
































Developmental Malformations
Walker-Warburg syndrome
Apr. 14, 2024
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
2-hydroxyglutaric acidurias belong to the organic acidurias. They represent inherited disorders, each with characteristic symptomatology, MR pattern, and biochemistry. Three of these disorders have been identified: L-2-hydroxyglutaric aciduria, D-2-hydroxyglutaric aciduria, and combined L-2- and D-2-hydroxyglutaric acidurias. Their genetic basis has been largely clarified in recent years.
|
• L-2-hydroxyglutaric and D-2-hydroxyglutaric acids are products of the tricyclic acid (TCA) cycle with different pathways. They are waste products without metabolic function, and accumulation of these products has toxic effects. Removal of these potentially toxic acids is mediated by specific dehydrogenases. | ||
|
• Three distinct disease entities are known: | ||
|
- L-2-hydroxyglutaric aciduria (L-2-HGA) | ||
|
• All three disorders profoundly and specifically affect development and functioning of the central nervous system. | ||
|
• Causative gene defects have been found for all 3, including subtypes of D-2-hydroxyglutaric aciduria. | ||
|
• Distinct neuroradiological profiles are associated with L-2- and D-2-hydroxyglutaric aciduria, allowing a presumptive diagnosis prior to metabolic and genetic testing. | ||
|
• No specific treatments are available. Antenatal diagnosis through gene testing is possible. | ||
L-2-hydroxyglutaric aciduria (L-2-HGA), D-2-hydroxyglutaric aciduria (D-2-HGA), and mixed D-2- and L-2-hydroxygluatic aciduria (D,L-2-HGA) form three distinct entities belonging to the group of genetic organic acidurias. Two of these disorders were discovered in 1980 (14; 24), whereas awareness of their association with defined neurologic conditions began in the 1990s. The first reported case of L-2-hydroxyglutaric aciduria was found in an intellectually disabled male child of consanguineous parents (24). Barth and colleagues profiled the specific clinical and radiological aspects as a slowly progressive subcortical leukoencephalopathy with magnetic resonance signal changes in the basal ganglia and cerebellar dentate nuclei (09). Van der Knaap and associates first profiled clinical and neuroradiological patterns of D-2-hydroxyglutaric aciduria (86; 87). Kranendijk and colleagues discovered that D-2-hydroxyglutaric aciduria was heterogenous, resulting from two mutated genes with different modes of inheritance (36). A third type, causing mixed L-2- and D-2-hydroxyglutaric aciduria, was proposed by Muntau and colleagues (49). Causative gene defects for these three disorders, including two subtypes of D-2-HGA, were discovered in recent years.
L-2-hydroxyglutaric aciduria (L-2-HGA). Onset is usually in late infancy with slow evolution (09; 10; 21; 96; 39; 81; 78; 70). Usually, no symptoms are noticed within the first 6 months of life. The second 6 months may witness nonspecific developmental delay and muscular hypotonia symptoms. Delayed unsupported walking, delayed speech acquisition, or both, are often the first symptoms between 1 year and 3 years. More clearly defined neurologic patterns are present in most affected individuals past the age of 3 years, with cerebellar symptoms predominantly manifesting as gait ataxia; in a case series of hereditary ataxia, L-2-hydroxyglutaric aciduria was found in 12% (13). Other cerebellar symptoms may be present, such as hand dysmetria, dysarthria, and tremor. A low-amplitude postural tremor is common and is reportedly associated with the cerebellar atrophy and is accompanied by negative myoclonus (ie, asterixis) in one third of cases (31). Spastic paresis or extrapyramidal symptoms with dystonia develop in approximately one third of the patients. Eventually, all affected individuals develop mental deficiency. Most of the patients have seizures, usually of the generalized type. Macrocephaly is a frequent, though variable finding (21; 96; 07; 18), which may be the presenting sign in the first year of life (20). Cases may uncommonly present with behavioral problems and attention deficit hyperactivity disorder (29).
Apart from epileptic seizures, usually no episodes of life-threatening metabolic derangement or sudden periods of functional regression occur (in contrast to the metabolic crises with glutaric aciduria type I). Rare exceptions to this rule may occur, such as an adult with chronic epilepsy and normal intelligence who experienced subacute ataxia (64); his MRI showed changes typically found in L-2-hydroxyglutaric aciduria.
Cerebral neoplasms, especially gliomas, have been reported during the course of L-2-hydroxyglutaric aciduria, apparently reflecting a specific tendency (47; 19). In a meta-analysis of 295 published cases, the incidence of brain tumors in L-2-hydroxyglutaric aciduria was estimated at 5%. The observed malignancies varied in type and included astrocytoma, primitive neuroectodermal tumors, medulloblastoma, and oligodendroglioma. All were supratentorial (55). Diffuse gliomatosis cerebri affecting both cerebral hemispheres and the upper brainstem was reported in an adult (43). Periventricular supratentorial cysts were reported in a single case (34).
As indicated by the name, there is increased urinary excretion of L-2-hydroxyglutaric acid. This compound is also elevated in CSF and, to a lesser extent, in plasma. Lactic acid is not increased.
In rare cases, onset of cognitive impairment, seizures, and ataxia may be delayed until grade school (42).
D-2-hydroxyglutaric aciduria (D-2-HGA). Manifestations of D-2-hydroxyglutaric aciduria are complex, more variable, and distinct from those of L-2-hydroxyglutaric aciduria (14; 30; 44; 16; 52; 77; 28; 05; 86; 87; 25; 92; 33; 94). The disorder typically presents as an epileptic disorder with neonatal or early infantile onset, ultimately developing into a generalized disorder of movement and cognition (30; 16; 77; 28; 05). Cardiomyopathy appears in most cases, especially in the severe early-onset type (52; 77; 05; 86; 87). In a female patient with dilated cardiomyopathy, histochemistry of her muscle biopsy demonstrated excessive glycogen and ultrastructural examination revealed subsarcolemmal cylindrical spirals and normal mitochondria (05). One male patient without epilepsy had prominent weakness of facial, shoulder, and pelvic muscles with retained tendon reflexes at 9 months, but he could pull himself up at 16 months and displayed cognitive and speech delay at 2 years (44). The second and third patients of Nyhan and colleagues were female siblings (52): the eldest had mild developmental delay, weakness, exertional myalgia, reduced muscle mass, and mild dysmorphia, whereas the youngest had speech delay at 3 years, but no other neurologic symptoms. Skin changes consisting of focal alopecia and tan-colored waxy plaques were described in one case (30; 16). The first patient ever reported with D-2-hydroxyglutaric had protein-losing gastroenteropathy without neurologic symptoms (14).
van der Knaap and colleagues reported clinical, biochemical, and neuroimaging features in 17 patients, including nine patients reported previously (86). This study confirmed the existence of at least two phenotypes of D-2-hydroxyglutaric aciduria. The more common phenotype presents with neonatal or early infantile onset encephalopathy characterized by severe epilepsy, marked hypotonia, cerebral visual failure, and little, if any, development. Variable features included cardiomyopathy, microcephaly, and macrocephaly. In the other phenotype mental retardation, hypotonia, and macrocephaly were the most frequently noted clinical signs. Neuroimaging findings were remarkably consistent: mild ventriculomegaly involved the occipital horns, enlarged frontal subarachnoid spaces, subdural effusions, and signs of disturbed and delayed cerebral maturation affecting gyral development, opercularization, and myelination. Two patients had occipital agyria. Seven of 10 patients had subependymal cysts of variable size in the head or body of the caudate nucleus.
In a further study, 25 patients were reviewed, 14 of the severe type and 11 of the mild type (87). In the severe type, the main findings were neonatal onset, severe epilepsy, hypotonia, and developmental failure, often accompanied by spasticity or dystonia; in the mild type, the main symptoms were epilepsy with infantile or later onset and mild or moderate developmental delay. When MRI was done before 6 months, ependymal cysts were typically noted. Multiple white matter lesions were seen in MRI studies at later ages. Multiple aneurysms in the middle cerebral arteries in one patient and middle cerebral artery infarctions in another patient suggested possible vascular involvement in this disorder.
The clinical spectrum has expanded since 2000. Eeg-Olofsson and colleagues reported a patient with D-2-hydroxyglutaric aciduria, cerebrovascular aneurysms, aortic insufficiency, and myopathy (25). Agenesis of the corpus callosum (94) and spondyloenchondromatosis (33) were added to the spectrum.
Patients with normal mental and motor development and increased excretion of D-2-hydroxyglutaric acid and a mutated D-2-hydroxyglutaric acid dehydrogenase gene were identified following screening for organic acids in an inbred family (74). The problem of variable phenotypic expression is further highlighted by the case of identical twins with D-2-hydroxyglutaric aciduria: one had multiple congenital anomalies, severe developmental delay, and abnormal neuroradiological findings, whereas the other was phenotypically normal (46).
Two siblings with D-2-hydroxyglutaric aciduria type 1 presented with mild intellectual disability and adult-onset seizures; both later developed early-onset dementia (79).
Patients with D-2-hydroxyglutaric aciduria have elevated levels of D-2-hydroxyglutaric acid, but not L-2-hydroxyglutaric acid in urine, plasma, and CSF; in addition, their total GABA concentration in CSF is elevated (30; 86).
Metaphyseal chondromatosis with D-2-hydroxyglutaric aciduria. Metaphyseal chondromatosis with D-2-hydroxyglutaric aciduria (OMIM 614875) is an extremely rare genetic disorder characterized by the unique association of enchondromatosis with D-2 hydroxyglutaric aciduria (80; 33; 12; 91; 98; 99). Enchondromatosis is characterized by benign growths of cartilage in the bones, typically in the bones of the hands and feet, but they may also occur in the skull, ribs, and spine. Clinical features associated with metaphyseal chondromatosis with D-2-hydroxyglutaric aciduria include enchondromatosis (with associated short stature, severe metaphyseal dysplasia, and mild vertebral involvement), elevated levels of urinary 2-hydroxyglutaric acid, and mild developmental delay. A wide spectrum of variable features is evident from the small number of reported cases, including dysmorphic features, macrocephaly, microcephaly, delayed myelination and gyration, ventriculomegaly, white matter atrophy, symmetrical hyperintensity of globus pallidus and central pontine tracts, cerebellar dysplasia, ectasia, marked tortuosity of intracerebral vessels, hypotonia, divergent squint with large corneas, alopecia, abnormal dental eruption, thoracolumbar structural scoliosis, and widespread and debilitating hemangiomas.
Combined D-2- and L-2-hydroxyglutaric aciduria (D,L-2-HGA). Muntau and colleagues reported the cases of three patients, a brother and sister from one family and a single girl from another family (49). All three had neonatal encephalopathy and severely impaired motor and mental development. They excreted both L-2-hydroxyglutaric acid and D-2-hydroxyglutaric acid in excess, together with an increase of 2-oxoglutaric acid, and intermittent excretion of other citric acid cycle intermediates (lactate, fumarate, 2-oxo-glutarate, and succinate) in one patient. In two patients, D-2-hydroxyglutaric acid, but not L-2-hydroxyglutaric acid, was elevated in CSF. MRI performed in two patients (one from each family) showed periventricular cysts and ventricular widening. In one patient, cerebellar hypoplasia was found. A severe neonatal-onset case featured lactic acidosis, mitochondrial complex IV dysfunction in muscle, and periventricular frontal lobe cysts (69). A previously reported case may also belong to this subtype (15).
Atypical 2-hydroxyglutaric aciduria. Barth and colleagues described a male patient with consanguineous parents with neonatal-onset seizures and severely impaired mental and motor development with L-2-hydroxyglutaric aciduria, but normal L-2-hydroxyglutaric acid in the CSF, elevated lactic acid in blood and CSF, and normal respiratory chain function in muscle and liver (11). MRI showed cerebrocortical and cerebellar atrophy and a supracollicular lipoma.
L-2-hydroxyglutaric aciduria generally has a slowly progressive course of mental regression. Ambulation may be maintained for an extensive period, in some cases even into adulthood (09). Multiple cases of primary brain tumor in patients with L-2-hydroxyglutaric aciduria have been described, suggesting another neurotoxic complication in this disease (02).
D-2-hydroxyglutaric aciduria has a more variable course, due to epilepsy and cardiomyopathy (86; 87).
Combined D-2- and L-2-hydroxyglutaric aciduria is a devastating neurometabolic disorder that is usually lethal in the first years of life (57).
L-2-hydroxyglutaric aciduria. The patient is the third male offspring of consanguineous parents whose first two children are normal. Development seemed normal during the first year of life when he learned to sit unsupported and communicate normally, but subsequently, he appeared to level off. When seen by a pediatric neurologist at age 14 years, verbal communication was at the level of a 3-year-old child. He displayed a shuffling, ataxic gait with impaired tandem walk, hand dysmetria, severe intentional tremor, and dystonic hand posturing. Plantar responses were flexor. His EEG showed nonspecific background slowing. Flash-evoked visual-evoked potentials showed symmetrically increased latencies and diminished amplitudes. Metabolic screening revealed L-2-hydroxyglutaric aciduria. MRI showed typical abnormalities with generalized subcortical loss of white matter, as well as cerebellar vermal atrophy, abnormal T2-signal in the region of the dentate nuclei, and involvement of the basal ganglia. Over the next 10 years, he made no further progress in mental development. The impression was one of slow mental and verbal regression, whereas motor impairment was stationary. He had occasional generalized epileptic seizures. A younger brother developed the same disease.
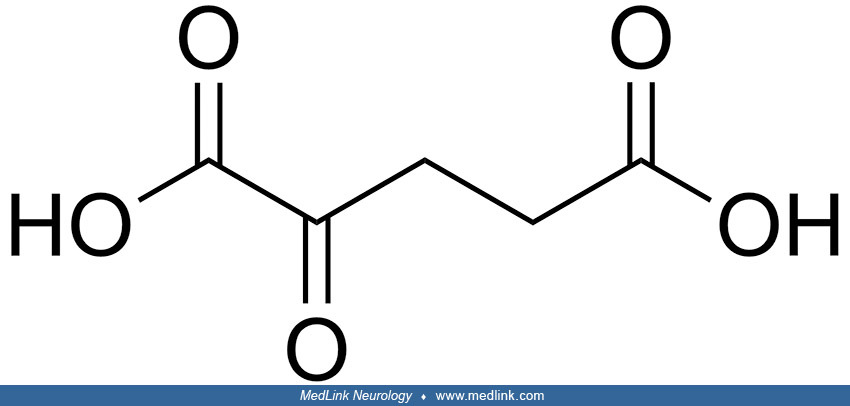
Biochemistry. 2-hydroxyglutarate (2-HG) is structurally similar to α-ketoglutarate (α-KG), an intermediate of the tricarboxylic acid (TCA) cycle; 2-hydroxyglutarate can be generated by reducing the ketone group of α-KG to a hydroxyl group. Because the C-2 position is a chiral carbon, 2-hydroxyglutarate has two enantiomers: L-2-hydroxyglutarate and D-2-hydroxyglutarate.
Inborn errors of metabolite repair. Enzymes of intermediary metabolism are commonly assumed to be extremely specific, precluding the production of useless and/or toxic side-products. However, enzymes are not strictly specific and instead often act weakly on other intracellular metabolites that resemble their physiological substrate ("substrate promiscuity") or slowly catalyze abnormal reactions on their physiological substrate ("catalytic promiscuity") (89). These "promiscuous" reactions produce nonclassical metabolites that are not efficiently metabolized by conventional enzymes (89). So-called "metabolite repair enzymes" serve to eliminate these nonclassical metabolites and prevent their accumulation (89).
Metabolite repair enzymes also eliminate nonclassical metabolites formed through spontaneous (ie, not enzyme-catalyzed) reactions (89).
L-2-hydroxyglutarate and D-2-hydroxyglutarate are normal but potentially toxic waste products from the tricyclic acid cycle. Both originate from the key intermediate 2-ketoglutaric acid (alpha-ketoglutarate, 2-KG).
2-Hydroxyglutarate (2-HG) derives from the promiscuity of lactate dehydrogenase A (LDHA), malate dehydrogenase 1 (MDH1), and PHGDH in the cytoplasm and malate dehydrogenase 2 (MDH2) in the mitochondria. Mutations of IDH1 and ID...
"Inborn errors of metabolite repair" result from genetic deficiencies in metabolite repair enzymes. L-2-hydroxyglutaric aciduria and D-2- hydroxyglutaric aciduria are prototypical inborn errors of metabolite repair (88; 61; 89; 23).
Mutations inactivating metabolite repair enzymes lead to various diseases. An example is L-2-hydroxyglutaric aciduria in which mutations in L-2-hydroxyglutarate dehydrogenase prevent the conversion of toxic L-2 hydroxy-glutarat...
L-2-hydroxyglutarate as a normal but toxic metabolite. The enzyme L-malate dehydrogenase, a constituent of the citric acid cycle, reversibly converts malate to oxaloacetate by oxidation and reduction, with the direction dependent on the redox couple NADH/NAD+).
L-malate dehydrogenase can also catalyze the conversion of ketoglutarate to L-2-hydroxyglutarate, but not the reverse reaction (63). In vitro studies using lymphoblasts and isotope-labeled precursors confirmed that ketoglutarate is a precursor of L-2-hydroxyglutarate (72).
The "metabolite repair” enzyme L-2-hydroxyglutarate dehydrogenase, which catalyzes the reverse reaction, is necessary to prevent the toxic accumulation of L-2-hydroxyglutarate (88; 89). This enzyme is absent in L-2-hydroxyglutaric aciduria.
D-2-hydroxyglutarate as a normal, but toxic metabolite. The enzyme hydroxyacid-oxoacid transhydrogenase oxidizes the terminal hydroxyl group from gamma-hydroxybutyrate, resulting in succinic semialdehyde at the expense of reducing 2-ketoglutarate to D-2-hydroxyglutarate. The reaction is cofactor-independent and unidirectional.
The "metabolite repair" enzyme D-2-hydroxyglutarate dehydrogenase, which catalyzes the reverse reaction, is necessary to prevent toxic accumulation of D-2-hydroxyglutarate (75; 89). Genetic deficiency of this enzyme causes D-2-OH-glutaric aciduria type I. A second cause of D-2-OH-glutaric acid accumulation is a gain-of-function mutation in isocitrate dehydrogenase.
The 2-hydroxyglutaric acidurias. There are three major forms of the 2-hydroxyglutaric acidurias: L-2-hydroxyglutaric aciduria, D-2-hydroxyglutaric aciduria (types I and II), and combined D-2- and L-2-hydroxyglutaric aciduria. Most forms of 2-hydroxyglutaric aciduria have an autosomal recessive pattern of inheritance.
L-2-hydroxyglutaric aciduria. L-2-hydroxyglutaric aciduria is an autosomal recessive disorder caused by mutations of the L2HGDH gene on chromosome 14q21.3 (62; 82; 70; 56; 84; 100; 50). A multicenter study revealed mutations in this gene in 106 patients from 83 families, confirming that all cases are related to defects in this gene (70).
Two independent groups identified the gene associated with L-2-hydroxyglutaric aciduria on chromosome 14q22.1 (62; 82). Rzem and colleagues purified and partially characterized a FAD-dependent enzyme from rat liver that catalyzes the oxidation of L-2-hydroxyglutarate to alpha-ketoglutarate and is associated with mitochondria (62). A candidate gene was found on chromosome 14q22.1. Patients with L-2-hydroxyglutaric aciduria were found to carry mutations in this gene. Topcu’s group, investigating inbred families with L-2-hydroxyglutaric aciduria by homozygosity mapping independently identified the same gene, L2HGDH, which they proposed to call “duranin” as a tribute to the pioneering work of Dutch geneticist Marinus Duran (24).
Experimental studies have identified in vitro and in vivo evidence of oxidative stress and oxidative damage to DNA and protein, which could be attenuated in vitro by L-carnitine (60).
In a knockout mouse model with homozygous mutation of the l2hgd gene (lhgd-/-) typical involvement of brain white matter was found, similar to human involvement in L-2-hydroxyglutaric aciduria with spongiosis and vacuolar lesions in oligodendrocytes and myelin sheaths (61). Malate dehydrogenase as the source of excess L-2-hydroxyglutarate was supported by the commensurate decrease in the formation of this dicarboxylic acid when downregulating this enzyme in mouse l2hgdh-/- embryonic fibroblasts. Male mice were more affected than female mice: male mice had a 30% higher excretion of L-2-hydroxyglutarate compared to female mice, caused by lactate dehydrogenase C, a poorly specific form of this enzyme exclusively expressed in testes. The high concentrations of L-2-hydroxyglutarate inhibited the classical lysine degradation pathway in brain : lysine-alpha-ketoglutarate reductase, which converts lysine to saccharopine, was inhibited by L-2-hydroxyglutarate and the concentrations of lysine and arginine were markedly increased in the brain of l2hgdh-/- adult mice (61).
The scheme shows how L-2-hydroxyglutarate is formed and degraded. It also shows the initial steps of the major lysine catabolic pathway (via saccharopine) present in mammalian tissues and of the minor pathway (via L-pipecolate)...
In this model, the accumulation of L-2-hydroxyglutarate exerted toxic effects by more than one mechanism, including enzyme inhibition and glial swelling (61).
Neuropathological studies in humans have shown a cavitating type of leukoencephalopathy, predominantly affecting the subcortical white matter. Significant involvement was also seen in the globi pallidi, putamina, and dentate nuclei of the cerebellum (39; 66).
Example 1. A protein truncating mutation in L2HGDH causing L‐2‐hydroxyglutaric aciduria in a consanguineous Pakistani family (50). Pedigree analysis in a consanguineous Pakistani family with a novel protein truncating mutation in L2HGDH (NM_024884.3 c.180 del G), causing L‐2‐hydroxyglutaric aciduria, shows an autosomal recessive mode of disease segregation (50).
Pedigree analysis shows an autosomal recessive mode of disease segregation. The genotype status is represented as -/- (homozygous deletion) or -/G (heterozygous carrier). (Source: Muzammal M, Ali MZ, Brugger B, et al. A novel p...
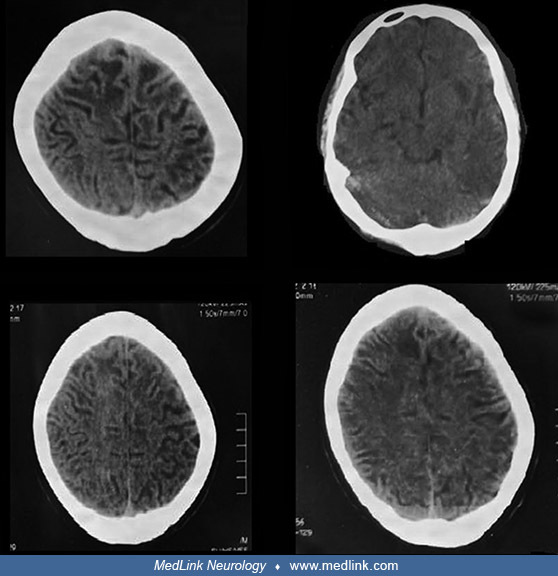
Representative clinical photographs of two affected family members (V4 and V8) are shown as are CT scan images from one of them (V8).
(Source: Muzammal M, Ali MZ, Brugger B, et al. A novel protein-truncating mutation in L2HGDH causes L-2-hydroxyglutaric aciduria in a consanguineous Pakistani family. Metab Brain Dis 2022;37[1]:243-52. Creative Commons Attribut...
(Source: Muzammal M, Ali MZ, Brugger B, et al. A novel protein-truncating mutation in L2HGDH causes L-2-hydroxyglutaric aciduria in a consanguineous Pakistani family. Metab Brain Dis 2022;37[1]:243-52. Creative Commons Attribut...
The images show extensive white matter abnormalities. (Source: Muzammal M, Ali MZ, Brugger B, et al. A novel protein-truncating mutation in L2HGDH causes L-2-hydroxyglutaric aciduria in a consanguineous Pakistani family. Metab ...
Molecular modeling showed that the identified frameshift mutation results in structural distortion of L2HGDH.
Key: (a) Normal L2HGDH protein model; (b) mutant L2HGDH protein model; (c) superimposed structure. Due to misfolding the 3D structures of wild type and mutated L2HGDH failed to overlap when superimposed, confi...
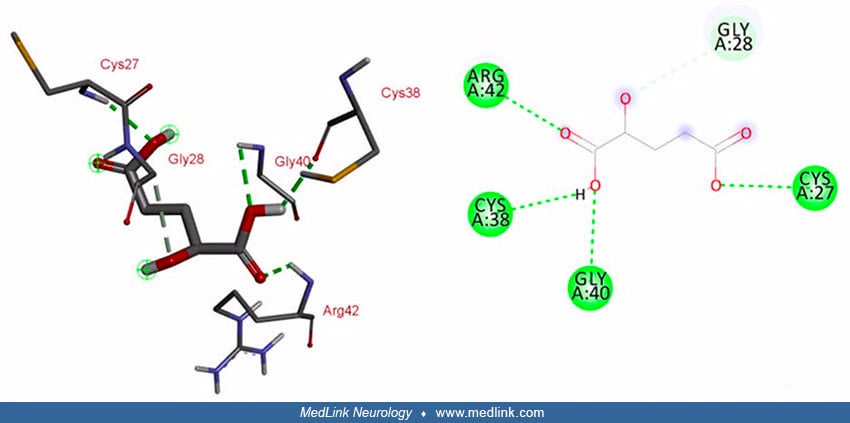
Interaction studies for L2HGDH and its substrate L-2-hydroxyglutarate predicted that five amino acids (ie, Gln-89, Tyr-195, Val-404, Ala-402, and Gly-403) of wild-type L2HGDH are involved in the interaction with its substrate via conventional hydrogen bonding. These binding sites are within the FAD-dependent enzyme domain (50).
Interaction studies for L2HGDH and its substrate L-2-hydroxyglutarate predicted five amino acids (ie, Gln-89, Tyr-195, Val-404, Ala-402, and Gly-403) of wild-type L2HGDH are involved in the interaction with its substrate via co...
In mutated L2HGDH, interacting sites within the FAD domain are lost due to the resulting frameshift and protein truncation. However, the mutant enzyme predictably showed interaction with its substrate at different positions (ie, Arg-42, Cys-38, Gly-40, and Cys-27) through conventional hydrogen bonds, and at Gly-28 through a carbon-hydrogen bond (50).
In mutated L2HGDH, interacting sites within the FAD domain are lost due to the resulting frameshift and protein truncation. However, the mutant enzyme predictably showed interaction with its substrate at different positions (ie...
Example 2. A novel homozygous mutation in L2HGDH causing L‐2‐hydroxyglutaric aciduria in a second consanguineous Pakistani family (84). In a large consanguineous Pakistani family, the index case (VI:4) presented with generalized tonic-clonic seizures, developmental delay, intellectual disability, and ataxia (84). T2-weighted MRI at 16 years of age showed diffuse hyperintense signal abnormality in the subcortical white matter and bilateral symmetrical T2 hyperintense signals in bilateral basal ganglia EEG in case VI:4, showing epileptiform changes.
T2-weighted MRI in case VI:4 shows diffuse hyperintense signal abnormality in the subcortical white matter and bilateral symmetrical T2 hyperintense signals in bilateral basal ganglia. (Source: Ullah MI, Nasir A, Ahmad A, et al...
EEG in case VI:4 shows epileptiform changes. (Source: Ullah MI, Nasir A, Ahmad A, et al. Identification of novel L2HGDH mutation in a large consanguineous Pakistani family--a case report. BMC Med Genet 2018;19[1]:25. Creative C...
A genome-wide SNP scan identified the candidate region on chromosome 14q22.1. DNA sequencing showed a novel homozygous mutation in the candidate gene L2HGDH (NM_024884.2: c.178G > A; p.Gly60Arg). The Gly60 residue of the wildtype protein resides in a helix region of the highly conserved FAD/NAD(P)-binding domain of the mitochondrial enzyme L2HGDH.
Such glycine residues commonly provide flexibility necessary for enzyme-active sites to change conformation (97). With the c.178G>A mutation, this short nonpolar glycine residue (ie, a hydrogen atom) is replaced in the mutant protein by a larger, more positively charged, and hydrophilic arginine residue. The native nonpolar glycine residue is only involved in intermolecular interactions with threonine at position 90; in contrast, the replaced basic arginine residue introduces an electrically charged, basic guanidium group, which can form multiple intermolecular hydrogen bonds (ie, with Thr90, Arg196, and Thr195). This produces a slight local perturbation of the helix conformation, disturbing cofactor binding and disrupting enzymatic activity.
(Source: Ullah MI, Nasir A, Ahmad A, et al. Identification of novel L2HGDH mutation in a large consanguineous Pakistani family--a case report. BMC Med Genet 2018;19[1]:25. Creative Commons Attribution 4.0 License, https://creat...
(Source: Ullah MI, Nasir A, Ahmad A, et al. Identification of novel L2HGDH mutation in a large consanguineous Pakistani family--a case report. BMC Med Genet 2018;19[1]:25. Creative Commons Attribution 4.0 License, https://creat...
(Source: Ullah MI, Nasir A, Ahmad A, et al. Identification of novel L2HGDH mutation in a large consanguineous Pakistani family--a case report. BMC Med Genet 2018;19[1]:25. Creative Commons Attribution 4.0 License, https://creat...
Example 3. Novel L2HGDH mutations identified in a Chinese family with L-2-hydroxyglutaric aciduria (56). T2-weighted images in the index case revealed symmetrical, high-signal changes in the subcortical white matter and basal ganglia, and the lateral ventricular wall showed multiple nodular, gray-matter heterotopia.
T2-weighted MR images in the index case of a Chinese family with L-2-hydroxyglutaric aciduria revealed symmetrical, high-signal changes in the subcortical white matter and basal ganglia, and the lateral ventricular wall showed ...
T2-weighted MR images in the index case of a Chinese family with L-2-hydroxyglutaric aciduria revealed symmetrical, high-signal changes in the subcortical white matter and basal ganglia, and the lateral ventricular wall showed ...
There were also symmetrical, high-signal changes in the cerebellar dentate nuclei, and the right cerebellar hemisphere and vermis showed dysplasia.
Mutational analysis of the L2HGDH gene in the index case and his parents revealed two novel mutations in exon 3: a homozygous missense mutation (c.407A> G, p.K136R) in both the maternal and paternal allele, and a heterozygous frameshift mutation (c.407A> G, c.408 del G; p.K136SfsX3) in the paternal allele. The mutation site p.K136R of the protein is in the pocket of the FAD/NAD(P)-binding domain and is predicted to be pathogenic.
D-2-hydroxyglutaric aciduria, types I and II. D-2-hydroxyglutaric aciduria is associated with two genes. Either there is an autosomal recessive defect in the D2HGDH gene (type 1) or a dominant change in the mitochondrial isocitric dehydrogenase (IDH2) gene (type 2) due to a germline mutation (01; 73; 36). IDH mutations are also involved in human cancer metabolism.
The mitochondrial enzyme D-2 hydroxyglutarate dehydrogenase catalyzes the irreversible oxidation of D-2-hydroxyglutarate; inactivation of this enzyme by mutations causes D-2-hydroxyglutaric aciduria type I. D-2-hydroxyglutarate...
D-2-hydroxyglutaric aciduria is characterized by the accumulation of D-2HG in human mitochondria. Increased levels of D-2HG are detected in humans exhibiting point mutations in the genes encoding isocitrate dehydrogenase, citrate carrier, the electron transferring flavoprotein (ETF) and its downstream electron acceptor ETF-ubiquinone oxidoreductase or D-2-hydroxyglutarate dehydrogenase (83). Addition of FAD to the catalytically inactive apo-proteins of the two disease-related variants (hD2HGDH-I147S and -V444A) failed to restore enzymatic activity of the variants, indicating that the cofactor binding site is compromised by the single amino acid replacements (83). Both variants form aggregates that are unable to bind the FAD cofactor (83). Because D-2-hydroxyglutaric aciduria may also result from a loss of function of either the ETF or its downstream electron acceptor ETF-ubiquinone oxidoreductase, ETF may serve as the cognate electron acceptor of reduced D-2-hydroxyglutarate dehydrogenase (83). D-2-hydroxyglutarate dehydrogenase directly reduces recombinant human ETF, establishing a metabolic link between the oxidation of D-2-hydroxyglutarate and the mitochondrial electron transport chain (83).
A large study performed in 50 patients identified mutations in 24 (36). Simultaneous enzyme assay of D-2-hydroxyglutarate dehydrogenase showed that all patients with mutations in D-2-hydroxyglutarate dehydrogenase had deficient enzyme activity. Conversely, patients without mutations (approximately 50%) had normal enzyme activity. Accordingly, the authors proposed dividing patients in two groups: type I with and type II without D-2-hydroxyglutarate dehydrogenase deficiency. Interestingly, plasma levels of D-2-hydroxyglutarate in type II patients were significantly higher than in type I, with only minimal overlap. The two subtypes of D-2-hydroxyglutaric aciduria appear to be equally frequent (37).
D2-hydroxyglutaric aciduria type I. D2-hydroxyglutaric aciduria type I is an autosomal recessive disorder caused by mutations in the D-2-hydroxyglutarate dehydrogenase gene (D2HGDH) (01; 73; 58). An enzyme isolated from the mitochondrial fraction of fractionated rat liver was found to catalyze the conversion of D-2-hydroxyglutarate to 2-ketoglutarate. This was followed by the finding of mutations in patients with D-2-hydroxyglutaric aciduria in mild and even in asymptomatic patients (74).
D2-hydroxyglutaric aciduria type II. Findings in cancer genetics lead to the discovery of the genetic background of type II D-2-hydroxyglutaric aciduria. A frequent mutation found in glioblastoma cells affects cytosolic isocitrate dehydrogenase (IDH1), whereas mutations were also detected in various malignancies in the gene encoding mitochondrial isocitrate dehydrogenase (IDH2). These mutations disable the normal ability to convert isocitrate to 2-ketoglutarate and instead alter the ability of the enzyme to convert isocitrate to D-2-hydroxyglutarate. Kranendijk and colleagues assumed the existence of a similar mechanism in D-2-hydroxyglutaric aciduria type II and did identify a heterozygous mutation in IDH2 (38). In 14 of 15 cases the parents did not carry this mutation in heterozygous form, indicating that the mutation must have arisen de novo as a germline mutation. None of these patients had affected siblings. In the remaining case, the mother, who did not excrete D-2-OH glutaric acid, had three affected children; germline mosaicism could explain this extraordinary finding.
Type II arises as a gain-of-function mutation, and the increased amount of D-2-hydroxyglutaric acid produced by the mutated enzyme reaches toxic levels, even though D-2-hydroxyglutaric acid dehydrogenase is unaffected.
Metaphyseal chondromatosis with D-2-hydroxyglutaric aciduria. Exome sequencing of blood DNA in four unrelated patients revealed no evidence for recessive mutations (91); instead, two patients showed mutations in the IDH1 gene (OMIM 147700), predicting R132H and R132S as apparent somatic mosaicism. Sanger sequencing confirmed the presence of the mutation in blood DNA in one patient, and in blood and saliva (but not in fibroblast) DNA in another patient.
In enchondromas and hemangiomas from patients with other forms of enchondromatosis (ie, Ollier disease [OMIM 166000] and Maffucci syndrome [OMIM 614569]), somatic heterozygous mutations were found in the IDH1 and IDH2 genes (03; 54).
Combined D-2- and L-2-hydroxyglutaric aciduria. Combined D-2- and L-2-hydroxyglutaric aciduria is an autosomal recessive disease caused by homozygous or compound heterozygous mutations in the SLC25A1 gene on chromosome 22q11.21, which encodes a mitochondrial citrate carrier (51; 53). Combined D-2-, L-2-hydroxyglutaric aciduria has been reported due to a hemizygous mutation in SLC25A1 in combination with 22q11.2 deletion, with clinical features including agenesis of the corpus callosum, cardiac anomalies, and a dysmorphic face (26). Biallelic mutation in the SLC25A1 gene can also cause a form of congenital myasthenia gravis with mild intellectual disability: congenital myasthenic syndrome-23 (CMS23; OMIM 618197) (06).
The patient had symmetrical facial weakness and ophthalmoparesis. This is case 1 from family 2. (Source: Balaraju S, Töpf A, McMacken G, et al. Congenital myasthenic syndrome with mild intellectual disability caused by a recurr...
The patient had ptosis and mild facial weakness. This is case 2 from family 2. (Source: Balaraju S, Töpf A, McMacken G, et al. Congenital myasthenic syndrome with mild intellectual disability caused by a recurrent SLC25A1 varia...
The patient had severe ptosis with compensatory over-activation of the frontalis muscle as well as mild facial weakness. This is case 1 from family 3. (Source: Balaraju S, Töpf A, McMacken G, et al. Congenital myasthenic syndro...
An intermediate phenotype between congenital myasthenic syndrome and D-2- and L-2-hydroxyglutaric aciduria has also been reported due to novel SLC25A1 variants (41); a Chinese boy presented with fatigable muscular weakness (partial responsive to pyridostigmine), myasthenic crisis, epilepsy, and developmental delay along with mild elevation of urinary 2-ketoglutarate and lactic acid levels. The distribution of 27 novel and previously published SLC25A1 variants can be compared for pathogenic variants in patients in whom a congenital myasthenic syndrome was the main feature, for pathogenic variants in patients diagnosed with D/L-2-hydroxyglutaric aciduria.
Combined D2-/L2-hydroxyglutaric aciduria is caused by a defect of the mitochondrial citrate carrier, localized on the inner mitochondrial membrane. It exchanges citrate and malate between the mitochondrial matrix and the cytosol. Its absence or dysfunction has been assumed to cause accumulation of citrate in the mitochondrial matrix, which in turn causes an accumulation of tricyclic acid cycle intermediates downstream from citrate, between citrate and malate, including α-ketoglutarate (48). α-Ketoglurtarate is the metabolic source of both L-2-hydroxyglutarate and D-2-hydroxyglutarate (48).
Although D-2- and L-2 hydroxyglutaric aciduria is a metabolic hallmark of the disease, the citrate carrier probably does not play a significant role in the removal of hydroxyglutarate from the cytosol for oxidation to oxoglutarate in the mitochondrial matrix (45). Instead, computer simulations show that the export of citrate from the mitochondrion cannot be fully compensated by other pathways, restricting the cytosolic production of acetyl-CoA that is required for the synthesis of lipids, sterols, dolichols, and ubiquinone (45).
Because of the limited number of patients diagnosed to date, no epidemiologic statements are warranted.
Primary prevention focuses on preventing disease before it develops, whereas secondary prevention attempts to detect a disease early and intervene early, and tertiary prevention seeks to optimally manage established disease to avoid further complications. Prenatal screening can be classed as secondary prevention only where its purpose is to lead to more successful surgical or other treatment, in utero or postnatally (22).
No approach is available for the primary or secondary prevention of the 2-hydroxyglutaric acidurias. No method is presently available to affect the course of the disease.
Regarding tertiary prevention, the high brain tumor risk in patients with L-2 hydroxyglutaric aciduria may warrant regular screening with brain MRIs (90).
Antenatal detection in at-risk pregnancies should be possible when a pathogenic mutation has been established in the family.
Clinical findings of a slowly progressive encephalopathy, with mental retardation and ataxia from childhood, together with subcortical leukoencephalopathy evidenced by MRI are strong presumptive evidence of L-2-hydroxyglutaric aciduria (09). Other findings include involvement of the basal ganglia and the dentate nuclei. Differential diagnosis of a leukoencephalopathy that affects the U-fibers includes Canavan disease (aspartoacylase deficiency) (27), mitochondrial encephalopathies (08), and leukoencephalopathy with swelling and a discrepantly mild clinical course (85). Patients with the latter disease show severe megalencephaly with white matter signal changes and cysts. Their mild course is explained by the absence of cavitation. Confirmation of this disease is by finding mutations in the MLC1 gene (40). Differential diagnosis of L-2-hydroxyglutaric acidurias includes all leukoencephalopathies associated with macrocephaly as discussed by Renaud (59). Findings in D-2-hydroxyglutaric aciduria present a varied clinical profile that may include facial dysmorphia, cardiomyopathy, severe epilepsy, and varied MR abnormalities including neuroepithelial cysts and neocortical dysplasia. In both diseases, gas chromatography and mass spectroscopy provide the means for screening. Separation of D- and L-types is possible through biochemical means.
Biochemical studies. D-2-hydroxyglutaric acid and L-2-hydroxyglutaric acid are 5-carbon dicarboxylic acids that have identical chemical and physical properties but differ because they have a hydroxyl group on the second carbon, which yields a chiral center. Therefore, two 3D structures present mirror images (37) with different properties requiring different enzymes for their disposal (37). The finding of increased 2-hydroxyglutaric acid in a sample, therefore, requires determination of its chirality (D2, L2, or combined D2,L2).
In the typical case of L-2-hydroxyglutaric aciduria, gas chromatography of the urine will reveal a solitary peak of 2-hydroxyglutaric acid. This excretion pattern only affects the L-stereoisomer. No increase of other organic acids has been found. Increased levels of L-2-hydroxyglutaric acid can also be detected in plasma and CSF. Typically, the metabolite concentration in CSF greatly exceeds the level in plasma. Lysine is also increased both in plasma and in CSF.
L-2-hydroxyglutaric aciduria may be missed in an adult neurologic practice where gas chromatography of the urine may not be a regular part of the diagnostic workup of a neurodegenerative disorder. The finding of subcortical leukoencephalopathy together with cerebellar atrophy in an adult with progressive intellectual deterioration and ataxia is sufficiently characteristic to warrant a search for L-2-hydroxyglutaric aciduria.
D-2-hydroxyglutaric aciduria patients do not excrete significant amounts of other organic acids, with the occasional exception of 2-oxo-glutaric acid or other citric acid cycle intermediaries (86; 87). D-2-hydroxyglutaric acid concentrations are greatly elevated in CSF in patients with an encephalopathic phenotype. In addition, total GABA is increased in CSF (30; 86; 87).
MRI in L-2-hydroxyglutaric aciduria. MRI in L-2-hydroxyglutaric aciduria characteristically demonstrates a subcortical leukoencephalopathy affecting the arcuate fibers and sparing the central hemispheric white matter. Typical MRI findings in L-2-hydroxyglutaric aciduria are a subcortical decrease of T1-weighted signal and increase of T2-weighted signal. Subcortical white matter often shows signs of swelling.
Other MRI findings include signal abnormalities in the globi pallidi, striatum, and the cerebellar dentate nuclei, along with cerebellar folial atrophy. The subcortical findings resemble those in Canavan disease (aspartoacylase deficiency), although the clinical severity of L-2-hydroxyglutaric aciduria is less than in Canavan disease.
These findings are progressive (09; 71).
Proton magnetic resonance spectroscopy in L-2-hydroxyglutaric aciduria. Proton magnetic resonance spectroscopy can identify peaks attributable to L-2-hydroxyglutaric acid in vivo (67; 04).
MRI in D-2-hydroxyglutaric aciduria. MRI findings in D-2-hydroxyglutaric can be divided into severe cases with neonatal seizures, and milder cases. In the former, a combined pattern of delayed gyration, delayed opercularization, delayed myelination, and ventricular dilatation is found; subependymal cysts overlying the caudate nucleus are often found. Findings in milder cases are variable (86; 87). In a single case of D-2-hydroxyglutaric aciduria, MRI findings were suggestive of a mitochondrial encephalopathy with bilateral involvement of the striatum (92). The following table summarizes the highlights of the major subtypes and their differences.
|
L-2-hydroxyglutaric aciduria | ||
|
• Onset: insidious during first years | ||
|
- dystonia: ± | ||
|
• Other neurologic abnormalities: | ||
|
- epilepsy: majority | ||
|
• MR abnormalities: | ||
|
- subcortical leukoencephalopathy: + | ||
|
• Cerebrospinal fluid: | ||
|
- 2-OH-glutaric acid increased | ||
|
• Cardiomyopathy: - | ||
|
D-2-hydroxyglutaric aciduria | ||
|
• Severe type: neonatal encephalopathy with seizures, later onset with mild developmental delay in mild type | ||
|
- dystonia: ± | ||
|
• Other neurologic abnormalities: | ||
|
- epilepsy: majority | ||
|
• MR abnormalities: | ||
|
- subcortical leukoencephalopathy: - | ||
|
• Cerebrospinal fluid: | ||
|
- 2-OH-glutaric acid increased | ||
|
• Cardiomyopathy: + | ||
|
- type I: autosomal recessive | ||
|
Combined L-2- and D-2-hydroxyglutaric aciduria (D,L-2-HGA) | ||
|
• Severe encephalopathy developing from birth | ||
|
| ||
No systematic treatment trials have been applied to any of the known types of 2-hydroxyglutaric aciduria.
L-2 hydroxyglutaric aciduria. Amelioration of spasticity and improved motor performance were reported in a single adult woman with L-2-hydroxyglutaric aciduria using FAD and levocarnitine (65).
The high brain tumor risk in patients with L-2 hydroxyglutaric aciduria may warrant regular screening with brain MRIs (90).
D-2 hydroxyglutaric aciduria. Because CSF GABA levels are usually elevated in D-2-hydroxyglutaric aciduria, vigabatrin should be avoided for the treatment of seizures.
A selective inhibitor to the mutant enzyme in a mouse model of D-2-hydroxyglutaric aciduria type II lessened the associated cardiomyopathy, leading to improved survival (93). The inhibitor has not been studied in the human disease.
Combined L-2- and D-2-hydroxyglutaric aciduria. Preliminary studies of citrate supplementation in a patient with combined L2/D2 hydroxyglutaric aciduria suggested a partial effect (48).
Epilepsy treatment in the case of D2-hydroxyglutaric acidurias must omit vigabatrin because of high levels of GABA due to metabolic causes.
Pregnancies of genetic carriers who had a previous child affected with either L-2-hydroxyglutaric aciduria or D-2-hydroxyglutaric aciduria type 1 carry a recurrence risk of 25%. Prenatal diagnosis is possible with mutation analysis of the respective genes.
D-2-hydroxyglutaric aciduria type II represents a de novo (germline) mutation; recurrence, though rare, has been recorded.
A normal outcome of pregnancy has been reported in a patient with L-2-hydroxyglutaric aciduria (35).
No anesthetic risks are known in either disorder. The usual precautions should be taken in the case of manifest epilepsy.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Douglas J Lanska MD FAAN MS MSPH
Dr. Lanska of the University of Wisconsin School of Medicine and Public Health and the Medical College of Wisconsin has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Developmental Malformations
Apr. 14, 2024
Developmental Malformations
Apr. 09, 2024
Childhood Degenerative & Metabolic Disorders
Apr. 07, 2024
Childhood Degenerative & Metabolic Disorders
Mar. 22, 2024
Developmental Malformations
Mar. 22, 2024
Developmental Malformations
Mar. 22, 2024
Developmental Malformations
Mar. 22, 2024
Developmental Malformations
Mar. 22, 2024