures. Although slow EEG activity is generally considered a nonspecific sign of functional disturbance, interictal regional slowing in temporal lobe epilepsy should be conceptualized as a distinct electrographic phenomenon directly related to the epileptogenic abnormality. There is a strong correlation between interictal regional slowing and lateral temporal hypometabolism (169). In hippocampal background EEGs recorded from patients with temporal lobe epilepsy, both delta and theta spectral components are present. Zaveri and colleagues found group differences in spectral measures of background hippocampal signals recorded from patients with mesial temporal sclerosis compared to patients with the so-called “paradoxical temporal lobe epilepsy,” which is characterized by minimal cell loss and comparatively poorer surgical outcome, suggesting that substrate differences in cellular composition and connectivity are reflected in hippocampal background EEGs (362). Significant diagnostic value is given to lateralized rhythmic delta activity. It is seen predominantly in temporal lobe epilepsy (224), and it differs from other focal or diffuse slowing, which are relatively nonspecific findings and are seen in a variety of focal or diffuse brain dysfunctions or structural damages.
Interictal typical epileptiform graphoelements in scalp EEG are grouped, blunt sharp-waves with or without slow waves that recur with a frequency of about one per second. They show a characteristic distribution with a maximum of their field in basal anterior electrodes (such as sphenoidal or true temporal electrodes). They may be unilateral, bilateral-dependent, or bilateral-independent (327; 330; 338).
Ictal scalp EEG findings typically consist of fairly regular theta-rhythms of about five per second with a crescendo-like amplitude increase paralleled by slowing of the discharge rhythms.
Seizure onset in scalp EEG might be characterized by regional temporal or generalized attenuation of background EEG rhythms with the disappearance of interictal “spikes.”
Direct recording from the temporal lobe, including the hippocampal formation, often shows that hippocampal spikes remain localized. However, they might propagate to the amygdala or lateral and anterior temporal areas.
Hippocampal seizure onset often is with the so-called “hypersynchronous hippocampal discharge pattern” followed by a low-voltage, fast-recruiting rhythm of more than 20 Hz (78; 338).
The other frequent seizure onset pattern of hippocampal seizures is the low-voltage, high-frequency discharge.
These two patterns may both predict hippocampal tissue loss but of different degrees and distribution (313; 287; 288; 317). Out of 478 seizures recorded in 68 patients with temporal lobe seizures who were undergoing diagnostic monitoring with depth electrodes, Velasco and colleagues found that the seizure onsets in 78% of these patients were either hypersynchronous onsets beginning with low-frequency, high-amplitude spikes, or low-voltage fast onsets increasing in amplitude as the seizures progressed (317). Nearly twice as many patients were having hypersynchronous seizure onsets as those having low-voltage, fast onsets. Patients with hypersynchronous seizure onsets had a significantly greater probability of having (1) focal rather than regional seizure onsets, (2) seizures spreading more slowly to the contralateral mesial temporal lobe, and (3) cell counts in resected hippocampal tissue showing greater neuronal loss. These results provide evidence that the most frequent electrographic abnormality associated with mesial temporal seizures is local hypersynchrony, a condition associated with major neuronal loss in the hippocampus. Low-voltage, fast seizure onsets more frequently represent widely distributed discharges, which interact with and spread more rapidly to surrounding neocortical areas.
Bartolomei and colleagues studied the pre-ictal synchronicity in limbic networks and found that the time interval that precedes the rapid discharge was characterized by significant cross-correlation values, indicating strong interactions among mesial temporal structures as compared to those seen during background activity (18). Interactions between the hippocampus and entorhinal cortex were predominant in 83% of patients.
Interactions between the entorhinal cortex and amygdala were observed in 50% and between the amygdala and hippocampus in 58% of patients.
Analysis of coupling directionality indicated that most of the couplings were driven either by the hippocampus or the entorhinal cortex. During rapid discharges, there was a significant decrease in cross-correlation values. At high-frequency tonic discharge seizure onset, significant interactions between the hippocampus and entorhinal cortex were observed. The entorhinal cortex was found to be the leader structure in most of the seizures with tonic discharge in the mesial structures, whereas the hippocampus was the leader in “type 1 transition” (similar to the hypersynchronous seizure onset pattern) in which a rapid discharge followed the emergence of pre-ictal spiking.
32-channel stereoelectroencephalographic referential recording of a spontaneous hippocampal onset seizure. The recording from the left hippocampus (inner contacts of depth electrode 2 = channel 5) is enlarged at the top of the ...
In 63% of patients, volumetric measurements of the entorhinal cortex demonstrated atrophy ipsilateral to the epileptic side. A significant correlation was found between the strength of entorhinal cortex-hippocampus coupling and the degree of atrophy. In patients with a normal entorhinal cortex volume, the entorhinal cortex was never the leader structure in entorhinal cortex-hippocampus coupling (16). Although with SEEG, we have found that amygdala onset seizures are relatively infrequent, ie, constitute about 3% to 5% of psychomotor seizures (327), Gotman and Levtova reported in their phase-coherence study that the amygdala was leading in 21% of focal-mesial and 53% of regional temporal lobe seizures, and the hippocampus was leading in 48.5% of focal mesial and in 27% of regional temporal lobe seizures (109). In the remaining seizures, discharges were synchronous in these two structures.
Bartolomei and colleagues also confirmed a strong unidirectional or bidirectional coupling between the amygdala and hippocampus in seizures of mesial temporal origin (17). In this seizure subtype, there was no significant coupling between medial and lateral structures at the beginning of the seizures.
Seizure propagation. Seizure spread is important in accounting for intermediate and late clinical signs and symptoms. In MTLE-HS the “symptomatogenic zone” might include insular and lateral neocortical temporal cortices. In general, the following aspects are important:
Compared to seizures in other brain areas, seizure spread is slow in MTLE-HS. Seizure spread is not random but follows preferred, stereotyped propagation pathways, which reflect at least to some degree the physiological connectivity of brain areas. A characteristic spread is into the posterior cingulate gyrus.
The mode of transhemispheric propagation is not entirely clear; it might be transcallosal after the ipsilateral frontal lobe is “ictally” activated. Spread to the ipsilateral neocortical temporal lobe might be seen as often as spread to the contralateral mesial temporal lobe structures without involvement of the ipsilateral neocortical temporal lobe. At present, there are no good estimates on the frequency of these two scenarios. Often, ipsilateral frontal or orbitofrontal cortices are invaded ictally (but this observation is based on depth recordings with the sampling error) (180; 331).
32-channel stereoelectroencephalographic referential recording of a spontaneous hippocampal onset seizure. The recording from the left hippocampus (inner contacts of depth electrode 2 = channel 5) is enlarged at the top of the ...
About 30% of hippocampal onset seizures appear first in the contralateral hippocampus before the ipsilateral or contralateral temporal neocortex. There is some evidence that ipsilateral frontal or orbitofrontal ictal involvement is not a necessary condition for seizure spread to the contralateral mesial temporal lobe. Contralateral propagation is usually accompanied by a marked disturbance or loss of consciousness. The mean interhippocampal seizure propagation time is around 15 seconds (332). A long interhippocampal seizure propagation time predicts surgical success (180) and was inversely correlated with cell counts in the CA4 region (289).
Some localized hippocampal discharges might occur without any noticeable clinical accompaniments, and others may produce “minor” symptoms only. The electrographic discharges without subjective or objective neurologic or somatic manifestations are known as subclinical seizures. They are commonly seen during long-term intracranial EEG recording and relatively rarely observed during scalp EEG monitoring. Most subclinical seizures have a hippocampal origin, though they can arise in extratemporal or temporal neocortical regions (361). Usually, they do not propagate beyond the area of origin and involve a restricted area of the brain, which is one of the reasons why these discharges do not cause any behavioral alterations. It is possible that not all the subclinical seizures are truly subclinical, and more sophisticated testing during subclinical seizures may reveal subtle cognitive or other behavioral alterations not apparent during routine testing. Isolated hippocampal discharges in the language-dominant hemisphere might express themselves in a marked decline of performance of tachistoscopically presented lexical decision tasks, and isolated discharges of the right hippocampus were shown to lead to a decline of performance in a tachistoscopic matching task of facial expressions (339).
A regional but unilateral mesial temporal lobe involvement of the discharges is usually experienced as a clearly recognized aura by the patient (332). Lack of aura experience strongly correlates with indicators of bitemporal dysfunction such as bitemporal interictal sharp waves and bitemporal ictal propagation in scalp EEG, as well as absence of lateralized MRI sclerosis in patients with mesial temporal lobe epilepsy (274).
In children with mesial temporal lobe epilepsy in the scalp EEG, ictal seizure discharges might be more wide-spread and consist of irregular, high-voltage, spike-and-slow-wave patterns (101).
Discharges confined to the hippocampus produce no scalp EEG rhythms. The regular 5-to 9-Hz subtemporal and temporal scalp EEG pattern requires the synchronous recruitment of adjacent inferolateral temporal neocortex.
Ebersole and Pacia defined seven patterns of early seizure discharges, grouped patients according to their seizure pattern, and correlated these with the site of seizure onset determined by intracranial EEG (74; 231). Categorization by seizure pattern was also compared with brain MRI findings and intracarotid amobarbital (Wada) testing. These authors found that an initial, regular 5- to 9-Hz inferotemporal rhythm was most specific for hippocampal-onset seizures. Less commonly, a similar vertex or parasagittal positive rhythm or a combination of these rhythms was recorded. Discharges confined to the mesiobasal temporal cortex produce a vertex-dominant rhythm due to the net vertical orientation of dipolar sources located there. A focal or regional, low voltage, fast-rhythm (20 to 40 Hz) is often associated with widespread background flattening. Seizures originating in the temporal neocortex were most often associated with irregular, polymorphic, 2- to 5-Hz lateralized activity. Seizures without a clear lateralized EEG discharge were most commonly of temporal neocortical origin. The authors conclude that the initial pattern of ictal discharge on scalp EEG can assist in distinguishing seizures of temporal neocortical onset from those of hippocampal onset. Visual and quantitative sublobar source analyses of scalp ictal EEGs can predict surgical outcomes in most cases after anterior mesial temporal lobe resection (08) and can complement noninvasive presurgical evaluation.
Fast frequency oscillations and ripples. Localizing information by EEG is usually provided by the region of onset of the seizure, the predominance of the discharge during the seizure, and postictal slow waves. Fast frequencies (15 to 30 Hz) are much more frequent during focal onset seizures than during seizures of widespread onset. High-frequency oscillations (80 to 200 Hz) appear to characterize small epileptogenic zones (110). Hippocampal high-frequency oscillations (200 to 500 Hz) have been associated with seizure onset in both human and experimental epilepsy. In rat hippocampal slices in vitro, the correlation between the action potentials of bursting pyramidal cells and local field potential oscillations suggests that synchronous onset of action potential bursts and similar intrinsic firing patterns among local neurons are necessary conditions for high-frequency oscillations. The similar association of high-frequency oscillations and epileptogenic areas was found in humans with microelectrode recording from the different parts of an affected hippocampus (227). Areas of the hippocampus with focal atrophy showed three times higher rate of high-frequency oscillations (150 to 500 Hz) than areas without atrophy, suggesting that neuronal loss and synaptic reorganization may contribute to the generation of high-frequency oscillations representing a neurophysiological marker of epileptogenic area. Blockade of ionotropic glutamate receptors desynchronized onset of action potential bursts in individual pyramidal cells and abolished fast ripples (73). Thus, high-frequency oscillations seem to be the product of pathological neuronal hypersynchronization associated with seizure-generating areas (294; 271). In a study by Jirsch and colleagues, no discrete high-frequency activity was present in poorly localized seizures, whereas it was found in regions of primary epileptogenesis and rarely in regions of secondary spread (149). Absent high-frequency activity seems to indicate poor localization, whereas the presence of focal high-frequency activity near the time of seizure onset may signify proximity to the epileptogenic focus in mesial temporal lobe and neocortical seizures.
On the other hand, the very high-frequency activity may not propagate as readily in hippocampal structures as relatively lower-frequency discharges. Pisciotta and colleagues, studying the initiation and propagation of epileptiform waves in the hippocampus by means of substrate 3-dimensional microelectrode arrays in hippocampal slices in vitro, showed that for CA3-generated activity, only the components of lower frequencies’ electrical activity of less than 95 Hz were completely coherent, whereas components at higher frequencies were not possibly suggesting ineffectiveness of the cellular mechanisms of the hippocampal structures for the propagation of very high frequency discharges (251). High-frequency events in the local field potential of CA1 cells, which are similar to ripples, play a role in the rapid storage of new memories (53).
Ictal onset slow potential shifts. We have recorded ictal DC shifts with foramen ovale electrodes and a special recording technique, including a subgaleally implanted reference electrode (299). In a study by Vanhatalo and colleagues, all seizures were associated with negative DC shifts at temporal derivations (30 to 150 µV relative to vertex), beginning at the electrical seizure onset and lasting for the whole seizure (315). In eight seizures (five patients) with documented mesial temporal lobe onset, the polarity of the DC shift was initially positive, followed by a negative shift after the lateral spread of seizure activity. In all cases, the side of the EEG shift agreed with other diagnostic tests and, at times, was more clearly lateralized than the conventional scalp EEG. Mader and colleagues studied 32 seizures recorded with hippocampal depth and subdural electrodes with the low-frequency filter “opened” (LLF=0.1 Hz, HLF=70 Hz, 3 dB/octave) and identified a slow potential shift at the onset of the seizure of longer than 1.5 seconds in duration (1.5 sec to 11.5 sec; 62% longer than 5 sec) and more than 100 µV in amplitude in 84% of the seizures (189). The maximum shift ranged from 139 µV to 2305 µV. Thus, a slow potential shift at the onset of depth-recorded seizures is likely to be a useful visual aid for localizing electrographic seizure onset.
Magnetoencephalography (MEG). Magnetoencephalography records brain magnetic fields with high temporal and spatial resolution and provides accurate real-time information about source localization. Unlike EEG, it registers tangential fields, and with that, it has the ability to detect sources within the brain sulci. MEG coregistered with MRI, commonly called magnetic source imaging, has emerged as a new noninvasive tool in epilepsy localization. Its greatest value is in establishing neocortical temporal and extratemporal epileptic focus localization. Stefan and colleagues, studying 455 epilepsy patients, detected epileptic activity in 70% of patients (297). The MEG localization findings were concordant in 89% of 131 surgically treated patients, with a higher degree of lateralization accuracy in extratemporal lobe patients. In 109 surgical temporal lobe epilepsy patients, the MEG correctly localized the affected lobe in 86%; in the remaining extratemporal patients, the accuracy rate for identification of the affected lobe reached 100% (297). When comparing epilepsy focus localizing value of MEG, MRI, scalp, and intracranial interictal and ictal recordings, Wheless and colleagues found MEG to be second only to ictal intracranial video-EEG in predicting the epileptogenic zone (325). Although MEG detects fewer hippocampal spikes captured by implanted subdural strips (208), when temporal spikes are recorded, it can significantly improve localization of mesial epileptic focus during nonlocalized scalp ictal EEG or nonlesional MRI (153). Magnetic source imaging has an additional use in guidance of implants. It can tailor electrode placement to improve the yield of the intracranial recording, leading to a higher rate of successful surgeries (164). One of the shortcomings of the MEG is the inability to perform long-term monitoring; therefore, the ability to capture spontaneous seizures is limited. Therefore, even though MEG can accurately establish the source of interictal spikes, the findings may not necessarily reflect the localization of the seizure onset, and, thus, MEG should not be expected to replace scalp or intracranial long-term EEG monitoring when seizure onset localization is to be determined (163).
Magnetic resonance imaging (MRI). High-resolution, thin-section T1-weighted MRI demonstrates hippocampal atrophy in a high percentage of patients with mesial temporal lobe epilepsy (171), and T2-weighted imaging techniques visualize hippocampal sclerosis by increased signals. Quantitative MR volumetry may reveal asymmetries and is useful to answers questions about the posterior extent of hippocampal abnormalities (47). Progressive volume loss in the mesial temporal lobe is related to duration of epilepsy and not a seizure onset age and is not limited to the hippocampus but also affects the entorhinal cortex and the amygdala (29). History of febrile seizure is another predictor of mesial atrophy, selectively affecting the hippocampus and sparing the entorhinal cortex and amygdala (29). Progression of hippocampal volume loss in patients with chronic temporal lobe epilepsy depends on seizure frequency rather than mere duration of the diseases. The seizure-free patients show no changes in hippocampal volume compared to those with continuing, frequent mesial temporal seizures, demonstrating further decline of hippocampal volume (95). Entorhinal cortex atrophy ipsilateral to the seizure focus appears to be specific to mesial temporal lobe structural damage associated with temporal lobe epilepsy (28). Furthermore, thalamic atrophy ipsilateral to the seizure focus is found in temporal lobe epilepsy but not in other forms of focal epilepsy or idiopathic generalized epilepsies. In temporal lobe epilepsy, thalamic atrophy is correlated with the disease duration. Patients with a history of prolonged febrile seizures had smaller thalamic volumes ipsilateral to the seizure focus than those without (218). Bonilha and colleagues evaluated extra-hippocampal brain involvement by voxel-based gray matter volume assessment in patients with mesial temporal sclerosis and found regional ipsilateral volume loss in the medial temporal lobe, occipitotemporal areas, cerebellum, cingulate cortex, insula, and thalamus (36). In conclusion, hippocampal atrophy proven by MRI volumetrics is highly predictive of significant neuronal cell loss and an excellent indicator of success (186). However, a normal MRI can accompany intracranial EEG evidence for hippocampal seizures with histopathological evidence of hippocampal sclerosis (314).
Diffusion tensor imaging. Diffusion tensor imaging is an MRI method that provides microstructural information about brain-connecting fibers. Its main application in clinical neurology and in research is to perform tractography of the white matter. Diffusion tensor imaging of the hippocampal formation revealed that patients with temporal lobe epilepsy had significantly increased diffusivity and decreased fractional anisotropy in the hippocampal formation ipsilateral to the seizure focus, as compared with values in the contralateral hippocampal formation. Compared to healthy subjects, patients had significantly higher mean diffusivity in the ipsilateral hippocampal formation (09; 360).
Thivard and colleagues performed diffusion tensor imaging and statistical parametric mapping of the entire brain in 35 well-defined mesial temporal lobe epilepsy patients and 36 healthy volunteers (310). They identified three abnormal areas: an increased diffusivity was detected in the epileptic hippocampus and the ipsilateral temporal structures associated with a decreased anisotropy along the temporal lobe; a decreased diffusivity was found in the contralateral nonsclerotic hippocampus, the amygdala, and the temporal pole; and finally, a decreased anisotropy was noted ipsilaterally in posterior extratemporal regions. Duration of epilepsy, age at onset, and frequency of generalized tonic-clonic seizures or partial complex seizures did not correlate with the presence of diffusion abnormalities. Region of interest analysis in the hippocampus and parahippocampus demonstrated a correlation between lower ipsilateral diffusivity values and the occurrence of epigastric aura as well as between higher anisotropy values in both hemispheres and history of febrile seizures. Rodrigo and colleagues performed uncinate fasciculus fiber tracking and compared its fractional anisotropy values between patients and controls, separately for the right and left uncinate fasciculus (261). The left-minus-right fractional anisotropy uncinate fasciculus asymmetry index was computed to test for intergroup differences. Asymmetries were found in the control group with right-greater-than-left fractional anisotropy. This asymmetrical pattern was lost in the patient group. Right fractional anisotropy values were lower in patients with right hippocampal sclerosis versus controls. These findings may be related to the preferential seizure pathway spread from the mesial temporal lobe to frontal and insulo-perisylvian areas.
Magnetic resonance spectroscopy. Magnetic resonance spectroscopy, in particular proton-MRS (1H-MRS) has proven to be able to indicate neuronal loss and hippocampal sclerosis by measuring reduced N-Acetyl-L-aspartate (NAA) and increases in the choline/NAA ratio (199; 174; 337). However, MRS abnormalities in temporal lobe epilepsy do not result solely from neuronal loss and gliosis, can extend the region of epileptogenesis, and can be reversible after postsurgical control of seizures (320). This implies that the NAA and Cr abnormalities in patients with temporal lobe epilepsy, at least in part, are dynamic markers of both local and remote physiologic dysfunction associated with ongoing seizures (49). Li and colleagues found that in patients with localization-related epilepsy, 40% to 50% have neuronal metabolic dysfunction that extends beyond the epileptogenic zone defined by clinical-EEG or the structural abnormality defined by MRI (178). Serles and coworkers extended these observations of postoperative NAA recovery of seizure-free patients by characterizing the time course of recovery as an exponential function with a half-time of approximately 6 months (278). The reversal of neuronal metabolic dysfunction remote from the epileptic focus may underlie the clinical observation of improvement of cognitive dysfunction after successful epilepsy surgery. Li and colleagues confirmed a linear correlation between the midtemporal NAA/Cr relative asymmetry ratio and surgical outcome (179).
Positron emission tomography. Functional deficits can be demonstrated in patients with mesial temporal lobe epilepsy using 18F-fluorodesoxyglucose (FDG) PET. Interictal hypometabolism is, as a rule, wide-spread and involves the ipsilateral lateral temporal lobe as well as the ipsilateral thalamus and other subcortical structures (81; 115; 126). The extent and degree of hypometabolism correlates with outcome. Extratemporal cortical hypometabolism outside the seizure focus, particularly hypometabolism in the contralateral cerebral cortex, is associated with a poorer postoperative seizure outcome in temporal lobe epilepsy (54). According to a study by Vinton and colleagues, the extent of resection of the region of hypometabolism on the preoperative FDG-PET is predictive of outcome following surgery for nonlesional temporal lobe epilepsy independent of the presence of hippocampal sclerosis and total brain volume of hypometabolism (319). Carne and colleagues found that the patients with well-lateralized EEG and concordant FDG-PET and with negative MRI for hippocampal sclerosis had similar surgical outcomes as the patients with concordant EEG, FDG-PET, and MRI findings of hippocampal sclerosis (46). The patients without hippocampal sclerosis revealed more diffuse temporal hypometabolism on FDG-PET than those with mesial sclerosis; they were thought to have lateral temporal neocortical rather than mesial structural involvement.
Salanova and coworkers correlated the volumetric head MRI and FDG-PET scan findings with the history, intracarotid amobarbital procedure, pathology, and outcome in 38 patients with medically refractory temporal lobe epilepsy treated surgically and found that FDG-PET scans and head MRIs were complementary: 95% of patients had either MRI-hippocampal sclerosis (MRI-HS) or temporal hypometabolism (262).
Kim and colleagues found that bilaterality of the EEG findings correlated with bilateral temporal hypometabolism on 18F-FDG PET (160). Symmetric or asymmetric interictal bitemporal hypometabolism may signal bilateral independent seizure onset in approximately half the patients. Patients with temporal lobe epilepsy and bitemporal hypometabolisms but without bilateral MRI changes may still be operated on successfully, but surgical suitability should be proven by comprehensive intracranial EEG studies and Wada tests (170).
Flumazenil-PET has demonstrated reduced benzodiazepine receptor binding (267), and 11C-carfentanil-PET an upregulation of mu-opioid receptor binding, in mainly the ipsilateral temporal lobe (94), but 18F-cyclofoxy studies (cyclofoxy binds to mu and kappa) and 11C-diprenorphine-studies (diprenorphine binds to mu, kappa, and delta receptors) did not give conclusive results.
Single photon emission computed tomography. SPECT studies have shown reduced blood flow interictally in the temporal lobe ipsilateral to the epileptogenic mesial temporal lobe and ictal SPECT in mesial temporal lobe epilepsy temporal hyperperfusion during the seizures, mesial hyperperfusion and lateral temporal lobe hypoperfusion in the immediate postictal period, and hypoperfusion in the entire temporal lobe in later postictal seizure phases (27). 99mTC-hexamethylpropyleneamineoxime (HMPAO) and 99mTc-L,L-ethyl cysteinate dimer (ECD) have advantages over 123I-labeled amines (HIPDM and IMP) because there is no redistribution in the brain. With 123I-Iomazenil SPECT, reduced benzodiazepine receptor binding in the area of the focus can be demonstrated (116; 334). SPECT with MAO-B-inhibitors (such as Ro 43-0463) might be able to visualize gliosis (43).
Memory: presurgical evaluation methods. Animal and human lesion studies have revealed the importance of the hippocampal formation and the amygdala for learning and memory. More precisely, the encoding of new information has been associated with the hippocampal formation and, in particular, with the perirhinal and entorhinal cortices (292; 291; 293; 211).
Disorders of learning and memory are clearly associated with temporal lobe dysfunction and have been studied in temporal lobe epilepsy before and after temporal lobe surgery (209; 210; 32; 255; 228; 108; 150; 151; 123; 122). Early investigators noted severe mnestic deficits following either bilateral mesial temporal resections or unilateral resections in the presence of occult lesions in the contralateral temporal lobe (275; 249; 248). These observations gave rise to the functional reserve model of hippocampal function in which postsurgical memory deficits were believed to be mediated by the capacity of the contralateral temporal lobe structures to support memory. However, material-specific learning and memory deficits, found in most patients with temporal lobe epilepsy (210), might be specifically related to the involved temporal lobe, and at least certain types of preexisting bilateral mesiobasal lesions, such as cysts, do not necessarily lead to an amnestic syndrome (124). When the language-dominant hemisphere is involved, memory deficits are usually more pronounced, and verbal memory, in particular, is insufficient. Verbal memory impairment has been found to correlate with hippocampal pyramidal cell loss (264). However, there is no doubt that dysfunction of structures other than the mesiobasal temporal structures (in particular, the diencephalic and probably also the thalamic structures) (135) also can produce severe memory deficits. For example, Kapur and colleagues have described anterograde, but not retrograde memory loss following combined mamillary body and medial thalamic lesions (155). Furthermore, there is evidence that the mesial temporal structures play an important role in the recognition of emotion in facial expressions (02) and in complex musical abilities, particularly in the recognition of musical consonances and dissonances (328).
Pre- and postoperative comparison of performances of patients with temporal lobe epilepsy have contributed much to the understanding of temporal lobe functions (152). There are, however, some limitations because most patients with a long history of temporal lobe seizures (1) have a preoperative memory deficit, and the degree of this deficit influences postoperative outcome; (2) take high-dosed antiseizure medications with known side effects regarding higher cognitive functions preoperatively and reduce or discontinue antiseizure medications postoperatively; (3) become seizure-free and, thus, escape the negative consequences of seizures on cognitive performance and motivation. Also, in lesional cases and in patients with resective surgery, compensatory mechanisms (ie, plasticity) play an important role.
Thus, although substantial evidence has been accumulated that the temporal lobe and, particularly, its mesiobasal and basal structures are important for memory, the exact role of the hippocampal formation is still a matter of research. Hippocampal N-acetyl-aspartate and T2 relaxation time, functional MRI, (O15)-H2O PET-studies, selective memory temporal lobe-amobarbital tests (which allow the temporary inactivation of restricted mesial temporal areas), and intrahippocampal cognitive-evoked potential studies have revealed some interesting results. In conclusion, these studies suggest that nonverbal stimuli activate the hippocampal region if the material is visually complex, if spatial relations are stressed in the instruction, and if the stimuli are novel. Moreover, there is evidence that the hippocampus and the parahippocampal gyrus are critical for establishing and storing associations between components of a learning event rather than items in isolation (123). The combined measurements of hippocampal N-acetyl-aspartate and T2 relaxation time can be used to examine the degree of ipsi- and contralateral hippocampal dysfunction or injuries and their relationship to memory performance (217).
Selective memory temporal lobe amobarbital tests. The temporary inactivation of the structures intended to be definitively resected is one method to predict possible deficits associated with the resection. The so-called intracarotid amobarbital test, pioneered by Juhn Wada, was first used for language lateralization and predicting postoperative memory outcome (323). Although in temporal lobe surgery, it predicts at least a certain degree of severe global memory deficits, it has considerable limitations to exactly predict the risks concerning the type and degree of material-specific memory deficits in amygdalohippocampectomy. The selective inactivation of the territory of the anterior choroidal artery (or of the P2 segment of the posterior cerebral artery in the case of a so-called “posterior” test or of the anterior choroidal artery and posterior communicating artery in the case of the technique with temporary balloon occlusion distal to the origin of the anterior choroidal artery) has improved the validity of this test (345). Nevertheless, the variability of the vascular supply and the fact that these arteries provide, as a rule, only a restricted territory of interest (the anterior choroidal artery supplies the amygdala and the anterior part of the hippocampus, and the posterior cerebral artery supplies the more posterior hippocampus) still present problems. In addition, the appropriate interpretation of the task-performance asks for the co-injection of a SPECT tracer and concomitant EEG recording from the structures of interest.
Selective temporal lobe memory Amobarbital tests in candidates for amygdalohippocampectomy have generally confirmed the material-specific memory role of anterior mesiotemporal structures. With a dual verbal- and nonverbal-coding recurrent memory task (so-called DOKO) with nonverbal motor responding (button-press), the prediction of postoperative memory performance is rather good for verbal memory but underestimates postoperative nonverbal (figural) memory performance.
Event-related potentials. In temporal lobe epilepsy patients, intracranial ERPs show alterations (increase in latency and decrease of amplitude) that correlate well with the Wechsler Memory Scale, and the hippocampal P300 correlates well with neuronal cell density. The N400 recorded from the anterior fusiform gyrus is thought to reflect access of semantic memory, whereas the P300 is thought to reflect the rules-based mapping of stimuli onto discrete covert or overt responses, ie, encoding processes (222; 200; 223). Late potentials around 500 to 900 msec account for the patient’s engagement in recollection processing, and ERPs differentiate priming and recognition to familiar and unfamiliar faces (19; 235). Several authors have described limbic correlates of the scalp P300 elicited in visual and auditory oddball paradigms using depth electrodes in epileptic patients (118; 117; 295; 184; 113) and to the scalp N400 in word recognition paradigms (282). Therefore, it can be assumed that the careful study of ERPs recorded from mesiobasal temporal structures in temporal lobe epilepsy patients has the potential to clarify the role of these structures in the various kinds of memory and to quantify preoperative deficits. Indeed, Grunwald and colleagues reported that the amplitude of the intrahippocampal N400 elicited in a word recognition paradigm is an excellent predictor of postoperative memory performance in patients suffering from temporal lobe epilepsy (113).
Hippocampal and parahippocampal seizures are usually detectable with scalp EEG unless the seizure is focal without impaired awareness. If precise localization is necessary (ie, if selective mesial resection is planned), stereotactic depth EEG is useful.
32-channel stereoelectroencephalographic referential recording of a spontaneous hippocampal onset seizure. The recording from the left hippocampus (inner contacts of depth electrode 2 = channel 5) is enlarged at the top of the ...
Other techniques are subdural recordings with strips and grids.
Intraoperative electrocorticography can be used to tailor the extent of the resection (229; 130).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

John M Stern MD
Dr. Stern, Director of the Epilepsy Clinical Program at the University of California in Los Angeles, received honorariums from Ceribell, Jazz, LivaNova, Neurelis, SK Life Sciences, Sunovian, and UCB Pharma as advisor and/or lecturer.
See Profile
Jerome Engel Jr MD PhD
Dr. Engel of the David Geffen School of Medicine at the University of California, Los Angeles, has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Epilepsy & Seizures
Mar. 22, 2024
Epilepsy & Seizures
Mar. 20, 2024
Epilepsy & Seizures
Mar. 07, 2024
Epilepsy & Seizures
Mar. 06, 2024
Epilepsy & Seizures
Mar. 05, 2024
Epilepsy & Seizures
Mar. 05, 2024
Epilepsy & Seizures
Mar. 04, 2024
Epilepsy & Seizures
Feb. 29, 2024