



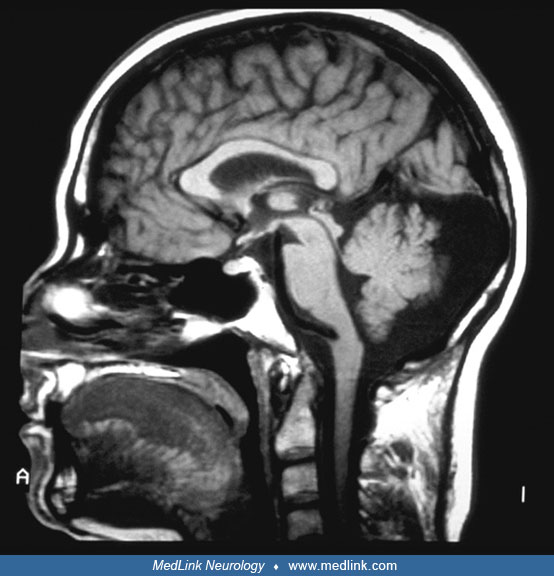
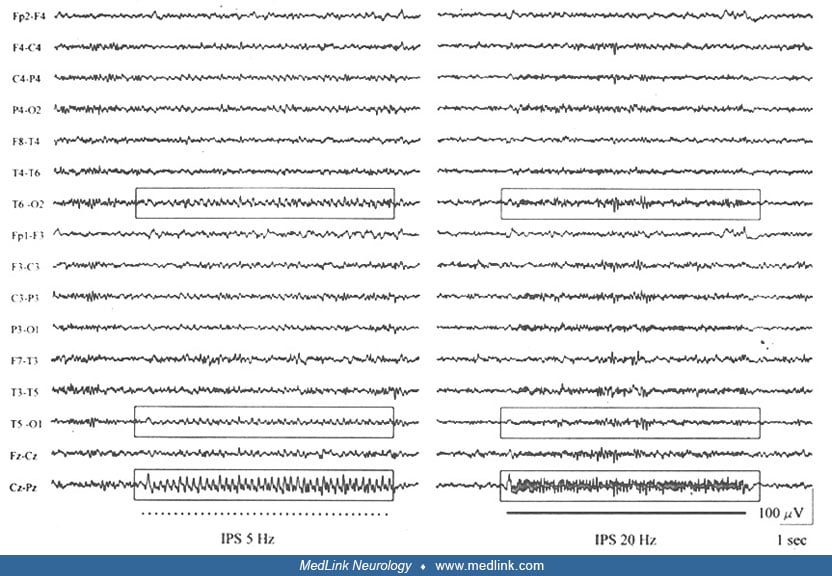
Developmental Malformations
Walker-Warburg syndrome
Apr. 14, 2024
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Periventricular nodular heterotopia is a malformation of cortical development due to impaired neuronal migration. Neurologists should become familiar with this brain malformation because periventricular nodular heterotopia are among the most frequent brain dysgenesis encountered in clinical practice and affected patients are frequently characterized by focal, drug-resistant epilepsy, varying degrees of cognitive impairment, as well as extracerebral findings. In this article, the author provides an update on the topic, including the expanding clinical and genetic spectrum, neurophysiological investigations, and current management options, including epilepsy surgery.
|
• Periventricular nodular heterotopia can be: (1) bilateral and symmetrical, (2) bilateral single-noduled, (3) bilateral and asymmetrical, (4) unilateral, or (5) unilateral with extension to neocortex. | |
|
• It is likely that genetic factors play a more important role in the bilaterally, as opposed to unilaterally, affected cases. | |
|
• There is extreme genetic heterogeneity underlying periventricular nodular heterotopia. Patients are at risk for seizures, intellectual impairment, and other systemic symptoms, depending on the underlying etiology. | |
|
• Due to its relative frequency and the risk of systematic complications it is important to carefully look for signs of FLNA-related, X-linked periventricular heterotopia. | |
|
• For patients with refractory epilepsy, surgery outcome may be good, with resections limited to the periventricular nodules, cortex, or both. |
During the embryonic development of the brain, neuroblasts migrate away from the germinal layer of the ventricular neuroepithelium, mainly along the tracks of radial glial fibers, which stretch from the ventricular zone to the external pial surface of the developing brain. Any impairment of the complex processes underlying neuronal migration may determine the presence of clusters of abnormally located neurons (eg, gray matter heterotopia) (14).
According to classic neuropathologic studies, two types of heterotopia can be distinguished: laminar and nodular (71). Laminar heterotopia is organized in laminar bands of neurons interposed between the cortex and the lateral ventricles. This is now termed "band heterotopia," according to MRI studies (11). Nodular heterotopia is characterized by the periventricular location of the heterotopic neurons close to the subependymal germinal matrix. For this reason, this is usually referred to as "periventricular nodular heterotopia." Periventricular nodular heterotopia is made up of round nodular masses of normal neurons and glial cells with no laminar organization, which cause bulging of the ventricular walls. They can be diagnosed in vivo by means of neuroimaging (12; 10), and they are, together with focal cortical dysplasia, the most common forms of human brain dysgenesis.
Patients affected by periventricular nodular heterotopia have been classified on the basis of MRI features (13). Five different groups of periventricular nodular heterotopia have been distinguished (17): (1) bilateral and symmetrical; (2) bilateral single-noduled; (3) bilateral and asymmetrical; (4) unilateral; and (5) unilateral with extension to neocortex. It is likely that genetic factors play a major role in the bilaterally affected patients. In particular, bilateral and symmetrical cases are characterized by a clear female predominance, positive family history for epilepsy, and familial occurrence (53). They have been identified as an X-linked dominant disease with linkage mapping to chromosome Xq28, determined by mutations of the filamin A or filamin1 gene (FLNA or FLN1), a gene originally isolated from blood cells (68). A multicentric study analyzed the filamin A gene in a large number of patients, revealing FLNA mutations in patients with bilateral and symmetrical periventricular nodular heterotopia, associated with Ehlers-Danlos syndrome in a few cases (139). Several other causative genes have been described for periventricular nodular heterotopia and will be discussed in this review. Unilateral periventricular nodular heterotopia, on the other hand, is not usually associated with preferential sex prevalence, familial occurrence, or FLNA mutations. Therefore, unilateral cases are most probably determined by vascular mechanisms that may impair in utero the perfusion of a limited part of the developing brain. In addition, a malformation complex has been described with periventricular nodular heterotopia in the trigones and the occipitotemporal horn associated with hippocampal, corpus callosum, and cerebellar dysgenesis (141).
Clinical presentation. As in other types of cerebral malformations, the clinical picture, particularly for neurologic and mental deficits, is related to the amount of the heterotopic tissue, the distribution of nodules, and extension to the overlying cortex. Patients affected by periventricular nodular heterotopia are individuals capable of conducting a normal social and working life. Often, periventricular nodular heterotopia is discovered as an incidental MRI finding.
Epilepsy. Patients with periventricular nodular heterotopia have a higher incidence of epilepsy than the general population. The spectrum of epilepsy phenotypes in periventricular nodular heterotopia is broad, seizure patterns are variable, and not all patients with periventricular nodular heterotopia develop seizures.
The age of onset of epilepsy is in the second decade of life in most cases (147; 53; 18), although early epileptic encephalopathy and infantile spasms have also been, albeit rarely, described (117). Infantile spasms are predominantly observed in children with bilateral disease (171).
Seizure semiology is variable in patients with PVNH. Multiple seizure types are present in most bilaterally affected patients, in agreement with the diffuse localization of the structural brain anomalies. By contrast, elementary seizures with visual or auditory onset are frequently observed in patients with unilateral periventricular nodular heterotopia located in the paratrigonal region, close to visual and auditory cortical areas. In all periventricular nodular heterotopia, generalized seizures are rare and easily controlled by therapy, whereas focal seizures are frequently resistant to treatment. Overall, drug-resistant epilepsy occurs in approximately one quarter of affected individuals with periventricular nodular heterotopia with variable genetic etiologies (135).
It is well established that among patients with PVNH, seizure generators can be variable. In some patients, the PVNH are the main epileptogenic zone, in which case removing or destroying the heterotopic nodules alone is sufficient to lead to seizure reduction or cessation (17; 160). In other patients, the aberrant connections formed between the PVNH and the cortical structures are both integral in the epileptogenesis, including the amplification and synchronization of epileptic activities, and the cortical regions involved may need to be identified and removed at the same time to achieve seizure control (98; 04; 179). These aberrant connections or vast networks involving both the heterotopic nodules and the cortex have been demonstrated in both imaging and electrophysiological studies.
The reason why only some individuals with periventricular nodular heterotopia develop epilepsy and others do not is not fully elucidated. One plausible explanation is related to the neuroanatomy of periventricular nodular heterotopia. There are no axonal projections from mature neurons in the nodules extending through the cerebral mantle into the cortex to join epileptic networks. Even if axons did project radially, they could not traverse the U-fibre layer because of keratan sulfate that would repel them (159). Therefore, these may be less epileptogenic. However, some periventricular nodular heterotopias in close proximity to the ventricular trigone or in the temporal horn could send axons into the amygdala, as this structure does not have a U-fibre layer. As the amygdala can be highly epileptogenic and involved with cortical epileptogenic networks, epilepsy may result. To further understand why some nodules are more epileptogenic than others, further studies that correlate the epileptogenic nodule location and anatomy to nodule metabolism (FDG-PET), connectivity (diffusion tensor imaging, fMRI, neurophysiology) and genetic/epigenetic studies are required.
Mild intellectual disability and neurologic deficits. It is believed that most heterotopic nodules are structurally connected to the overlying cortex, especially in those with epilepsy of longer duration. This has been demonstrated with diffusor tensor imaging and fMRI studies. Altered connectivity is likely the substrate for other neurologic dysfunction (40). Felker and colleagues found that reading dysfluency and behavioral issues can be evident in children with nodular heterotopia, even in the absence of epilepsy (62). Symptoms of dyslexia appear to be unrelated to the anatomy or location of the heterotopia (149). In an observational study, 40 subjects with bilateral periventricular nodular heterotopia were found with significantly smaller white matter volume, correlated with reading disability, in comparison to 40 matched healthy controls (109). A dyslexia susceptibility gene, KIAA319L, has been discovered that is also associated with neuronal migration. This supports the association of abnormal neuronal migration with developmental dyslexia (142).
In patients characterized by periventricular nodular heterotopia not associated with other brain malformations, psychomotor development and mental level are normal in most cases, even if IQ scores may be within the lower limits of normality (18). Neurologic examination is also usually normal, but mild motor, sensory, or visual defects have been reported, most likely related to the involvement of the neocortical areas overlying the heterotopic nodules (53; 15; 18; 17). A case with periventricular heterotopia with megalencephaly and normal intelligence has been reported (01). This patient had no other features of cortical dysplasia and hydrocephalus. Less frequently, in cases of periventricular heterotopia associated with diffuse brain abnormalities, the clinical picture is much more severe and characterized by mental retardation, neurologic deficits, and dysmorphic features (51; 78; 164; 189; 139).
Other presentations. A case of chronic headaches due to periventricular nodular heterotopia leading to obstructive hydrocephalus has been described (169). In this case, evidence of heterotopia in the cerebral aqueduct and floor of the fourth ventricle led to the symptoms. An association between nodular heterotopia and Chiari malformations and myelomeningocele and encephalocele has been described (111; 99). There is a case report of a patient with periventricular nodular heterotopia, Chiari I malformation, and congenital hemiplegia. The patient likely also had an additional dysplasia in the motor cortex (94). In one study, a high percentage of patients with Chiari II malformation had periventricular nodular heterotopia, suggesting an association (84).
FLNA mutations and other spectrum of diseases. Bilateral and symmetrical periventricular nodular heterotopia cases are characterized with mutations in the FLNA gene (68).
Skeletal manifestations. Periventricular nodular heterotopia may also present clinical features of Ehlers-Danlos syndrome (165). Mutations in FLNA are associated with a wide spectrum of diseases also including disorders of the otopalatodigital syndrome spectrum (including OPD1 and OPD2 syndromes, Melnick-Needles syndrome and frontometaphyseal dysplasia, terminal osseous dysplasia, familial cardiac vascular dysplasia, chronic idiopathic intestinal pseudo-obstruction, and thrombocytopenia (Robertson et al 2003; 176; 127). An association of FLNA mutations and craniosynostosis has also been noted (63). Therefore, the need to consider a filaminopathy in the differential diagnosis of craniosynostosis, especially in the presence of atypical cranial or skeletal features, is important. Frontonasal dysplasia associated with bilateral periventricular heterotopia has been identified by pre and postnatal MRI (148). The unique constellation of observed syndromic features of cleft palate and syndactyly of the second and third toes may aid in the differential diagnosis to FLNA-associated periventricular nodular heterotopia (55).
Connective tissue manifestations. Periventricular nodular heterotopia may also present clinical features of Ehlers-Danlos syndrome, a group of connective tissue disorders (165). Vascular abnormalities, including aortic aneurysm, patent ductus arteriosus, bicuspid aortic valve, peripheral arterial dilation, and atresia have been reported in patients and families with known FLNA mutations (68; 162; 150). Thoracic aortic aneurysms most frequently involve the aortic root and ascending aorta, and many patients require surgery. These patients have a risk of devastating cardiovascular complications, such as spontaneous aortic rupture, which highlights the importance of early genetic diagnosis and close follow-up (37). A deletion of smooth muscle FLNA in adult mice demonstrates the vascular phenotype of human bilateral periventricular nodular heterotopia, culminating in aortic dilatation (151). It has been proposed by some that all x-linked forms of periventricular nodular heterotopia should be screened for connective tissue disease (150). Platelet dysfunction and thrombocytopenia in FLNA mutations have been noted (21).
Pulmonary manifestations. Clinicians should also be aware of the risk for lung disease. There have been reports of FLNA mutations resulting in pulmonary hyperinflation during the neonatal period or early infancy leading to a diagnosis of FLNA-associated progressive lung disease (192). Respiratory failure secondary to progressive obstructive lung disease during infancy may be the presenting phenotype of FLNA-associated periventricular nodular heterotopia. Six female infants were described to present with respiratory failure, requiring lung transplants by 11 months of age (30). A premature neonate with a FLNA mutation had lung disease mimicking bronchopulmonary dysplasia (112). A 1-year-old female child with respiratory distress at birth had recurrent lower respiratory tract infections, bilateral lung emphysema with basal atelectasis, bronchospasm, pulmonary artery hypertension, and oxygen and mechanical ventilation dependency. They had a new pathogenic variant of one copy of c.3153dupC in exon 21 in the FLNA gene (56). It is suggested that patients with pathogenic FLNA variants should be studied actively for lung involvement, even in the absence of pronounced respiratory symptoms, as they can be mild and occur later than expected. In addition, any patient with unexplained panlobular emphysema should be analyzed for pathogenic FLNA variants (177).
Cardiovascular manifestations. In a report, a patient with Ehlers-Danlos symptoms, seizures, and a Moyamoya disease-like vascular malformation was described with FLNA and RNF213 mutations (86). It is postulated that a gene abnormality in FLNA, a target of RNF213-mediated proteasomal degradation, may promote Moyamoya disease-like vascular formation as a possible second-hit gene in individuals having the RNF213 p.R4810K variant.
Gonadal manifestations. The mutational inactivation of the FLNA gene induces insufficiency of translocation and activation of the androgen receptor. Consequently, it causes a developmental disorder of the male gonad and hypogonadism, similar to those observed in partial androgen insensitivity. This association recommends investigating a possible mutational inactivation of the FLNA gene in patients with cryptorchidism and epididymo-testicular dissociation. This is especially indicated when the family history, more often that of the mother, is suggestive. Likewise, growth and gonadal development of all male patients carrying this genetic trait should be monitored since childhood (34).
Psychiatric manifestations. Psychiatric complications are often underrecognized. There have been reports of schizophrenia, depression, anxiety, and autism. In one report, two patients with periventricular nodular heterotopia presented with neuropsychiatric presentations. One had the FLNA mutation (72). A case of Graves hyperthyroidism-induced psychosis in a patient with periventricular nodular heterotopia has been described. More information is needed to determine the etiology and if aberrant brain networks were involved leading to the psychosis (133). Sometimes, epilepsy, especially arising from the frontal or temporal lobes, has been misdiagnosed as a psychiatric disorder in patients with periventricular nodular heterotopia (58).
Other types of presentation.
Bilateral posterior periventricular nodular heterotopia. This is a distinct malformation that consists of a continuum of brain malformation, including posterior or infrasylvian structures (116). Posterior periventricular nodular heterotopia usually has no gender predilection and is often negative for a filamin A gene mutation (50). Hyperphagia and morbid obesity have been described in bilateral posterior nodular heterotopia. This association suggests that periventricular nodular heterotopia is part of a more diffuse process of posterior or infrasylvian brain developmental abnormalities other than just a disorder of neuronal migration (80).
Pediatric presentation of periventricular nodular heterotopia. It has been described that the phenotypic spectrum can differ in the pediatric and adult populations. It is likely that milder forms go unrecognized until seizure onset in adulthood. In a study of 31 children with periventricular nodular heterotopia, 71% died in the neonatal period or childhood, 80% had additional cerebral malformations, and 74% had systemic malformations. Most patients had cognitive impairment, developmental delay, and intractable epilepsy. It has been proposed that lung and brain involvement, in association with left ventricular outflow obstruction and persistent patency of ductus arteriosus, should be considered highly suggestive of FLNA gene abnormalities in female newborns (118).
Unlike in the adult population, bilateral periventricular nodular heterotopia due to FLNA mutations do not commonly present in children, whereas other chromosomal abnormalities and metabolic disorders are important potential causes that should be adequately investigated. Therefore, clinicians should be aware that in contrast to adult series, developmental delay and mental retardation are present in the majority (171).
Periventricular nodular heterotopia can be associated with seizures, a wide range of neurodevelopmental disorders and syndromes, and other system disorders. There is a large genetic heterogeneity underlying periventricular nodular heterotopia. A genetic diagnosis enables proper counseling of prognosis and recurrence risks, and enables individualized patient management. Patients affected by periventricular nodular heterotopia are usually not characterized by motor deficits or mental impairment and can, therefore, lead a normal social and working life. In one study, looking at a cohort of 47 patients with FLNA mutations, the vast majority had normal psychomotor and cognitive development (100). The prognosis essentially depends on the severity of the associated epilepsy and systemic symptoms, such as cardiovascular or connective tissue disease. Some patients develop epilepsy, which can be refractory. A diagnostic latency of 17 to 20 years between seizure onset and the genetic diagnosis was described in one large study (100).
Long-term medical surveillance for potentially life-threatening cardiovascular, pulmonary, and connective tissue complications as well as genetic counselling is of utmost importance in FLNA mutations (100). In a review of patients with FLNA, loss-of-function, periventricular nodular heterotopia, half of the patients did not present with neurologic symptoms (22). Most patients presented a syndromic association combining cardiovascular and connective tissue features. Cardiovascular anomalies, mostly aortic aneurysm and/or dilation, were present in 75% of patients. This emphasizes the importance of routine screening of these disorders, despite the absence of neurologic symptoms.
In the pediatric population, in contrast to adults, developmental delay and mental retardation are the general rule (171). Diffusor tensor imaging (DTI) has shown that perilesional white matter shows significantly decreased fractional anisotropy and elevated mean and radial diffusivity, suggesting microlesional white matter abnormalities. The more diffuse pathology may be one of the reasons that children with periventricular nodular heterotopia often remain refractory to medical and surgical interventions and have cognitive impairments (66). In addition, anaplastic ganglioglioma and beta-amyloid plaques have been reported in the heterotopic nodules of affected patients (47; 89).
Genetic counselling and educating families about the phenotypic variability within a single family of individuals with periventricular nodular heterotopia is important. For example, a report describes three generations of individuals with FLNA mutations: a fetus with absence of the corpus callosum and periventricular nodular heterotopia on imaging, an asymptomatic mother, and a maternal grandmother with a long-standing history of epilepsy (54).
Case 1. Bilateral and symmetrical periventricular nodular heterotopia. The patient was a 37-year-old woman with drug-resistant focal epilepsy. Three cases of epilepsy were reported in her family. Prenatal and perinatal histories were uneventful, and the psychomotor development was normal. She achieved a junior high school diploma and maintained a job. The patient developed tonic-clonic and focal complex seizures at 19 years of age. Convulsive seizures disappeared after antiepileptic treatment was started. The focal seizures were characterized by loss of contact, head flexion, and gestural automatisms and were followed by loss of speech. They persisted despite different antiepileptic treatments. The neurologic examination and psychometric evaluation were normal. MRI demonstrated bilateral and symmetrical heterotopic nodules bulging into the walls of the lateral ventricles. The EEG recordings showed bilateral asynchronous interracial epileptiform activities.
Case 2. Unilateral periventricular nodular heterotopia. A male patient developed tonic-clonic seizures during sleep at 40 years of age. No family history of epilepsy was reported. Prenatal and perinatal histories were uneventful, and his psychomotor development was normal. He achieved a high school diploma and worked as a librarian. The neurologic examination, including psychometric evaluation, was normal. MRI demonstrated unilateral periventricular nodular heterotopia in the right posterior paratrigonal region extending into the parietal cortex. The EEG recordings demonstrated interictal epileptiform activities on the right temporal leads. There was no recurrence of seizures after antiepileptic treatment was started.
Like other brain dysgeneses, periventricular nodular heterotopia may be the result of different etiologic factors. Bilateral and symmetrical nodules may be found in different members and generations of the same family (129). A family history of epilepsy, febrile convulsions, or spontaneous abortions could be present in bilateral and symmetrical cases, whereas prenatal risk factors for brain damage have been reported in unilaterally affected patients.
Bilateral periventricular nodular heterotopia. Periventricular nodular heterotopia could arise due to the inability of newly postmitotic neurons to exit the ventricular zone because of the inability to properly crosslink and mobilize actin, as in the case of FLNA mutations (153). Fetal human periventricular nodular heterotopia cases exhibit disrupted radial glial architecture along the ventricular wall (156). Therefore, periventricular nodular heterotopia might be, in part, attributable to abnormalities of the radial glial scaffolding on which postmitotic neurons depend to migrate away from the proliferative zones (144). In addition, periventricular nodular heterotopia could be related to over 60 molecules known to interact with or affect FLNA (153). For example, inhibition or disruption of molecules that control the subcellular distribution of FLNA, such as BIG2 (vesicular and membrane trafficking protein encoded by ARFGEF2), might contribute to the pathogenesis by disrupting normal migration and weakening the neuroependymal lining (164; 113). Furthermore, FLNA binds multiple adhesion molecules, such as integrins, and mutations of such adhesion molecules might collectively impact the rate of cell migration and/or the adhesiveness of radial glial endfeet and contribute to periventricular nodular heterotopia (174; 143). Control of FLNA phosphorylation is important for cytoskeletal reorganization and potentially for cell motility. Dysregulation of molecules that control FLNA phosphorylation could also impact both neuronal migration and the integrity of the neuroependymal lining (183). In the largest case series of FLNA negative patients with seizures and bilateral periventricular nodular heterotopia, a direct correlation was observed between high heterotopia burden, female gender, and trigonal location. Quantitative MRI measurements indicated that this correlation is based on the diffuse nature of the heterotopic nodules rather than on the total volume of abnormal heterotopic tissue (59).
FLNA gene. The X-linked-dominant form of bilateral periventricular nodular heterotopia, related to mutations in FLNA encoding filamin A, is the major cause of bilateral nodular heterotopia, causing prenatal and neonatal lethality in males and explaining the excess of affected women. The female predominance and familial occurrence indicate a genetic origin for bilateral periventricular nodular heterotopia. Genetic studies have revealed in all familial cases of bilateral and symmetrical periventricular nodular heterotopia mutations of the FNLA gene on Xq28, encoding filamin 1 (or filamin A), an actin-cross-linking phosphoprotein involved in cellular locomotion (68; 139). Few living males have been described with this condition. A novel no-stop FLNA mutation has been described in an affected male (130). Two additional males have been described with exon skipping of FLNA, which partially restored its protein function and could have contributed to milder clinical courses (128). A case of paternal inheritance has also been described (92). In addition, mutations in FLNA have been exceptionally associated with unilateral nodular heterotopia (64). However, only sporadic patients with bilateral periventricular nodular heterotopia carry FNLA mutations (162; 139). A case has been described of germline mosaicism in FLNA-associated periventricular nodular heterotopia. This case must now be considered when providing genetic counseling to families where a proband presents as an isolated case and parental investigations are unremarkable (101). FLNA mutations account for the majority of X-linked inherited periventricular heterotopia and approximately 25% of all cases of periventricular nodular heterotopia. In sporadic cases of classical bilateral periventricular nodular heterotopia, approximately 50% of patients have a FLNA mutation (139). In addition, studies indicate a more complex scenario of the genetics of bilateral cases. FLNA mutations not interfering with the production of full-length filamin A lead to a group of congenital malformations collectively termed otopalatodigital spectrum disorders (154), and mosaic FLNA mutations have been reported in patients with periventricular nodular heterotopia and mild clinical phenotype (138). There are reports of an FLNA mutation in a family of seven individuals over two generations with Ebstein anomaly, a rare cardiac condition (07; 119). The family members also had various skeletal abnormalities. However, neurologic symptoms were not described in these individuals.
ARFGEF2 gene. In addition, other genes are most probably involved; families with an autosomal recessive mode of inheritance have been described (166), and other causative genes, like the ARFGEF2 gene, and disease loci in different chromosomes have been reported (102; 145; 164). There have been reports of at least nine genes that have been linked to periventricular nodular heterotopia in at least more than one individual, including FLNA, FAT4, DCHS1, ARFGEF2, C6orf70, AKT3, INTS8, MCPH1, and NEDD4L (83). ARGREF2 mutations are a rare cause of autosomal recessive periventricular nodular heterotopia. ARFGEF2 is a member of a family of exchange factors that likely regulates protein trafficking in neuronal progenitor cells. BIG1 and BIG2 proteins are encoded by ARFGEF2 and are responsible for interior trafficking in the trans-Golgi network (28). These patients have a more severe phenotype than those with FLNA mutations. Microcephaly, enlarged ventricles, and delayed myelination have been described. There is also more severe epilepsy, cognitive disability, and motor deficits, such as quadriparesis (107). With more genetic testing, the phenotype continues to expand. Mutations in the ARFGEF2 must be considered in the presence of bilateral periventricular nodular heterotopia and putaminal hyperintensity in children presenting with movement disorder, severe developmental delay, and microcephaly. In cases of ARFGEF2 mutations, screening for cardiomyopathy may be indicated (191). A mutation in axon 3 of ARFGEF2 has been associated with a severe phenotype of dystonic quadriplegia, developmental delay, obstructive cardiomyopathy, infections, and feeding difficulties (178; 09).
Other genes.
|
Gene/chromosomal deletion |
Reference | |
|
PQB1 |
(167) | |
|
22q11.2 deletion |
(96) | |
|
ARX |
(131) | |
|
YWHAE |
(120) | |
|
Monosomy 1p36 |
(48; 168) | |
|
6p25 deletion |
(35) | |
|
6q terminal deletion |
(42) | |
|
11q microdeletion |
(170) | |
|
6q27 terminal deletion |
(193) | |
|
DCHS1, FAT4 |
(31) | |
|
17q21.31 microdeletion, KANSL1 |
(124) | |
|
7q11.23 heterozygous deletion of 1.5-1.8 Mb |
(125) | |
|
TMTC3 |
(60) | |
|
NEDD4L |
(26; 93; 115; 175) | |
|
ENG, ALK1, SMAD4 |
(134) | |
|
KLH7 |
(91) | |
|
SCN1A |
(08) | |
|
MEKK4 |
(157) | |
|
MOB2 |
(132) | |
|
17p11.2 deletion |
(32) | |
|
LAMC3 |
(45) | |
|
KCNB1 |
(85) | |
|
SZT2 |
(114) | |
|
ARF1 |
(73) |
Pathogenic mechanisms. All types of periventricular nodular heterotopia are determined by impairment of the mechanisms that govern the migration of postmitotic young neurons during brain ontogenesis from the ventricular zone to the developing cortical plate. The timing of this impairment should be located during early ontogenesis, given the total failure of migration of some neurons. However, different pathogenic mechanisms may be responsible for the different types of periventricular nodular heterotopia.
FLNA. Families with bilateral and symmetrical nodular heterotopia are characterized by expression in females and prenatal or perinatal lethality in males. Most families are X-linked with linkage mapping to chromosome Xq28, and they are determined by mutations of the filamin A gene (68). Filamin A is a member of a family of actin-binding proteins. It is a high molecular weight (2647 amino acid residues) protein, with three major functional domains allowing its homodimerization and heterodimerization with actin and possibly with filamin B (163). Filamin A is extensively regulated via phosphorylation (157), and it has a number of essential functions in hemostasis, vascular remodeling, and the migration of different cell types (68). It has been proposed that filamin A is part of either the motor system that allows the migration of young neurons or the system that make neurons competent for migration. This may explain why, in patients with loss of function of the filamin A protein, some neurons fail to migrate but still retain the capability of normal cellular differentiation. Filamin A is also shown to regulate neural progenitor proliferation and cortical size (106). It increases G2 to M phase entry, leading to prolonged cell cycle, compromised neural progenitor proliferation, and reduced brain size.
Genetic studies addressing the correlation between genotype and phenotype in bilateral symmetrical periventricular nodular heterotopia demonstrated that in all pedigrees considered, FLNA mutations were inherited from female to female with no surviving male offspring, probably reflecting a severe loss of function of the protein (162). The difference in the clinical expression between females and males was likely due to the random inactivation in females of 1 X chromosome in single neurons. Affected females, therefore, could be the expression of a cell autonomous mosaic phenotype, by which neurons expressing mutant X chromosome failed to migrate, whereas neurons expressing normal X chromosome migrated properly. Affected males could be unable to survive gestation for the presence of a single mutant X chromosome (162). However, mosaic FLNA mutations (138) or point mutations compatible with the preservation of partial protein functions (162; 79) have been described in a few male patients with mild clinical phenotype. Two male patients have been described with exon skipping causing atypical phenotypes associated with a loss-of-function mutation in FLNA by restoring its protein function (128).
In addition, FLNA mutations not interfering with the production of full-length protein but determining gain-of-function of FLNA cause a broad range of congenital malformations (otopalatodigital syndrome types 1 and 2, frontometaphyseal dysplasia, Melnick-Needles syndrome) called otopalatodigital spectrum disorders (154). The otopalatodigital spectrum results in hearing loss, craniofacial dysmorphism (hypertelorism, prominent supraorbital ridges, ear abnormalities, broad nasal bridge, small mouth, down-slanted palpebral fissures, and cleft palate), and skeletal abnormalities leading to shortened digits and short stature. There are few reports of patients with Melnick-Needles syndrome and periventricular nodular heterotopia (167; 137; 152). The co-occurrence of periventricular nodular heterotopia and Melnick-Needles syndrome is the likely consequence of a single mutational event resulting in co-occurring gain-of-function and loss-of-function effects.
Other genes (refer to Table 1). A metaanalysis resulted in 105 periventricular nodular heterotopia gene/locus associations (186). Therefore, there is extreme genetic heterogeneity underlying periventricular nodular heterotopia. Identification of a genetic etiology enables proper counseling of prognosis and recurrence risks, and enables individualized patient management (186).
Genes other than FLNA may determine bilateral and symmetrical periventricular nodular heterotopia. Indeed, in rare cases a recessive mode of inheritance was described, mutations of the ARFGEF2 gene have been reported, and new disease chromosome loci have been suggested (102; 145; 166; 164). Because only a minority of sporadic patients carries FLNA mutations (162; 139), it is clear that bilateral periventricular nodular heterotopia is a heterogenous disorder and that different, still to be defined genetic factors are likely involved. It has been reported that transgenic null mice for MEKK4, a protein kinase that regulates downstream the activity of different proteins involved in neuronal migration, including filamin A and doublecortin, frequently develop nodular heterotopia resembling those observed in human patients (157). A patient with periventricular nodular heterotopia with a MOB2 gene mutation has been described. A MOB2 gene knockout mouse model demonstrated impaired number and positioning of cilia within migrating neurons and increased phosphorylation of filamin (132). Thus, data from experimental animals may provide useful insights into the pathogenesis of this brain malformation in humans.
Two patients with deletions of the 17p11.2 region in whom the Smith-Magenis syndrome typical phenotype was associated with had bilateral periventricular nodular heterotopia. This finding expands the spectrum of chromosomal rearrangements associated with periventricular nodular heterotopia and indicates that abnormal neuronal migration may contribute to the neurocognitive phenotype of Smith-Magenis syndrome (32). Xq26.1-26.2 gain has been identified on array comparative genomic hybridization in bilateral periventricular nodular heterotopia with overlying polymicrogyria (02). A case was published of extensive posterior periventricular nodular heterotopia with biallelic variants in LAMC3, which encodes the laminin subunit ɣ3 (45). This is novel, as LAMC3 mutations are typically associated with polymicrogyria and pachygyria of the occipital cortex.
Mutations in SCN1A are known to be associated with epileptic encephalopathies. It has been found that SCN1A mutations can coexist with cortical dysplasia and bilateral periventricular nodular heterotopia. Although epidemiology does not support a causative role for SCN1A mutations, loss or impaired protein function combined with the effect of susceptibility factors and genetic modifiers of the phenotypic expression of SCN1A mutations might play a role (08).
One research group performed whole exome sequencing from primary DNA sources or lymphoblastoid cell line on 202 probands with sporadic periventricular nodular heterotopia and their parents (83). Two hundred and nineteen de novo variants were found in this set of samples. As expected, multiple de novo mutations were identified in filamin A. When assessing for loss of function genes, after filamin A, MAP1B was the most significant gene. MAP1B gene encodes a neuronal microtubule-associated protein that plays a role in neurogenesis and neuronal migration. Three of the four patients with MAP1B mutation showed bilateral symmetric anterior periventricular nodular heterotopia. They had variable clinical presentation for seizures, cognitive impairment, and dysmorphic features.
There is growing evidence indicates that the FLNA-dependent actin dynamics and regulation of vesicle formation and trafficking by activation of ADP-ribosylation factors (ARFs) can play an important role in this cortical malformation. The first inherited variant of ARF1 in a girl with intellectual disability and periventricular nodular heterotopia who inherited the variant from the father with previously undiagnosed single nodular heterotopia and mild clinical expression is reported (73).
Unilateral periventricular nodular heterotopia. In contrast, pathogenesis of unilateral periventricular nodular heterotopia is probably different and more likely related to acquired mechanisms. This is suggested by the following: (1) the presence of prenatal risk factors for brain damage in the history of unilaterally affected patients; (2) the possible association of single periventricular heterotopic nodules and polymicrogyria, a condition for which a pathogenic vascular mechanism is postulated (12); (3) the frequent location in the posterior paratrigonal region, which is a vascular watershed area (185); and (4) the anatomical continuity with structural abnormalities of the hippocampus (146). In unilateral cases, therefore, acquired insults, possibly of ischemic nature (158), may determine the failure of neuronal migration and the structural abnormalities of a limited region of the brain during early ontogenesis. It is possible, however, that combined genetic and epigenetic factors may contribute to determine unilateral periventricular nodular heterotopia (17).
Nodule characteristics and pathogenesis. Neuropathologic analyses have been reported in autopsic and surgical specimens (15; 90; 180; 179). The nodules were made up of neurons of normal size and morphology, without any evidence of cortical lamination, and they were richly innervated. Pyramidal neurons with randomly oriented apical dendrites were present within the nodules, and laminar fusiform neurons were located at the heterotopia borders. The neocortical areas close to unilateral periventricular nodular heterotopia may be modestly disorganized and slightly thinner than normal (15), and diffuse microvascular abnormalities have been found in the neocortex overlying bilateral nodules of a single patient with filamin A mutation (90). Reciprocal anatomical connections between the periventricular nodules and the neocortex have been described, suggesting the existence of functional circuits relevant for the propagation of epileptogenic discharges (82; 90; 95).
The intrinsic organization of nodular formations and of the overlaying neocortex was assessed by analyzing mRNA expression of three different layer-specific markers: Ror, Er81, and Nurd. It was found that in periventricular nodular heterotopia, the different gene expression profiles revealed a rudimentary laminar organization of the nodules. Also, in their overlaying cortex, fewer cells expressed the three genes in the appropriate layers, compared to controls. This shows the potential role of layer-specific markers in the neuropathology of malformation of cortical development and insight into ontogenetic mechanisms of periventricular nodular heterotopia (74). In a study, one group found a de novo heterozygous pathogenic variant in MEN1 from trace tissue adherent to the poststereotactically placed electroencephalographic depth electrodes (122). This suggests a possible novel candidate gene and a potential role for integration of electrophysiological and genetic approach.
Regarding the mechanisms underlying the genesis of the malformation, the presence of disorganized radial glial fibers around periventricular nodules of autopsic fetal brains suggested that disruption of radial glia could contribute to failure of migration from the ventricular zone (156). However, radial glia abnormalities could be the consequence rather than the origin of impaired neuronal migration. Indeed, neurogenetic studies in rats prenatally treated with methylazoxymethanol-acetate, an experimental model of this brain malformation (41), have indicated that the failure of the migration of early generated cortical neurons is sufficient to set the base for the formation of cerebral heterotopia (20). Data from this model also provided a neurogenetic scheme that may explain the genesis of the malformation in human patients (16). It is thought that periventricular nodular heterotopia arises from a final common pathway involving disruption of vesicle trafficking, leading to impaired cell adhesion and loss of neuroependymal integrity. The reason for the periventricular neurons is that the progenitor neuroepithelial cells cannot attach to the radial glia that guide them to the cortex; hence, they remain in their primitive position but mature as individual cells to become mature neurons in the abnormal fetal position in the brain. These neural progenitor cells are disrupted (65). Regulators of calcineurin 1 (RCAN1), a Down syndrome-related gene, plays an important role in radial migration of rat cortical neurons. RCAN1 acts upstream from FLNA in regulating radial migration, and rat studies suggest that impairment of RCAN1-FLNA pathway may underlie periventricular nodular heterotopia pathogenesis (105).
Endothelin-converting enzyme-2 (ECE2) is a metallopeptidase that has previously been associated with nonclassical production of neuropeptides and removal of intracellular amyloid-b in Alzheimer disease. One study examined the neurodevelopmental role of ECE2 for which two biallelic variants have been identified in two separate patients with periventricular nodular heterotopia (27). Buchsbaum and colleagues demonstrated that manipulation of ECE2 levels in human cerebral organoids and in the developing mouse cortex leads to ectopic localization of neural progenitors and neurons. Therefore, ECE2 plays a role in neurogenesis and the generation and secretion of extracellular matrix proteins, which can result in periventricular nodular heterotopia (27).
Pathogenesis of epileptic discharges. Alterations of the alpha subunit of the Ca2+/calmodulin-dependent kinase II and the NMDA receptor complex have been found in the epileptogenic cortex and nodules of human patients with unilateral nodules (19; 67). Studies in the methylazoxymethanol-acetate–treated rat model have demonstrated a selective impairment of the NMDA receptor complex in the cerebral heterotopia (75), thus confirming the data obtained in surgical samples from human patients. Altered distributions of GABAergic interneurons within the nodules also have been reported (90; 180; 179). These abnormalities could together contribute to the intrinsic hyperexcitability associated with this brain malformation. Furthermore, a study showed significant hypoactivation of insula in a resting-state fMRI of 38 patients with periventricular nodular heterotopia related epilepsy. This may suggest that the insula may be the cortical hub of whole-brain networks (108).
The incidence of periventricular nodular heterotopia is not known. The reported cases in the last decade indicate that the condition is equally distributed geographically and among ethnic groups. In selected clinical series, periventricular nodular heterotopia was found in 2% of epileptic patients studied by MRI, it and accounted for about 20% of all cortical dysgeneses.
Periventricular nodular heterotopia has not been associated with in utero exposure to toxic agents. In patients with bilateral and symmetrical nodules, particularly in the presence of a family history of epilepsy or spontaneous abortions, genetic analysis of the filamin A gene should be considered (68; 162; 138). FLNA mutations have also been described so far in most familial and some sporadic cases of bilateral periventricular nodular heterotopia, but not in unilaterally affected patients (139).
Neuroimaging. Diagnosis rests on neuroimaging techniques. A CT scan may reveal the presence of nodules, but it may not be adequate and may lead to incorrect diagnosis. MRI is the method of choice, allowing the recognition of nodules, the definition of their size and number, and the extension to the overlying neocortex. The heterotopic nodules are isointense to cortical gray matter in all imaging sequences. In patients with bilateral and symmetrical heterotopia, multiple nodules symmetrically line the entire extension of the ventricular walls. Mega cisterna magna and cerebellar hypoplasia are frequently associated findings.
The morphology of the overlying cortex is usually normal. However, in rare, mostly male patients, diffuse gyral abnormalities may be associated (51). One group performed a multimodal MRI phenotyping study using various MRI markers of development, structure, and function and compared patients with periventricular nodular heterotopia to healthy controls (46). They observed widespread structural and functional alterations in the brains of patients with periventricular nodular heterotopia with differential interaction of the nodules with the overlying cortex and the hippocampus.
In the unilateral cases, the heterotopic nodules can be of different size and number in different patients. The nodules are most frequently confined to or located in the paratrigonal region of the lateral ventricles.
The nodules may extend from the periventricular region into the white matter and may involve the overlying neocortex. Or they may extend to the anterior edge of the temporal horn, associated with structural abnormalities of the hippocampal formation (146). In patients with nodules located in the paratrigonal region, a mild posterior cerebral hypoplasia is frequently associated (18; 17). Imaging study should always include the evaluation of the hippocampus, given the frequent finding of hippocampal abnormalities. Double inversion recovery acquisition suppresses the white matter signal, which may enhance visualization of abnormal features at the gray-white matter interface such as periventricular nodular heterotopias (190).
Transmantle bands are frequently associated with both unilateral and bilateral cases of periventricular nodular heterotopia and visualized with conventional 3T MRI sequences. The band highlights the underlying neuronal migration issues at play in the pathogenesis of this disorder, but its underlying role in the complex, patient-specific epileptogenic networks needs to be determined (81).
Imaging studies have also looked at predictors for epileptogenic periventricular nodular heterotopia. In one pediatric study, periventricular nodular heterotopia patients were divided into two groups: with epilepsy and without epilepsy (182). Radiological characteristics of laterality, regionalization, largest dimension, and number of nodules were compared between the two groups. Notably, only periventricular nodular heterotopia spreading across several regions was associated with a statistically higher chance of epilepsy. Other features including laterality, individual region, number, and largest dimension did not reliably predict epileptogenicity. A large retrospective cohort study demonstrated how drug resistance of epilepsy patients could be predicted to an extent by the pattern of periventricular nodular heterotopia (135). Patients were subdivided as follows: isolated periventricular nodular heterotopia single nodule or multiple nodules (PVNH-only) and periventricular nodular heterotopia with additional brain malformations (“PVNH-plus”) single nodule or multiple nodules. Of PVNH-only single nodule, none had drug-resistant seizures. Amongst PVNH-plus, 55% with multiple unilateral nodules were pharmacoresponsive, compared to only 21% with bilateral nodules. PVNH-plus with bilateral nodules demonstrated the highest proportion of drug resistance (39%) (135).
Alterations in functional connectivity have also been performed. One study compared functional connectivity in patients with periventricular nodular heterotopia and epilepsy to healthy controls (110). Periventricular nodular heterotopia patients exhibited significantly higher functional connectivity in the parietal lobe, cingulum, and thalamus, as well as significantly lower functional connectivity in frontoparietal, hippocampal, and precentral regions. Seed-based functional connectivity analysis confirmed disruption of activities and interregional connectivity in remote epileptic networks of patients. The cerebellum and limbic system of patients showed altered topology, suggesting that these regions or hubs may contribute to whole-brain circuits in periventricular nodular heterotopia and epilepsy (110).
Diffusor tensor imaging. A novel MRI postprocessing technique has been described for detection of periventricular nodular heterotopia (140). It is a further development of voxel-based morphometric analysis, which focuses in the region around the ventricles to increase the sensitivity and specificity of detecting nodules. As nodules can be missed with conventional imaging, this is a promising additional diagnostic tool. Differences in corpus callosum midsagittal area and subregional area changes were measured using an automated software-based method. Reduced corpus callosum area was found to be strongly associated with periventricular nodular heterotopia. The changes were noted primarily in the posterior corpus callosum (136). Diffusor tensor imaging has mainly been used as an experimental tool up to now. Whole-brain diffusion MRI tractography revealed abnormal fiber projections in nodular tissue suggestive of abnormal organization of white matter (with abnormal fibers both within nodules and projecting to the surrounding white matter) in patients with bilateral peri. Therefore, this technique has expanded our knowledge of the pathology and extent of these lesions (66; 61). If family history for epilepsy is reported, MRI evaluation of all potentially affected members should be performed to help determine the risk of genetic transmission. Prenatal ultrasonography and MRI have been reported to detect heterotopic nodules in utero at early gestational stages (121). Detection of nodules can be challenging on antenatal ultrasound. However, indented ventricular borders on axial views and irregular square-shaped lateral ventricles on coronal views are suggestive (23).
Genetic assessment. Diagnostic workup should also include a genetic testing, which can guide long-term screening of comorbidities, such as cardiac disease with FLNA mutations. Genetic testing includes sequencing and array-comparative genomic hybridization. Using a customized panel of known and candidate genes associated with brain malformations, one group applied targeted high-coverage sequencing (depth ≥ 200×) to leukocyte-derived DNA samples from 158 persons with brain malformations, including periventricular nodular heterotopia. A significant number were found to have causative somatic mutations. Therefore, high-coverage sequencing panels can provide an important complement to whole-exome and whole-genome sequencing in the evaluation of somatic mutations in patients with brain malformations, including periventricular nodular heterotopia (87). Similarly, another group examined array-comparative genomic hybridization in 106 patients with brain malformations, including 42 patients with periventricular nodular heterotopia. They showed that genomic imbalances, either small cryptic copy number variants or large rearrangements, are frequently associated with periventricular nodular heterotopia. The authors suggest array-comparative genomic hybridization should be considered as a first-line diagnostic genetic test in patients with sporadic, nonclassical periventricular nodular heterotopia (36). In any individual with imaging and/or clinical features suggesting a specific diagnosis, targeted testing can be pursued. In any other case, it is suggested to follow a broad approach with a chromosomal copy number analysis, a targeted gene panel, and trio exome sequencing (186).
Assessment of epilepsy. The diagnostic workup should always include the assessment of epilepsy.
Connectivity analysis using diffusion tensor imaging or fMRI has further provided insights into the epileptogenic network, including connections between the nodule and overlying or distant cortices (15, 38), as well as among nodules (17, 38). In a study analyzing fMRI and iEEG in patients with multiple nodules, we have demonstrated that fMRI can detect functional connectivity between nodules and that strongly connected nodules are more epileptogenic than the others. For instance, simultaneous seizure onset and faster seizure spread are more prevalent between strongly connected nodules (17••). However, how or why this connectivity develops is not fully understood at present.
Scalp EEG recordings during wakefulness and sleep and video-EEG monitoring define ictal and interictal epileptiform activities. The interictal EEG abnormalities are mainly focal and consistent with the anatomical location of the nodules. However, generalized discharges of 3 to 4 Hz spike-and-wave activity mimicking primary generalized epilepsy have been reported (76). Three Hz generalized spike-wave discharges with typical and atypical absence seizures have also been described. This suggests that migration disorders, such as periventricular heterotopia, may influence the formation and excitability of the striato-thalamo-cortical network involved in the generation of 3 Hz spike-waves (49). This was confirmed in an fMRI study of patients with absence seizures and periventricular nodular heterotopia. In one patient, there was connectivity of nodules to the thalamus and in another patient, to the parietal cortex (33). In addition, a photic driving of the posterior background activity, characterized by bilateral harmonic alpha rhythm frequencies, has been reported (18; 17).
Stereo-EEG recordings should also be performed before considering epilepsy surgery for the management of intractable focal seizures, due to the possible seizure onset from the neocortex, hippocampus, and heterotopic nodules itself. Depth EEG recordings have variably found seizures to arise in nodular tissue, overlying cortex, or simultaneously in both (179; 184).
Recently, the first in vivo microelectrode recording of heterotopic neurons in humans was completed. Highly consistent interictal patterns were identified within the nodules (70). Among the different interictal patterns, “sporadic discharges plus fast activity” pattern was found only in the nodules that were actively involved in seizure generation, but not in nodules that did not take part in ictal discharges. Therefore, interictal patterns may be useful predictors of epileptogenic nodules (70).
The simultaneous acquisition of continuous EEG and functional MRI (EEG-fMRI) recordings allows whole-brain visualization of changes in neural activity during interictal epileptiform events (187). Functional connectivity analysis detects brain regions showing temporally correlated fluctuations in fMRI activity, to display networks of brain regions presumed to be involved in a common function.
One study looked at the relative time at which fMRI-involved regions became active. Additionally, nodule-cortex interactions were explored using fMRI functional connectivity. There was EEG-fMRI activity in specific periventricular nodules and overlying cortex. In both nodules and cortex, the peak BOLD response to epileptiform events occurred earlier than expected from standard fMRI hemodynamic modeling. Functional connectivity showed nodule-cortex interactions to be strong in these regions, even when the influence of fMRI activity fluctuations due to spiking was removed. Nonepileptogenic, contralateral nodules did not show connectivity with overlying cortex. Therefore, EEG-fMRI and functional connectivity can help identify which of the multiple abnormal regions are epileptogenic in periventricular nodular heterotopia (05). In addition, combined EEG/fMRI analysis have revealed blood oxygenation level dependent changes within the heterotopia temporally related to interictal spiking activity (97).
F-fluorodeoxyglucose positron emission tomography/computed tomography (F-FDG) PET/MRI allows simultaneous evaluation of morphology and function in periventricular nodular heterotopia patients with epilepsy. On FDG PET, the heterotopic tissue in these patients manifests with different metabolism levels, ranging from ametabolism to hypermetabolism, and many patients with bilateral PVNH can have unilateral cortical hypometabolism. F-FDG PET/MRI can be a useful presurgical tool as studies have shown that F-FDG PET/MRI findings are mostly concordant with the clinical epileptogenic zone (29; 06). Quantitative MRI fingerprinting measures have also been demonstrated as a supplementary noninvasive presurgical tool. The T1 and T2 tissue property changes carry electrophysiological underpinnings relevant to the epilepsy, as shown by their significant positive associations with power changes during the stereo-EEG seizure onset. Qualitative MRI fingerprinting measures, especially T1 sequences, can provide additional noninvasive information to identify nodules that are part of the seizure-onset zone (39).
Magnetic source and diffusion tensor imaging may be of value in the assessment of surgical strategy (172). It should be kept in mind that periventricular nodules may be functionally involved not only in seizure onset and propagation but also in physiologic brain activity. Indeed, functional studies demonstrated metabolic activity and blood flow in periventricular nodular heterotopia similar to studies of normal cortex (123) and reported visual, sensorimotor, and speech-related activation of heterotopic nodules in some, but not all, patients (88).
Future risk assessment. Finally, because in X-linked periventricular nodular heterotopia patients the risk for stroke and other vascular and coagulopathic complications is increased (68; 162), careful cardiologic and hematologic assessment should be considered, particularly if FLNA mutations are present or suspected. Screening for connective tissue disease and lung disease should also be considered.
Epilepsy control.
Medical intervention. Given the high incidence of epilepsy, seizure control is the most relevant clinical aspect. Epilepsy is not necessarily characterized by high seizure frequency, but seizures may require several drugs and are often difficult to control.
Surgical intervention. Surgical outcome has been reported as poor (104). For patients with periventricular nodular heterotopia, surgical intervention is often limited due to the depth of the lesions and their close anatomical relations with essential functional brain connectome (25). However, data have demonstrated that surgery outcome may be good, even after resections limited to the periventricular nodules, after proper stereo-EEG assessment of the epileptogenic areas (69; 04; 160; 179). In some reports stereoelectroencephalography-guided radiofrequency thermocoagulation (SEEG-guided RF-TC), which makes it possible to investigate nodular tissue involved in epileptic networks and disable it in a single procedure, is considered to be a promising interventional option (25). The benefits of SEEG-guided RF-TC include the following: 1) it is performed using the same electrodes employed for SEEG recording, thus, eliminating the risk for new implantations with traditional lesioning electrodes; 2) the treatment plan is tailored according to data provided by SEEG recordings; 3) functional information obtained by intracerebral electrical stimulation is used to avoid injury to eloquent cortical and subcortical areas; 4) in cases of failure to control seizures, radiofrequency thermocoagulation lesions do not prevent eventual resective surgery (43). A meta-analysis of SEEG-guided RF-TC in patients with periventricular nodular heterotopia reported 38% seizure-free patients and 83% of responders (24). SEEG-guided RF-TC seems like a promising treatment option for drug-resistant epilepsy related to periventricular nodular heterotopia for cases in which patients are not eligible for epilepsy surgery (24).
Many patients with bilateral periventricular nodular heterotopia have epilepsy, and there is likely to be a complex interaction between epileptogenic nodular tissue and overlying cortex. A model was created to examine this relationship by examining c-fos activation in organotypic hippocampal slice cultures generated from an animal model of periventricular nodular heterotopia, which was created by treating pregnant rats with methylazoxymethanol acetate. In this model, the nodule does not appear to be responsible for enhanced excitability or seizure initiation. Excitability is different in tissue that contains a nodule, suggesting altered network function, perhaps reflecting the abnormal developmental pattern that gave rise to the nodule (52). Most have an electroclinical pattern of temporal lobe epilepsy, but routine temporal lobe resections have had limited success. Depth EEG recordings have variably found seizures to arise in nodular tissue, overlying cortex, or simultaneously in both (179; 184). There are some reports of seizure reduction in patients with focal periventricular nodular heterotopia following tailored wedge resections of heterotopic nodules and overlying brain (179). Others have found success with resection of the periventricular nodule alone, although in the presurgical work-up, the seizures did not originate from the nodule (03). There are no uniform guidelines for the management of heterotopia-based seizure disorder. However, treatment generally follows standard guidelines involving focal epilepsies. Surgical resection can be offered for patients with intractable seizures. At present, intracranial EEG evaluation remains essential for the vast majority of cases of periventricular nodular heterotopia prior to surgery. These recordings must methodically sample nodular tissue along with overlying neocortex and mesial temporal structures, so as to accurately delineate the epileptogenic network(s).
Radiosurgery. Gamma Knife-based stereo-radiosurgery may serve as a reasonable option for patients with heterotopia-induced intractable seizures who are deemed not to be surgical candidates and do not have other treatment options available (161). Stereotactic MR-guided laser interstitial thermal therapy has become available for focal ablation, promising minimally invasive techniques. This is especially promising for those with deep lesions not amenable to conventional surgery (57). Alternative treatments such as radiofrequency ablation and MRI-guided laser ablation enable the lesioning of multiple areas, consequently disrupting epileptogenic networks; however, this may be limited in cases where nodular tissue is associated with epileptogenic structural abnormalities or with eloquent cortex (126). Focal resections/ablations may be successful if the role of the periventricular nodular heterotopia within the epileptogenic networks is understood. The literature demonstrates that surgical outcomes are generally better when nodules are at least partially ablated. The role of nodular tissue within epileptogenic networks is likely not the same in every case, as demonstrated by the varied success with nodular ablation alone (181).
Responsive neurostimulation. Responsive neurostimulation is an emerging therapy for drug-resistant focal epilepsy. A prospective clinical trial demonstrated that eight patients with periventricular nodular heterotopia (mean follow-up of 10.1 years) had a mean reduction of 85.7% in disabling seizures. Seven patients had greater than 50% of seizure reduction from chronic ambulatory electrocorticograms analysis and two patients were completely seizure-free in the final year of analysis. This study suggests that responsive neurostimulation may be an effective treatment option in drug-resistant periventricular nodular heterotopia (126). A case report described a patient with bilateral periventricular nodular heterotopia treated with hippocampal deep brain stimulation, leading to major reduction in seizure frequency (44).
A small study reviewed five patients who underwent laser interstitial thermal therapy procedures for epilepsy with periventricular nodular heterotopia (188). The majority of patients had good seizure outcome and none of the patients experienced motor deficits, dysphasia, infection, or mortality. Therefore, laser interstitial thermal therapy may be a safe and promising option for patients with refractory epilepsy and periventricular nodular heterotopia that otherwise may not be surgical candidates.
Cardiac complications control. Due to the risk of cardiac complications with FLNA mutations, it is important to ensure good blood pressure control. Standard treatment for aortic or carotid dissection, congenital heart disease, and valvular disease is recommended. Echocardiogram and cardiac MRI may be used to screen for cardiovascular problems. One case study demonstrated that for individuals with intractable epilepsy and ictal asystole who are poor surgical candidates, pacemaker implantation is indicated to prevent injury and any potential contribution of ictal asystole in sudden unexpected death in epilepsy (77). Clinicians should also be aware of the risk for congenital heart disease, valvular abnormalities, and dilatation of the ascending aorta. Vascular disease can be present in neurologically asymptomatic individuals. Therefore, it is appropriate to screen at-risk relatives of an affected individual to identify those who would benefit from treatment and preventive measures (38).
Outcome and prognosis, particularly when related to cognition and seizures, depend on various factors. These include the underlying etiology of the periventricular nodular heterotopia, the amount of heterotopic tissue, the distribution of the nodules, extension in overlying cortex, and coexisting brain malformations. Earlier onset or more severe epilepsy can have a significant impact on neurodevelopment. Use of antiseizure medications and timely presurgical evaluations are important to obtain better control of the epilepsy and improve neuropsychiatric outcomes. Offering early genetic counseling is important, especially because various genes can be involved in periventricular nodular heterotopia. Determining a genetic etiology may be important for screening and preventing systemic complications in some individuals.
Given the identification of the FLNA as responsible for bilateral and symmetrical periventricular nodular heterotopia (68), genetic counseling should be offered to bilaterally affected patients, particularly those who have a family history of epilepsy or spontaneous abortions (103). Periventricular nodular heterotopia secondary to a FLNA mutation is inherited in an X-linked manner. The condition is lethal in most males; and the majority of affected individuals are female. About 50% of affected females inherit the pathogenic variant from their mother, and at least 50% have a de novo pathogenic variant. For women, the risk of passing the pathogenic variant to each child is 50%. Most sons born to women with FLNA-related periventricular nodular heterotopia are unaffected.
Prenatal diagnosis by molecular genetic testing is possible if the pathogenic variant has been identified in an affected relative. As early as 24 weeks gestation, periventricular nodules may be visualized by imaging. However, the sensitivity of imaging for the prenatal detection of periventricular nodular heterotopia is not known. In the early third trimester, irregular ventricular walls on axial view and irregular square-shaped lateral ventricles on coronal view are suggestive of periventricular nodular heterotopia. It is important to keep in mind the possible coexistence of periventricular nodular heterotopia with brain malformations such as ventriculomegaly, posterior fossa anomalies, or agenesis of corpus callosum (155). There is emerging evidence that 3-tesla (3T) MRI may be more sensitive at detecting periventricular nodular heterotopia. One such case was demonstrated in a woman who underwent 3T MRI after fetal ultrasound was suggestive of agenesis of the corpus callosum. 3T MRI showed subependymal heterotopia that was confirmed on postmortem pathology after the pregnancy was terminated (173).
The teratogenic risk to the fetus associated with the use of antiseizure medication during pregnancy depends on the type and dose of the seizure medication. There are currently no guidelines regarding the most appropriate surveillance for and management of cardiac, vascular, and connective tissue problems during pregnancy.
In the authors’ experience, no particular risk is associated with any type of anesthesia for patients with periventricular nodular heterotopia.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Anita Datta MD
Dr. Datta of the University of British Columbia has no relevant financial relationships to disclose.
See Profile
Harvey B Sarnat MD FRCPC MS
Dr. Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Developmental Malformations
Apr. 14, 2024
Developmental Malformations
Apr. 09, 2024
Developmental Malformations
Mar. 22, 2024
Developmental Malformations
Mar. 22, 2024
Developmental Malformations
Mar. 22, 2024
Developmental Malformations
Mar. 22, 2024
Developmental Malformations
Mar. 13, 2024
Developmental Malformations
Mar. 11, 2024