Abnormalities of tetrahydrobiopterin metabolism
Apr. 07, 2024
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Tyrosine hydroxylase deficiency is an autosomal recessively inherited inborn error of metabolism that involves the biosynthesis of catecholamines (dopamine, epinephrine, and norepinephrine). The clinical presentation ranges from severe infantile parkinsonism to a phenotype reminiscent of dopa-responsive dystonia. Diagnosis is based on the pattern of monoamine metabolites in CSF and the molecular analysis of the TH gene.
• Tyrosine hydroxylase deficiency is an autosomal recessively inherited disorder that leads to a deficient production of catecholamines (dopamine, epinephrine, and norepinephrine). | |
• Patients usually exhibit developmental delay, infantile parkinsonism, dystonia, oculogyric crises, and features of autonomic dysfunction. Milder phenotypes may also occur. | |
• Tyrosine hydroxylase deficiency is diagnosed by detection of decreased CSF concentrations of the downstream metabolites of catecholamine degradation, homovanillic acid, and 3-methoxy-4-hydroxyphenylglycol. | |
• The treatment of choice is levodopa; alternatively, patients are treated with other dopaminergic drugs, mainly dopamine agonists and monoamine oxidase inhibitors. |
Tyrosine hydroxylase deficiency was first reported in 1971 in 2 brothers with early-onset progressive dopa-responsive dystonia (03). Subsequently, young infants with a more severe phenotype were recognized (12). In 2010, Willemsen and colleagues described the largest series reported to date (36 patients) and proposed 2 phenotypes: type A, which has an onset in infancy or childhood, is less severe, has a better prognosis, and is L-dopa responsive; and type B, which has an earlier onset (neonatal period, early infancy), is more severe, and is poorly L-dopa responsive (32). This classification was widely used by clinicians and researchers, although the authors acknowledge that the phenotype of tyrosine hydroxylase deficiency has a spectrum with overlap of clinical features between both groups (32). A standardized deep phenotyping study from the registry of the International Working Group on Neurotransmitter Related Disorders did not find clear differences in perinatal abnormalities, postnatal problems, and achievement of gross motor milestones; drug response varied regardless of the age of initial symptoms (16). Consequently, the authors proposed to abandon this classification. Currently, more than 70 cases have been reported worldwide (32; 07).
In 1996 the first mutations in the tyrosine hydroxylase gene were reported (21). The spectrum of mutations is heterogenous with no hot spots detected (15; 07). Two common mutations due to founder effects have been detected, one in the Greek population (c.707C> T) (25) and another in the Dutch population (c.698G> A) (31).
The age of presentation of tyrosine hydroxylase deficiency ranges from the neonatal period to childhood, and the clinical phenotype ranges from a severe phenotype of encephalopathy and infantile parkinsonism-dystonia to a moderate phenotype reminiscent of dopa-responsive dystonia (Segawa disease). This phenotypic continuum of severity also applies to other monoamine neurotransmitter disorders (15).
The majority of patients became symptomatic during infancy (between 1 and 15 months) and manifest with severe developmental delay and hypotonia. On examination, patients show features of infantile parkinsonism characterized by hypokinesia, bradykinesia, and dystonia of variable severity. They often suffer oculogyric crises, which are episodes of dystonia characterized by intermittent or sustained upgaze deviation with variable association of axial and/or limb dystonic posturing lasting minutes to hours. Less severely affected patients present later in childhood with gait difficulties associated with dystonia or parkinsonian features (09; 16).
Patients with a more severe phenotype exhibit symptoms during the neonatal period or in early infancy and are profoundly hypokinetic; they have minimal or no motor developmental acquisitions and suffer dystonic crises with regular intervals every 4 to 5 days. They are often irritable and can suffer “lethargy-irritability crises” that are characterized by features of autonomic dysfunction (excessive drooling, sweating, body temperature instability) especially during periods of dystonia (32; 15; 26).
Atypical features and presentations have also been described and include early-onset spastic paraplegia, dopa-responsive myoclonus-dystonia, and acute episodes of motor deterioration (08; 23; 28; 11; 13).
Signs of autonomic dysfunction, including excessive sweating, temperature instability and nasal congestion, often occur in infants with tyrosine hydroxylase deficiency. Sleep disturbances during infancy are also reported often in the form of excessive sleep (32; 15; 26). Hormonal manifestations that may occur in tyrosine hydroxylase deficiency include growth retardation and delayed bone age (12). Furthermore, a high frequency of these patients are small-for-gestation age and have birth lengths below the 10th percentile (16b). In addition, hyperprolactinemia is also a characteristic feature because dopamine inhibits prolactin secretion. Galactorrhea has been reported in several patients with tyrosine hydroxylase deficiency (33).
The response to L-dopa therapy in tyrosine hydroxylase deficiency depends on the severity of the phenotype, which in turn is reflected by the concentrations of homovanillic acid and the HVA/5-HIA ratio in CSF (32). Response to treatment varies from minimal or no response in severe phenotypes to slow and gradual responses with escalation of L-dopa dose in moderate phenotypes and dramatic favorable response within days in phenotypes reminiscent of Segawa disease. Responders show improvement in hypokinesia, tremor, and dystonia, and oculogyric crises decrease in frequency and severity and may eventually disappear (32; 34; 26). It is generally accepted that early diagnosis and prompt treatment improves the final motor and cognitive outcome in tyrosine hydroxylase-deficient patients (32; 15).
Some patients with phenotypes of mild and moderate severity and favorable response to L-dopa therapy reach full normalization of neurologic condition, whereas others continue having some mild motor impairments. Patients can experience dyskinesias induced by L-dopa, which tend to improve with time, and, in certain cases, they may subside with no need for further management (32; 34; 26). Response to L-dopa generally remains stable over the years. However, a report on long-term outcome of patients with phenotypes reminiscent of dopa-responsive dystonia showed that 15% of patients manifest motor symptoms and paroxysmal events related to dopamine insufficiency (20).
Patients with severe phenotypes show a worse but variable outcome. A number of them show minimal or no response in motor function, whereas others show a slow and gradual response and are able to gain motor milestones over the course of months to years. They usually show variable degrees of motor impairment, and their condition remains stable over the years with no evidence of progressive disease. Although these patients generally show more problems with L-dopa-induced dyskinesias, their tolerance improves over time (32; 34; 26; 18).
Cognitive development is impaired in many individuals with tyrosine hydroxylase deficiency (32; 18). Although systematic assessment of cognitive profiles is lacking, Willemsen and colleagues reported mild to moderate intellectual disability in 91% of patients with severe phenotypes, and in 33% with mild to intermediate phenotypes. Long-term follow up did not reveal any signs of further cognitive decline in any of the tested patients (32).
The patient, a female, was the product of an uncomplicated pregnancy and an unremarkable perinatal period. At 2.5 months of age, she exhibited hand and leg tremors when crying. At 3 to 4 months of life, she started having episodes of eye rolling upwards associated with limb posturing (oculogyric crises). These episodes lasted 2 to 6 hours and occurred mainly in the late afternoon every 4 days. EEG recording during these paroxysmal episodes did not show any epileptiform activity, but she was treated with antiepileptic drugs with no favorable response. Her brain MRI was normal.
At the age of 2 years, she had not achieved any motor development. She had no head or trunk control. She was able to hold an object in her hand, but she could not reach. Although her motor performance was minimal, her parents noted that she was more active in the mornings and after a nap. She understood simple commands and babbled occasionally. On review of systems, she had increased sweating, a chronically stuffed nose, and constipation.
On exam, her vital signs were stable. Her head circumference was 50 cm. She had a narrow and long face, tented mouth, small hands, and mild gingival hypertrophy and was perspiring profusely. She was alert and appeared oriented and aware of her surroundings. She had bilateral ptosis with full extraocular movements and poor facial expression. Her cry was strong. She had no head control, and her spontaneous movements were minimal. Mild twitching of fingers and face was noted consistent with chorea. Although she appeared hypotonic when in her mother’s arms, she developed dystonic posturing and stiffness of limbs and neck when manipulated. Her deep tendon reflexes were hyperactive, and she had spontaneous up-going toes. No tremor was noted. The parents stated that the tremor previously noted had disappeared gradually during the first year of life.
Neurotransmitter metabolites were measured in CSF. A marked reduction of homovanillic acid concentrations with normal serotonin metabolites as detected. Sequencing analysis of the tyrosine hydroxylase gene revealed a homozygous mutation in exon 6 (c.707T> C).
She was started on L-dopa-carbidopa at 1 mg/kg-1 d-1, and the dose was increased gradually every month. Despite the gradual increase in the L-dopa dose, she developed jaw opening dystonia and generalized dyskinesias. Amantadine (4 mg/kg-1 d-1) was added at 3.5 years of age, and this was followed by a reduction of dyskinesias, which allowed further increases in the L-dopa dose.
Her motor development gradually improved. She walked independently at the age of 5 years, and she speaks with dysarthria and attends special education classes at the age of 10 years. She is fidgety, her gait is mildly dyskinetic, and she is independent in all her daily activities.
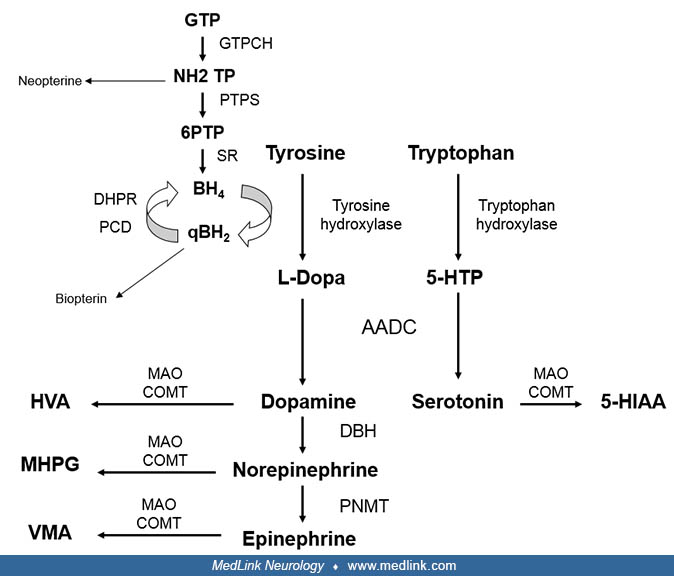
Tyrosine hydroxylase deficiency is caused by mutations in the tyrosine hydroxylase gene on chromosome 11p15.5. Tyrosine hydroxylase is a specific tetrabiopterin-dependent amino acid hydroxylase that converts tyrosine to dihydroxyphenylalanine (L-dopa), the rate-limiting step in the synthesis of catecholamines, dopamine, epinephrine, and norepinephrine (15). In the brain, tyrosine hydroxylase is mainly expressed in dopaminergic neurons in the ventral tegmental area and substantia nigra pars compacta and in the noradrenergic neurons of the locus coeruleus. In the periphery, tyrosine hydroxylase is mainly found in sympathetic neurons and in the adrenal medulla (07).
Monoamine neurotransmitters include serotonin and catecholamines. They play important roles in modulation of psychomotor function and hormone secretion as well as cardiovascular, respiratory, and gastrointestinal control, sleep mechanisms, body temperature, and pain.
The starting substrate for the formation of catecholamines is tyrosine and for serotonin is tryptophan. Tyrosine is converted to L-dopa by tyrosine hydroxylase and tryptophan to 5-hydroxytryptophan by tryptophan hydroxylase. L-dopa and 5-hydroxytryptophan undergo decarboxylation through the action of aromatic amino acid decarboxylase, leading to the formation of dopamine and serotonin, respectively. Within noradrenergic neurons, dopamine is converted to norepinephrine, and within the adrenal medulla, norepinephrine is methylated to form epinephrine. Dopamine and serotonin are released and rapidly catabolized. Through the action of monoamine oxidase and catechol-O-methyltransferase, 5-hydroxyindolacetic acid (5-HIAA) is produced from serotonin, homovanillic acid from dopamine, and 3-methoxy-4-hydroxyphenylglycol from norepinephrine.
Disorders of monoamine biosynthesis lead to reduced synthesis of specific neurotransmitters, and their clinical manifestations are caused by the deficiency of these neurotransmitters in the brain and peripheral nervous system. Tyrosine hydroxylase deficiency leads to reduced synthesis of all catecholamines. Reduced concentrations of dopamine are thought to be the cause of the motor symptoms, given dopamine’s role in coordination and regulation of movement. This has been shown in homozygous tyrosine hydroxylase knock-in mice that exhibited a gradual loss of central catecholamines and severe motor impairment, including rigidity, dystonia, impaired motor coordination, and bradykinesia (14). Norepinephrine and epinephrine deficiency are responsible for the manifestations of autonomic sympathetic dysfunction. Given the role of monoamines on hormonal function, endocrine dysfunction may also occur. Some clinical manifestations, such as cognitive disturbances, cannot be attributed to the deficit of a specific neurotransmitter. The early effects of dopamine deficiency in the developing brain may account for these disturbances. This is supported by animal models that have shown structural abnormalities in brain development, particularly in the growth and laminar organization of the cerebral cortex (19). Further support comes from the study of a tyrosine hydroxylase-deficient human fetus that demonstrated a decrease in the expression of developmental markers for synapses, axons, and dendrites (30).
Further pathophysiological mechanisms in tyrosine hydroxylase deficiency include changes in receptor sensitivity implicated in the drug-induced dyskinesias that are often observed in these patients (26).
Tyrosine hydroxylase deficiency is a rare disorder. Currently, more than 70 cases have been reported worldwide (32).
Only parents who are asymptomatic heterozygous carriers for tyrosine hydroxylase deficiency have a 1 in 4 chance of having an affected fetus with each pregnancy. Genetic counseling for a family with a previously documented proband is recommended.
Tyrosine hydroxylase is not expressed in amniotic fluid cells or in chorionic villus; therefore, prenatal diagnosis by measurement of enzyme activity is not possible. Prenatal diagnosis by sequencing the tyrosine hydroxylase gene for mutations in chorionic villus has been performed successfully (22).
Differential diagnosis of tyrosine hydroxylase deficiency includes other disorders of monoamine synthesis, such as aromatic amino acid decarboxylase deficiency and pterin disorders. These disorders lead to severe dopamine deficiency from early infancy, which manifest with infantile parkinsonism, oculogyric crises, and autonomic dysfunction. In moderate phenotypes of tyrosine hydroxylase deficiency that present in childhood with gait disorders and dystonia, differential diagnosis also includes Segawa disease. Associated clinical manifestations and biochemical findings, including blood phenylalanine concentration and CSF pattern of monoamine catabolites and pterins, allow the differentiation of these defects. Measurement of enzyme activity in some cases and molecular studies can confirm the diagnosis (15).
Epileptic encephalopathy is also considered in the differential diagnosis of severe cases of tyrosine hydroxylase deficiency because oculogyric crises are associated with prominent dystonic posturing that may be misdiagnosed as seizures. Unresponsiveness to antiepileptic drugs and the presence of infantile parkinsonism and autonomic dysfunction together, with the lack of EEG correlate during episodes, should suggest a defect in monoamine synthesis (15).
Because severe forms of tyrosine hydroxylase deficiency can be associated with perinatal complications, a differential diagnosis with neonatal infectious, hypoxic ischemic, epileptic, and metabolic disorders must be considered. Given the profound hypotonia and hypokinesia associated with ptosis and hypomimia that these patients show, neuromuscular disorders are also considered in the differential diagnosis. Finally, moderate forms of tyrosine hydroxylase deficiency can also mimic cerebral palsy and hereditary or acquired forms of juvenile parkinsonism. Medical history, clinical manifestations, and biochemical and genetic investigations can confirm the diagnosis (15).
Concentrations of the monoamine neurotransmitter catabolites reflect the turnover of these neurotransmitters in the brain. Therefore, the diagnosis of tyrosine hydroxylase deficiency relies on the measurement of the monoamine catabolites homovanillic acid, 3-methoxy-4-hydroxyphenylglycol, and 5-HIAA in CSF. The typical biochemical CSF pattern is a decreased homovanillic acid and 3-methoxy-4-hydroxyphenylglycol concentrations and low HVA/5-HIAA ratios with normal concentrations of pterins metabolites (26). It is important to note that collection and handling of CSF must be performed under appropriate conditions given the rostro-caudal gradient of these metabolites and the tendency for oxidation of some of them (15).
Measurements of dopamine precursor amino acids or catecholamines in blood or urine are in general noninformative. However, if differential diagnosis with other disorders of monoamine synthesis are considered, measurement of phenylalanine concentrations and analysis of tyrosine and phenylalanine concentrations after a load of phenylalanine may be necessary (phenylalanine loading test). Plasma prolactin measurement is an indicator of dopamine status because dopamine downregulates prolactin synthesis by the hypothalamus; thus, hyperprolactinemia is expected when dopamine deficiency occurs (15).
Neuroimaging is nondiagnostic. Brain MRI is normal in most patients with tyrosine hydroxylase deficiency, but approximately one third of patients show brain atrophy and, rarely, delayed myelination (16). Some children with severe phenotypes may also show nonspecific white-matter abnormalities and increased extracerebral CSF spaces (15).
Molecular analysis of the tyrosine hydroxylase gene confirms the diagnosis. To date, 40 different disease-related missense mutations, 5 nonsense mutations, 3 mutations in the promoter region, and 2 deletions have been reported (07; 23; 01). Most mutations are private, except for the 707T>C mutation in patients of Greek origin, and the 698G>A mutation in Dutch patients (31; 25). Functional studies of some missense variants revealed specific kinetic anomalies and altered substrate binding (p.Arg233His, p.Gly247Ser, p.Phe375Leu, and p.Cys359Phe) (07). Genotype-phenotype correlations have been difficult to demonstrate because patients are often compound heterozygotes except for patients with mutations in the promoter region of the TH gene, which is associated with milder phenotypes (23; 28).
The mainstay of treatment in tyrosine hydroxylase deficiency is restoring dopaminergic neurotransmission by administering L-dopa, the primary metabolite that is deficient in this condition. Response and tolerance to L-dopa therapy is variable and depends on the severity of the phenotype. For this reason, therapy in tyrosine hydroxylase-deficient patients needs to be tailored on an individual basis. The starting dose of L-dopa, its escalation, and dosing frequency and the need for the addition of other agents to potentiate dopamine transmission are different in each patient (26).
The second most often used pharmacological strategy to potentiate dopaminergic neurotransmission involves inhibition of dopamine catabolism via the selective type B monoamine oxidase inhibitor, selegiline. Other inhibitors of dopamine degradation, such as catechol-O-methyl transferase inhibitors, and dopamine receptor agonists, ropinirole, pramipexole, bromocriptine, have only been tried sporadically in tyrosine hydroxylase-deficient patients (06; 32; 28).
Some of the first patients with tyrosine hydroxylase deficiency underwent serial lumbar punctures in order to monitor treatment efficacy biochemically. Irrespective of the clinical response, homovanillic acid concentrations increased with treatment but rarely reached normal concentrations (32; 34). Currently, serial monitoring of metabolites in CSF is performed in selected cases. Determination of prolactin concentrations has been proposed as a functional parameter to tailor therapy in dopamine deficiency disorders. However, there are marked fluctuations of prolactin during the day that cannot be attributed to variations in L-dopa dosing. Thus, determination of prolactin is used as an indicator of insufficient dopamine production (12).
L-dopa. L-dopa is coadministered with a peripheral decarboxylase inhibitor carbidopa or benserazide in order to prevent conversion of L-dopa to dopamine peripherally, thus, increasing L-dopa brain bioavailability and decreasing its peripheral dose-related side effects. Tolerance to L-dopa treatment usually refers to the appearance of dyskinesias that may manifest with small doses of L-dopa even at the initiation of treatment, especially in children with the severe infantile disorder (32; 26). On the other hand, common dose-dependent adverse effects of L-dopa, such as nausea, vomiting, and hypotension, are rarely seen in these patients. Disabling L-dopa-related panic attacks were reported in one case (27).
In general, older children with phenotypes resembling Segawa disease are treated with L-dopa at 150 to 250 mg/d (08; 27), whereas younger patients with infantile parkinsonism are treated with starting doses of L-dopa at 0.1 to 1 mg/k/d divided in 3 to 6 doses a day. L-dopa is then increased gradually according to its tolerance. The final L-dopa dose is variable among patients, ranging from 0.8 mg/k/d to 30 mg/k/d (32; 29; 34; 04; 10; 26; 18).
Monoamine oxidase inhibitors. In patients with poor response and tolerance to L-dopa, treatment with selegiline, a selective monoamine oxidase B inhibitor, provides benefit and may allow further increases, even doubling of the L-dopa dose (06; 12; 34; 04; 10; 18). It has been postulated that the early introduction of selegiline in poor L-dopa-responder patients improves the overall treatment outcome (34). However, despite some favorable reports, selegiline has been ineffective in other patients (05; 35).
Selegiline is usually administered once daily, with reported dose ranges from 2.5 to 15 mg daily or 0.1 to 0.4 mg/k/d. Dose-dependent side effects reported in tyrosine hydroxylase deficient patients include hyperkinesia, rigidity, and sleep disorder (04). Additional side effects include nausea, dizziness, dry mouth, confusion, and orthostatic hypotension.
Monoamine oxidase hypertensive crisis precipitated by food rich in tyramine do not occur with selegiline doses below 10 mg/d. Because patients with tyrosine hydroxylase deficiency rarely take more than 10 mg/d, they do not need to be on a tyramine-restricted diet; it can be given safely in combination with L-dopa (26).
Management of L-dopa induced dyskinesias. Poor tolerance to L-dopa manifesting in the form of L-dopa-induced dyskinesias is a common feature of defects of dopamine synthesis; such movements may be intolerable and preclude treatment. Dyskinesias can also be induced by other dopaminergic agents (15).
In tyrosine hydroxylase-deficient patients, L-dopa-induced dyskinesias are of variable intensity and appear early in the course of treatment regardless of the age at which treatment was initiated. They have been characterized as chorea of the face, limbs, and trunk of variable intensity, and more rarely by prominent ballism. They are precipitated by increases in the dose of L-dopa and also by febrile illnesses and stress. Occasionally, they can be intolerable, especially in those cases presenting in the form of ballism (26).
L-dopa-induced dyskinesias in tyrosine hydroxylase-deficient patients are initially managed by slowing down the escalation of L-dopa dose either by decreasing the L-dopa to the previously tolerated dose or by postponing further increases of L-dopa until dyskinesias decrease or recede. When these measures are insufficient, amantadine may be added (26).
Amantadine is used in patients with Parkinson disease suffering from motor fluctuations and dyskinesias. Its mechanism of action is not completely clear. Side effects include nausea, vomiting, and livedo reticularis. Amantadine at 4 to 6 mg/k/d has been tried in 2 tyrosine hydroxylase-deficient patients with L-dopa-induced dyskinesias that were severe enough to prevent escalation of their L-dopa therapy. Patients showed a favorable response with no adverse effects, and the dose of L-dopa could then be further increased (26).
Symptomatic treatment. Treatment with anticholinergics and benzodiazepines has been used in some patients with tyrosine hydroxylase deficiency for the symptomatic management of dystonia and oculogyric crises. However, the response to these drugs is often disappointing (12; Zafiriou et al 2009; 04; 28).
In addition to pharmacological therapy, patients are placed on a rehabilitation program including physical, occupational, and speech therapy.
Management of autonomic dysfunction has not been necessary in these patients because initiation of L-dopa treatment, even at very small doses, usually corrects sympathetic dysfunction (26).
Other therapeutic strategies. Bilateral subthalamic nucleus deep brain stimulation was performed in a 6-year-old child with tyrosine hydroxylase deficiency exhibiting prominent dystonia and infantile parkinsonism that was medically intractable. After surgery, his condition improved significantly and his L-dopa treatment could be reduced (29). Although it is too soon to know, deep-brain stimulation may be an option for tyrosine hydroxylase-deficient patients with minimal or no response to dopaminergic treatment or with intolerable L-dopa-induced dyskinesias.
In a report of a series of adult patients with tyrosine hydroxylase deficiency with moderate phenotypes reminiscent of dopa-responsive dystonia, 2 patients experienced pregnancy. Although in one patient levodopa treatment was maintained in the low range and led to a safe delivery, levodopa was discontinued and led to aggravation of symptoms and forced the termination of pregnancy in the other patient. Based on this experience, the authors suggest that levodopa should be maintained at a low dosage (50 mg) during pregnancy in tyrosine hydroxylase deficiency (20).
Although dietary management is not indicated, a tyrosine hydroxylase patient with insufficient response to L-dopa therapy was placed on a protein-redistribution diet (normal protein content, including both animal and vegetable proteins, exclusively for dinner) that led to improvement in his body mass index and motor status (02). The rationale of this diet is to optimize L-dopa efficacy by reducing the competition between L-dopa and dietary amino acids.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Roser Pons MD
Dr. Pons of the University of Athens in Greece has no relevant financial relationships to disclose.
See Profile
Barry Wolf MD PhD
Dr. Wolf of Lurie Children's Hospital of Chicago has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Childhood Degenerative & Metabolic Disorders
Apr. 07, 2024
Childhood Degenerative & Metabolic Disorders
Mar. 22, 2024
Developmental Malformations
Mar. 22, 2024
Childhood Degenerative & Metabolic Disorders
Mar. 22, 2024
Childhood Degenerative & Metabolic Disorders
Mar. 15, 2024
Childhood Degenerative & Metabolic Disorders
Feb. 27, 2024
Childhood Degenerative & Metabolic Disorders
Feb. 27, 2024
Childhood Degenerative & Metabolic Disorders
Feb. 27, 2024