


Developmental Malformations
Walker-Warburg syndrome
Apr. 14, 2024
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
The author reviews the clinical and pathological features of sacral agenesis, with emphasis on the resulting neurologic deficits and the association in many cases with maternal diabetes mellitus. The demonstrated wide spectrum genetic defects are reviewed in the context of molecular genetic regulation of ontogenesis of bony and neural spinal structures. Differential diagnosis from meningomyelocele, diastematomyelia, and congenital sacrococcygeal teratoma; an association with more extensive congenital anomalies; and management of the neurologic complications are discussed. The most serious complications are lack of bowel and bladder control due to anorectal atresia and flaccid neurogenic urinary bladder. The autosomal dominant Currarino triad of sacral dysgenesis, anorectal malformations, and anterior meningocele is discussed, as well as caudal regression syndrome or syringomyelia.
|
• Agenesis of the sacrum and sometimes lumbar and even thoracic vertebrae is a congenital malformation that occurs sporadically, associated with other complex genetic syndromes and most frequently in infants of diabetic mothers (1% of all such infants). Rarely, it may be asymmetrical as hemi-sacral agenesis. | |
|
• Because of lack of neural tube (ie, floor plate) induction by the deficient notochord, at the level of the bony dysgenesis there is a severe dysplasia of the spinal cord, often with fusion of the ventral horns, an incomplete central canal, and abnormal architecture of white matter tracts. | |
|
• Clinical neurologic expression at birth is hypoplasia or aplasia of muscles innervated by the defective ventral roots, autonomic defects, and neurogenic bladder, but relative preservation of somatic sensory function of the dorsal roots. | |
|
• In its most extreme forms there may be a “caudal regression syndrome” of sirenomelia (fusion of the lower limbs), imperforate anus and other lower intestinal and urinary collecting system anomalies or the “Currarino triad” of anorectal malformation or ectopic anus, coccygeal and partial sacral aplasia, hypoplasia or dysplasia but preservation of the first sacral vertebra, and a presacral mass that often is an anterior meningocele, dermoid cyst, midline sacral lipoma, or a neuroendocrine tumor. | |
|
• The mechanism is abnormal segmentation of the embryonic sclerotomes with secondary failure of induction of the caudal neural tube and surrounding mesodermal structures. | |
|
• A strong association with maternal diabetes mellitus is of unknown pathogenesis, but suppressed genetic expression by insulin is suspected. | |
|
• Various genetic mutations are demonstrated in sacral agenesis not associated with maternal diabetes mellitus, in both familial and sporadic cases. |
Partial or complete absence of the sacral and coccygeal bones was first described in 1852 by Hohl (71); in 1857, Wertheim published a case of complete sacrococcygeal agenesis. Over the next hundred years, about 50 additional cases were published singly or in small series. In 1959, Blumel and colleagues reported 50 more cases, many of which included other malformations such as meningoceles, anal and bowel anomalies, and abnormalities of the lower limbs often associated with dysfunction of the bowel and urinary bladder. Sacral meningocele, often anterior, was noted by Coller and Jackson in 1943 to often be associated with sacral agenesis or sacral hypoplasia (37), and this association was subsequently confirmed by other authors. Case reports confirming the early descriptions continue to appear (141; 188; 166; 16; 52; 75; 156). An association of sacral agenesis with congenital tumors in the sacral region also is described, but this situation remains rare.
The frequent occurrence of sacral agenesis in infants of diabetic mothers was pointed out by Kucera in a survey that reviewed 48 papers published between 1930 and 1964 on the topic of congenital anomalies associated with maternal diabetes mellitus during gestation (92). The total incidence of all anomalies was 4.8%, and the incidence of sacral agenesis in infants of diabetic mothers is now estimated at about 1%, a more significant association than with any other predisposing condition, including genetic factors. Other case reports continue to be published, further confirming this association (132). Preexisting maternal diabetes mellitus, type 1 more than type 2, was redocumented as a strong risk factor for nonsyndromic fetal sacral agenesis in a large National Birth Defects Prevention Study (123).
Caudal regression syndrome. The term "syndrome of caudal regression" was first used by Duhamel to describe a spectrum ranging from simple sacral agenesis to severe lower limb anomalies, including fusion as "sirenomelia," often with imperforate anus and major malformations of the anus and rectum, omphalocele, and anomalies of the genitourinary system, but sacrococcygeal agenesis or hypoplasia was the constant feature (44; 23; 56; 19). Rarely, there is duplication of the rectum rather than atresia or ectopia (90; 131). Children born with the syndrome or caudal regression also may lack pelvic bones and have agenesis of the fibulae and of the external genitalia, and they often live only a few hours (04). Rare cases exhibit a vestigial tail despite sacral agenesis (64), suggesting lack of induction at the sacral level and overinduction at the coccygeal level, in terms of pathogenesis. Many inconstant anomalies of multiple organ systems include single umbilical artery in one third of cases and congenital heart disease in more than half of cases as well as renal agenesis, genital anomalies, popliteal webbing, and cleft lip and palate (06; 25). Asymmetrical sacral agenesis associated with hemivertebrae occurs rarely (57). The spinal cord may terminate at midthoracic levels, with a few bony vertebrae still developed caudal to that site (15). Deficient sensory and motor innervation of muscle corresponding to the level of the dysplastic spinal cord may sometimes be improved in caudal regression syndrome by extended use of growth hormone combined with physiotherapy rehabilitation (41). Some authors use the term “caudal regression syndrome” to refer to patients with lumbosacral agenesis, imperforate anus and unilateral or bilateral renal agenesis, and neurologic deficits of the lower extremities but not fusion (142), but this is a semantic issue of how broad the clinical spectrum should be.
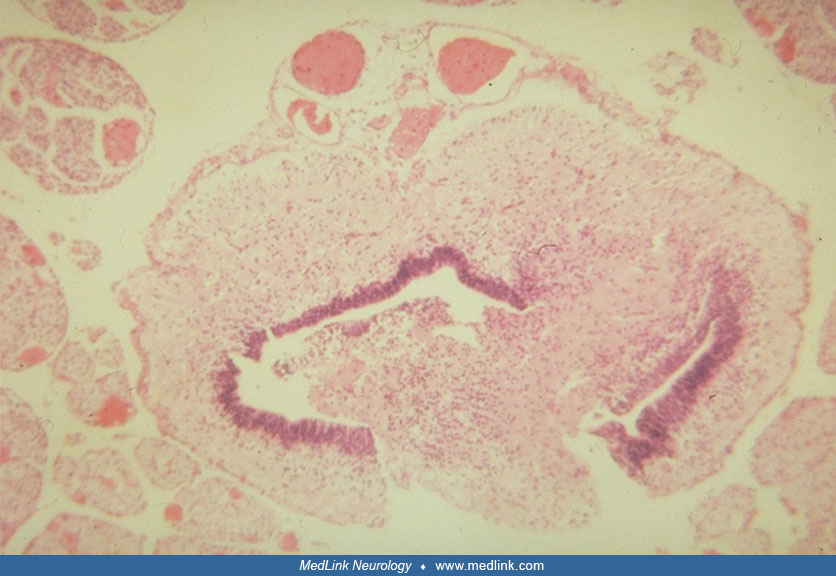
The clinical neurologic and neuropathologic findings in sacral agenesis were described by Sarnat and colleagues following fragmentary descriptions in the earlier literature and confirmed by subsequent observations (156; 122; 47; 46). Plain radiographic features had been described by many authors, and the MRI findings were systematically described by Nievelstein and colleagues (125). Radiographic and imaging findings of the bony spinal anomalies can be diagnosed at birth and prenatally as early as 26 weeks’ gestation (23; 57; 122). Though not generally familial, caudal regression syndrome has been found in one of dizygotic twins (91). Hemi-sacral agenesis occurs at times (112); in one case, it was discovered as an incidental finding in a (99m) Tc-MDP bone scan performed for other reasons in a 69-year-old woman (83). In addition to imaging for the vertebral defects, presacral masses may be demonstrated in Currarino syndrome that are not necessarily evident on physical examination of the patient (136).
Thin, flaccid lower limbs are evident in the neonatal period in cases of complete sacral agenesis or if the defect also involves lumbar segments, but minor defects of only the coccyx and lowest one or two sacral vertebrae may not show obvious clinical deficits at birth. Meningomyeloceles are usually small or occult and are generally associated only with extensive defects. The meninges and nerve roots may herniate through the region of the defective vertebral centra and, thus, may extend anteriorly, rather than posteriorly, and not form an external cystic mass over the lower spine (27; 37; 160). Lipoma of the conus medullaris and filum terminale, fusion of nerve roots and ganglia, and aberrant sensory ganglia are additional defects in severe cases (178). Palpation of the spinous processes of the newborn back may disclose a level below which only soft tissue is felt. About 17% have spina bifida bony deformities (43). The wings of the ilia may be rotated toward the posterior midline in the absence of an intervening sacrum. Half of the children have congenital subluxation or dislocation of one or both hips (43; 117).
The bony defect usually consists of total absence of the coccyx and of the lower two or three sacral vertebrae, hypoplasia of the one or two vertebral bodies immediately above the aplastic segments, and normally formed vertebrae at more rostral levels. Rarely, the sacral and lumbar articular facets are absent, but the centrum or vertebral body is formed (84). In some cases the spinal bony defect may be extensive and involve the entire sacrum, lumbar, and even thoracic vertebrae. Displaced hypoplastic sacral vertebral bodies may lie posterior to the lower lumbar vertebrae as a rostrocaudal dislocation in rare cases.
More severe and extensive vertebral defects may be accompanied by sirenomelia or fusion of the lower limbs in a "mermaid" configuration; this condition is nearly always associated with an ectopic or imperforate anus, malrotation of the bowel and sometimes omphalocele, genitourinary anomalies, and often hydronephrosis, rectovaginal fistula, and other visceral anomalies. The combination in sirenomelia is sometimes called the "syndrome of caudal regression" (44; 134; 107; 187; 61; 178; 74; 108). Visceral anomalies of the gastrointestinal and genitourinary systems are not found exclusively with sirenomelia but also may accompany some cases of usually extensive vertebral defects without lower extremity fusion (16; 11; 88; 174). Imperforate anus, anteriorized anus, and other anal anomalies occur in some cases (30). Aphallia was reported with urorectal septal anomalies and sacral agenesis in a male infant of a diabetic mother (60). Unilateral pulmonary atresia and tracheoesophageal fistula may occur rarely with total sacral agenesis (13). Klippel-Feil syndrome of cervical vertebral fusion is associated with sacral agenesis in some cases, and neural tube defects may extend along the entire spinal axis (158).
All children with complete sacral agenesis have abnormal bladder and urethral function that not only causes incontinence but puts their kidneys at risk for progressive nephropathy; early detection is essential to prevent chronic hydronephrosis and indolent urinary tract infection (189; 46). Complete neurogenic bladder may occur with secondary renal impairment from hydronephrosis (34). Anatomical anorectal malformations may cause rectovesical fistula and recurrent urinary tract infection (182). Pudendal neuromodulation is possible to monitor bladder and bowel dysfunction, and rectovesical fistula in particular, in sacral agenesis (162).
Neurologic manifestations. The neurologic deficits in sacral agenesis are present at birth and involve motor functions more commonly than sensory functions, in a lumbosacral root distribution (156). Autonomic involvement with a flaccid neurogenic bladder and hydronephrosis is variable and does not necessarily follow the extent of the somatic motor deficits (156). Flaccid neurogenic bladder is a serious complication in many cases, and it is difficult to treat and to prevent hydronephrosis and recurrent lower urinary tract infections (151; 179). Exstrophy of the bladder is present in rare cases (102). Neurogenic bladder also may be associated with imperforate anus (179; 80). Other gastrointestinal complications range from malrotations of the bowel and omphalocele to functional constipation probably due to poor peristalsis from defects in autonomic innervation of the gut (68). Isolated neurenteric cysts in the lumbar region are reported in sacral agenesis (109). A rare clinical presentation in the neonate is abdominal distension in an infant at risk for necrotizing enterocolitis (126).
Marked sensory deficits usually indicate an occult meningomyelocele that may be accompanied by a Chiari malformation and hydrocephalus. Spinal dorsal root ganglia and the dorsal horns of the spinal cord are usually preserved in sacral agenesis (156) but occasionally may be defective or absent even without open neural tube defects (178). Impaired parasympathetic and somatic motor innervation of the lower rectum and anal sphincter may be present (115).
The motor deficits are due in part to nerve root hypoplasia or dysplasia with lack of complete innervation of fetal muscle, in part to dysgenesis of the spinal cord and a deficient number of motor neurons, and in part to primary amyoplasia. Entire muscle groups, such as the gastrocnemius-soleus group or the anterior compartment muscle groups of the lower leg, may be replaced by fat with little or no residual thin strips of normal or atrophic muscle fibers (155).
At times, the dermis also is hypoplastic and only adipose tissue with a few small wandering nerves is interposed between the usually (except in sirenomelia) normally formed long bones and the epidermis. More extensive neuropathies accompany more extensive vertebral defects to higher levels of the spine, and extension into the thoracic spine may be associated with diaphragmatic hernias and respiratory distress at birth. The neurologic deficits are generally symmetrical. Rarely, incomplete sacral agenesis may be associated with terminal lipomyelocystocele (145).
The spinal cord may be tethered in sacral agenesis (47; 158), and syringohydromyelia or syringomyelia may be present as demonstrated by MRI (125; 128). Because of the tethered spinal cord, the neurologic deficits may be slowly progressive as the child grows (47; 46). Particularly in Currarino syndrome, presacrococcygeal mass lesions may occur with malignant neuroendocrine transformation (35). The conus medullaris may be elongated (59).
Orthopedists classify distal spinal agenesis into categories (Renshaw sacral agenesis types 1 to 4) that correlate with ambulatory and other motor functional abilities (180). Acetabular dysplasia of the hip may accompany unilateral or bilateral sacral agenesis (117). Rib grafts can be used to promote spinopelvic fixation in some patients with sacral agenesis (50).
Rarely, malformations of left-right asymmetry of visceral organs, including anomalous superior and inferior vena cavae, are present in infants with sacral agenesis who are born to diabetic mothers (101).
The prognosis for neurologic improvement is poor. Survival and quality of life depends largely on the management of the major visceral complications, such as hydronephrosis and anomalies of the gastrointestinal tract and anus. Occult meningomyeloceles should be demonstrated and treated as indicated. They may be accompanied by Chiari malformation and hydrocephalus. A longitudinal study of bowel function in children with sacral agenesis showed successive spontaneous improvement with age, though the children did not achieve normal bowel control (20). Fecal incontinence is a difficult complication of sacral agenesis (105; 173; 42).
The most common predisposing factor in the etiology of sacral agenesis is not chromosomal, but rather maternal diabetes mellitus. The majority of infants with sacral agenesis have diabetic mothers, and approximately 1% of all infants born to diabetic mothers have agenesis of at least the coccyx and the lower one or two sacral vertebrae (150; 134; 171; 39; 73; 82; 60; 94; 54). Pregestational diabetes invokes a higher risk than gestational diabetes (175). Sacral agenesis should be regarded as a part of a spectrum in the programming of the entire caudal region of the embryo, rather than as an isolated anomaly (32). Partial sacral agenesis associated with a dorsal enteric sinus and spina bifida is attributed to “split notochord syndrome” (124) but really is more likely a form of neurenteric cyst. Rarely, rather than just dysplastic spinal cord at the level of the bony hypoplasia of lumbar and sacral vertebrae, there may be absence of a segment of spinal cord altogether, but with preservation of the conus medullaris, the most caudal portion that forms by secondary, rather than primary, neurulation, termed “junctional neural tube defect” (161). Some authors regard sirenomelia as a “polytopic field defect” (19).
No constant genetic or chromosomal disorders are associated with sacral agenesis, but isolated cases are described. Some anterior spinal defects appear to be inherited as an autosomal dominant trait (191). A gene for autosomal dominant sacral agenesis maps to the holoprosencephaly region at 7q36 (103), though holoprosencephaly is not usually complicated by sacral agenesis. Mutation in the HLXB9 transcription factor also causes an autosomal dominant form of sacral agenesis (32). Furthermore, in five families with mirror polydactyly and sacral agenesis, no mutations were demonstrated (183). This gene is the primary genetic defect in Currarino syndrome as a specific form of sacral agenesis, but it is not involved in the pathogenesis of the caudal regression syndrome (110). Several cases of sacral agenesis are reported with terminal deletion of the long arm of chromosome 7, with 7q35 and 7q36 deletions in particular appearing as an important genetic etiology (07). A locus for sacral agenesis with anorectal malformations has been mapped to 6q25.3 in a 0.3 Mb interval region (176).
A whole exome sequencing and copy number pilot study concluded that despite great genetic diversity and complexity of the phenotype, common genetic features would be identified in patients with sacral agenesis (138). In addition to identifying several variants in infants, exome sequencing also identifies a possible association between inhibitor of DNA binding-1 (ID1) and nonsyndromic sacral agenesis (137).
There is no significant gender difference (43). In some cases, the occurrence of sacral agenesis in siblings suggests an autosomal recessive trait (121; 154), but autosomal dominant inheritance without expression in a parent is possible. X-linked dominant inheritance of partial absence of the coccyx and sacrum also is described rarely in other families (36). An infant with a 7q36-->7qter terminal deletion had sacral agenesis and also holoprosencephaly (116), but midline cerebral malformations are not generally associated with defects of the lower spine. Another fetus also is reported with 7q36.1-->qter monosomy and sacral agenesis with severe intrauterine growth retardation, but this fetus did not have holoprosencephaly (159). Structural rearrangements of the long arm of chromosome 7 with terminal deletions of chromosome 7 (q36-qtr) (185; 170), ring chromosome 7 (147), or deletions at band 7q36 (including the Sonic hedgehog gene), or 7q36-qter terminal deletion in infants with sacral agenesis and anterior myelomeningocele, are demonstrated (181; 146). Combination duplication/deletion of distal 7q also is demonstrated in some children with the Currarino triad (135). Trisomy 19q with monosomy 7q is reported with sacral agenesis (59).
Missense mutation of the human T-gene (brachyury), a transcription factor essential for the normal development of posterior mesodermal structures and defective in vertebral malformations, is suggested as a possible cause of sacral agenesis (58). A murine form of T-gene mutation is known, but this gene is not confirmed as an etiology of sacral agenesis in the mouse (133).
Currarino triad. A specific type of sacral agenesis of autosomal dominant transmission is identified as “Currarino triad or syndrome” or “caudal regression syndrome.” It consists of a unique complex of congenital caudal anomalies that include (1) anorectal malformation or ectopic anus, (2) coccygeal and partial sacral aplasia, hypoplasia, or dysplasia, and (3) a presacral mass that often is an anterior meningocele, dermoid cyst or lipoma (104; 87; 93; 97; 28; 135). The spinal cord may be tethered, and the brain may be micrencephalic but with a normal convolutional pattern (135; 94). Anorectal stenosis may be present, and bacterial meningitis may result from neurenteric fistulas with the spinal canal (172). A presacral mass may be present in some cases, and Hirschsprung disease may also occur (86). Neonatal lumbar teratoma is reported (65). Higher anomalies in the GI tract, such as esophageal stenosis or atresia, are also described (04). Though generally diagnosed in the neonatal period, infancy, or early childhood, the diagnosis of the Currarino triad sometimes is not established until adult life (12).
Constipation is the most frequent presenting clinical symptom of the Currarino triad (29). Presacral lipomeningocele and teratoma may be present. The most common anorectal malformation is the anal stricture, which may be associated with fecal incontinence; recurrent meningitis may result from unrecognized recto-meningeal fistulas, and the mortality rate of 56% is high (29). Complex congenital cardiac malformations are another possible complication of caudal regression (Currarino) syndrome, not directly associated anatomically with the lower spinal defects (195).
Mutations or microdeletions are demonstrated in the homeobox gene MNX1 (formerly HLXB9) that encodes the nuclear protein HB9; these genetic alterations are found in the majority of, but not all, patients with the Currarino syndrome phenotype and various novel mutations are demonstrated (149; 67; 87; 110; 55; 197; 111). A de novo 7q36.1-qter deletion is reported in Currarino syndrome with sacral agenesis and without structural malformations of the brain (72), but de novo nonsense and frameshift mutations also are described, generally in association with presacral tumors (197). In other cases a distal 7q chromosomal imbalance may involve micrencephaly with intellectual disability, sensorineural deafness, and somatomedin C deficiency (135). Another locus at 6q25.3 is demonstrated in sacral agenesis with anorectal malformations (176). A murine model of genetic knockout of this gene is described, and the affected mice also have sacral agenesis and visceral anomalies, including agenesis of the dorsal pancreas (100).
Sacral agenesis also has been reported rarely with presacral and intraspinal tumors, particularly extradural lipomas, dermoids, sacrococcygeal teratomas, and carcinoids (85; 66; 79; 153; 163; 38), but some of the benign tumors are part of the Currarino triad as described above.
All primary and associated defects in sacral agenesis can be dated to before the seventh gestational week (113). The basic defect occurs during the formation of somites, probably due to a mutation of an early regulatory homeobox gene, and is followed by defective induction of the somite and of the neural tube.
Cnot is a homeobox gene expressed early by cells of Henson node and the notochord, but also by the postnodal neural plate caudal to the future hindbrain (169). If this gene is defective, the notochord does not form normally. Pax-1 is another homeobox gene that is expressed in sclerotome cells, but only in the presence of an intact notochord (89). It is a mediator of notochordal signals for the dorsoventral specification of sclerotomes that will form the ventral parts of vertebrae. The ventralizing gradient effect of the notochord on the somite is mediated by an inductive gene: Sonic hedgehog (49). Mutant genes or deletions of nucleotide sequences are not proved in sacral agenesis, hence, defective regulator genes are still hypothetical, but these are examples of genes that, if incompletely expressed, could potentially explain the failure of the sclerotome and perhaps also of the myotome and dermatome of the somite to form over a restricted number of segments. A mutation in the T (brachyury) gene produced a syndrome of sacral agenesis and persistent embryonic notochordal canal in three consanguineous families (139). In all familial cases of sacral agenesis and in about 30% of sporadic cases in Korean patients, the MNX1 (motor neuron and pancreas homeobox-1) gene exhibits a mutation (99). Somitogenesis and vertebral development require several growth factors and enzymes in the extracellular matrix, without which sacral agenesis or more extensive lesions of the vertebral column may appear (02). A rare spinal dysraphism, myelocystocele, may result in a split and tethered spinal cord, syringomyelia, and sacral agenesis (09; 106).
The failure of the notochord to form properly in the lumbosacral region results in deficient induction of both the floor plate of the overlying neural plate and of the somites, particularly the ventral part of the somite that contains the sclerotome (155; 140). The myelodysplasia involves mainly the ventral half of the neural tube; the ventral horns and roots are defectively formed or incomplete, and the central canal is dysplastic (155; 74). The neural crest tissue that separates from the dorsal midline at the time of neural tube closure is not primarily involved; hence, the sensory function is preserved. However, autonomic axons of dorsal and ventral roots may be defective, in part secondary to involvement of the intermediolateral columns of the spinal cord where the preganglionic neurons may be deficient. Sacral anomalies often are a clinical clue, both pre- and postnatally, of associated spinal cord malformations, particularly of the conus medullaris (114; 119; 98).
Sacral agenesis may be experimentally induced in chickens by the injection of insulin into incubating eggs in early gestation (96; 45), and sacral agenesis also occurs in the pups of diabetic rats but may be prevented by insulin therapy (48). Insulin in certain concentrations might interfere with the differentiation of the caudal chorda-mesoderm, but fetal insulin secretion does not begin until after the critical period of teratogenesis and maternal insulin does not enter the placental circulation (03; 81; 113). On the other hand, structurally unique insulin receptors are found on cerebral neurons, but not other cells in the brain, and these receptors play a role in neuronal growth and differentiation that is unrelated to glucose homeostasis (69). Rarely, human patients with adrenal and growth hormone insufficiency are associated with sacral agenesis (62). Sacral agenesis occurs on a genetic basis in a strain of mice (53) and may be induced in rodents by streptonigrin and certain other teratogens (186).
Multiple additional spinal anomalies may accompany sacral agenesis (130). Some, such as kyphosis and scoliosis, may develop secondarily during infancy or childhood (08). Lack of a sacrum is reported in the fetal amniotic band syndrome with umbilical cord entrapment diagnosed by antenatal ultrasound studies at midgestation (63), but the role of amniotic bands early in gestation when sacral agenesis develops is uncertain.
Maternal diabetes mellitus is the most important predisposing factor (123). Both genders and all ethnic groups are affected.
Toxin- or drug-induction of sacral agenesis in early gestation may be an etiology in rare cases: one infant is reported with severe aplasia of the entire lower body pole and visceral anomalies, putatively related to the maternal use of minoxidil to prevent hair loss, prior to and during gestation (148).
Failure to account for terminations of pregnancy may represent a 4-fold underestimation of observed risks of stillbirth (13/1000) in fetuses with sacral agenesis (70). According to a large database, the prevalence of sirenomelia is about 1 in 100,000 live births (129).
Good control of blood sugar in early gestation in diabetic women may minimize the risk, but this is not proved. Prenatal diagnosis by ultrasound studies before mid-gestation is possible in some cases (168). Avoidance of known teratogenic drugs and toxins during early gestation, including valproic acid and certain other antiepileptic medications, and the use of prenatal folic acid supplements in the first trimester of pregnancy are other preventive measures of several types of neural tube defects. Thalidomide embryopathy may cause anterior sacral meningocele as well as phocomelia (40). Maternal smoking at conception and early gestation also increases the risk of sacral agenesis (123).
The major differential diagnosis is spina bifida and other defects of the lower spine, such as diastematomyelia, which are associated with neural tube defects, but these conditions also can occur together with sacral agenesis (158). Carcinoid transformation of a presacral dermoid cyst in Currarino syndrome is reported but is rare (38). Rare congenital tumors of the sacral or presacral region, particularly teratomas, should also be considered, as well as more remote tumors of the posterior fossa such as medulloblastoma (167). Ultrasound often is able to detect sacral agenesis in utero (143). Prenatal genetic studies may be helpful if a sacral lesion is discovered by ultrasound (97).
Plain roentgenograms of the lower spine reveal the bony defect, though ossification of the neural arches is incomplete at birth. MR imaging of the sacral region not only provides more detail but also discloses occult meningomyeloceles, lipomas, lipomeningoceles, and neurenteric cysts, and may demonstrate the small size and aberrant course of nerve roots (109; 125; 127; 30). An abnormal bulbous or club-shape of the conus medullaris may be demonstrated by MRI, as well as tethering of the spinal cord (77; 22). An abnormal conus medullaris can also be demonstrated prenatally by fetal sonography and MRI (118). Anterior sacral meningoceles may be asymptomatic but diagnosed by MRI (14). CT imaging provides less detail of the type needed. Associated anomalies of the gastrointestinal and genitourinary systems also may be seen by MRI. Additional radiological studies of the visceral anomalies may be helpful in such cases. Imaging of the brain may be indicated to reveal malformations of the forebrain or of the posterior fossa associated with maternal diabetes mellitus. Roentgenograms and CT of the thorax may demonstrate unilateral or bilateral pulmonary atresia in a minority of cases of total sacral agenesis (13). Closed neural tube defects in children with caudal regression syndrome may be detected by various modalities of imaging (78). Ultrasonography also may be of diagnostic value in various spinal anomalies in children with neurogenic bladder (05). Prenatal diagnosis by fetal ultrasound and MR imaging of caudal regression syndrome is now feasible for early diagnosis and planning for postnatal management (10; 190; 118).
The EMG reveals neuropathic changes in the external sphincters of the bladder and anus. Denervation of muscles of the pelvic floor correlates poorly with the level of the sacral bony defect. No EMG activity is detected in the legs of patients with complete sacral agenesis (24), probably because of amyoplasia or end-stage fetal neurogenic atrophy. Postoperative EMG including perineal-evoked potentials may be useful following surgical repair of spinal dysraphism including some cases of sacral agenesis, particularly for urodynamic assessment in patients at risk of neurogenic bladder (177). Muscle biopsy is rarely indicated.
Urinary tract studies, including renal ultrasound, voiding cystography, renal nuclear scan, and urodynamic studies, are essential because irreversible damage may be prevented and function improved (17; 30; 47; 46; 165). Recurrent urinary tract infections may present in children with urological complications of sacral agenesis (46). In the neonate, anal patency should be determined soon after birth. Rectal manometry in patients with sacral agenesis may be useful in quantitating changes in anorectal function association with impaired parasympathetic innervation and in analyzing fecal incontinence in older children (115). All infants and children with sacral agenesis, lower spinal cord anomalies, and Currarino triad should have urinary bladder and anorectal functions investigated early because of the high incidence of impairment that can lead to chronic complications (21). Bowel dysfunction in infancy improves spontaneously in most children with sacral malformations (20).
Though rare, hypopituitarism is reported in sacral agenesis, so that an endocrinological workup may be indicated (62). Investigation for congenital cardiac lesions in neonates may be indicated in some cases (195).
Genetic studies of chromosomal defects or specific genetic mutations may be helpful in some cases, even prenatally: haploinsufficiency of HLXB9 by FISH analysis is associated with the triad of presacral mass (teratoma or anterior meningocele), sacral agenesis, and anorectal malformation - the defining features of the Currarino syndrome (97; 72). Mutation analysis and genetic counseling are essential, particularly in Currarino syndrome (35; 197). A homozygous mutation in the T (brachyury) gene causes sacral agenesis but also preservation of the notochord and typical neurologic deficits of caudal regression syndrome in the lower extremities, diagnosed prenatally by sonography (51).
Visceral anomalies such as malrotation of bowel, omphalocele, imperforate anus, or meningomyelocele not covered by skin all require surgery in the first 24 hours after birth. Hydronephrosis and (rarely) hydrocephalus may require urgent attention within the first week. If the infant is born to a diabetic mother, neonatal hypoglycemia is a common complication that must be watched vigilantly. Chronic nephropathy secondary to anomalies of the lower urinary tract is important to recognize early and treat (189). The urological outcome of patients with sacral agenesis may be good if neurogenic bladder is recognized early, but many patients are not diagnosed until late childhood when they present with urinary incontinence; all require lifelong surveillance (01; 46).
Reconstructive procedures to compensate for the bony defects, separate fused lower limbs, create a new anal opening, or revise the genitalia are done electively after the neonatal period (127). In asymmetrical or hemi-sacral agenesis, transiliac lengthening with posterior lumbar-iliac percutaneous fusion may be successful (112). Surgical correction of spinopelvic instability in children with sacral agenesis, including stabilization of kyphoscoliotic deformities, is feasible by solid bony fusion using allografts for reconstruction of sagittal alignment (192; 184). Postoperative “coronal imbalance” present an additional risk factor that can be minimized by sufficient bone grafts at the sacroiliac joint (194). The neurologic deficits and amyoplasia cannot be improved by surgery. Neurogenic bladder is one of the most frequent complications requiring continuous urological care and may be accompanied in some cases by bilateral vesiculo-ureteral reflux (18; 17; 30; 165). Early diagnosis, including urodynamic studies, and treatment may prevent irreversible damage to the urinary tract and kidneys and may achieve urinary continence if partial function is preserved. In addition to surgery of the urinary tract, urinary continence may be restored in some cases by surgical correction of intraspinal lesions such as lipomeningocele, dorsal lipoma, and tethered spinal cord (120). The release of tethered spinal cord also prevents the progression of other neurologic deficits (127). Associated hydromyelia and syringomyelia do not generally require surgical intervention (128; 127). The resection of dorsal hemivertebrae associated with sacral agenesis prevents progressive spastic paraplegia of residual motor function in the lower extremities (196). Sacral hemi-agenesis has been found as an incidental finding in bone scans as well (83).
Imperforate anus and other anal anomalies should be diagnosed at birth and may require early treatment (17). Fecal incontinence may be an issue in older children (115). In studying children with fecal incontinence and anorectal malformations, the “sacral ratio,” calculated from anteroposterior and lateral roentgenograms, does not correlate and, hence, is not predictive in infancy for identifying children who will later have fecal incontinence (105). Pelvic widening may alleviate the mechanical component of constipation in caudal regression syndrome in children with an extremely narrow pelvis (157). Laparoscopic diversion into augmentation cystoplasty was performed in a child with sacral agenesis (144).
Intraoperative computerized tomography with sacral nerve stimulation (O-arm guided navigation) can overcome difficulties of lead placement in patients with sacral agenesis and fecal incontinence (31). Electrical stimulation of the sacral nerve by an implanted pulse generator may be useful therapeutically in fecal incontinence as a complication of sacral agenesis (173; 26). Pudendal or sacral “neuromodulation” in sacral agenesis may be beneficial for bowel and bladder dysfunction (162; 95).
Tethering of the caudal spinal cord must be treated surgically as early as possible to treat and avoid further progressive neurologic deficits (127; 47). The results of surgery in Currarino triad can be rewarding (76). Tethered cord may be due to more complex spinal anomalies than just sacral agenesis, such as Klippel-Feil anomaly, diastematomyelia, and lipomyelomeningocele (158). Presacral meningocele and teratoma also may cause tethered spinal cord, requiring complex surgical treatment (33). Junctional neural tube defects with segmental absence of spinal cord do not exhibit tethering (161). Distal spino-pelvic fixation may be superior to paraspinal rod fixation as a surgical stabilization of the lower spine (193). Bone grafts at the sacro-iliac joint are recommended to prevent postoperative coronal imbalance (194).
In caudal regression syndrome, chronic administration of growth hormone with rehabilitative measures may improve distal innervation over time (41).
Neuromodulation of postnatal patients with lower spinal defects including sacral agenesis by implantation of a pulse generator and sensor in the lower urinary tract is not yet an approved standard means of management but shows promise in managing refractory neuropathic lower urinary tract dysfunction (164).
Because the diagnosis of sacral agenesis can be made early by prenatal ultrasound and fetal MRI, counseling may be important in some cases in the first and early second trimesters in which termination of pregnancy is a consideration. As well, in pregnancies carried to term or near-term, the choice of vaginal or caesarean section delivery and anticipated special needs of the neonate at birth can be anticipated (152).
There are no specific contraindications.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Harvey B Sarnat MD FRCPC MS
Dr. Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Developmental Malformations
Apr. 14, 2024
Developmental Malformations
Apr. 09, 2024
Developmental Malformations
Mar. 22, 2024
Developmental Malformations
Mar. 22, 2024
Developmental Malformations
Mar. 22, 2024
Developmental Malformations
Mar. 22, 2024
Developmental Malformations
Mar. 13, 2024
Developmental Malformations
Mar. 11, 2024