













Abnormalities of tetrahydrobiopterin metabolism
Apr. 07, 2024
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Sialidosis is an autosomal recessive lysosomal storage disorder caused by deficiency of neuraminidase 1 due to NEU1 mutations. Sialidosis type I is characterized by the development of ocular cherry-red spots and generalized myoclonus in the second or third decade of life. Seizures, hyperreflexia, ataxia, and progressive atrophy on brain MRI may also occur. Sialidosis type II is more severe than type I and is distinguished by the early onset of a progressive, mucopolysaccharidosis-like phenotype with visceromegaly, dysostosis multiplex, and intellectual disability. In severe cases, hydrops fetalis may occur.
• Sialidosis is a disorder of sialic acid metabolism. | |
• It is often associated with myoclonic epilepsy in juveniles or young adults, sometimes without visual impairment. | |
• Eye and skeletal abnormalities may be helpful for the diagnosis. | |
• Macular cherry-red spots may occur in otherwise asymptomatic patients or be absent in symptomatic patients. |
Sialidosis is a disorder resulting from an inherited, isolated deficiency of neuraminidase 1 (sialidase) (36; 52; 05; 15). Because the first patients shown to suffer from this enzyme abnormality had originally been included in a group of patients given the designation "mucolipidosis" (70), this disorder is also sometimes referred to as mucolipidosis I (69). The enzyme defect was first demonstrated in this group of patients in 1977 (13; 69). Within a year, several investigators independently described an adult group of patients with deficiencies of neuraminidase 1 activity (18; 51; 59; 75). The late-onset form of the disorder, originally labeled the cherry-red spot-myoclonus syndrome, is now referred to as sialidosis type I.
Sialidosis (mucolipidosis I) is not to be confused with mucolipidosis II (I-cell disease) or mucolipidosis III (pseudo-Hurler polydystrophy). Although patients with these latter disorders are also characterized by neuraminidase deficiencies, the defect is neither a primary nor an isolated abnormality. Rather, the neuraminidase deficiencies in mucolipidosis II and III are the secondary result of defective post-translational modification involving many lysosomal enzymes (50). Mucolipidosis IV, although sharing the same name, is also a distinct and genetically unrelated disorder. It is caused by mutations in MCOLN1 that codes for a protein called mucolipin 1, thought to be a cationic ion channel (62).
The classification of sialidosis is also complicated by the existence of a combined neuraminidase and beta-galactosidase deficiency, generally referred to as galactosialidosis (53). Various studies, such as somatic cell hybridization and biochemical and genetic mapping, have demonstrated that sialidosis and galactosialidosis result from mutations of different genes and are, thus, two distinct disorders.
Sialidosis must be distinguished from sialuria, the term given to patients who excrete excessive amounts of free sialic acid due to the defective feedback control of UDP-GlcNAc 2-epimerase, leading to hypersialylated O-glycans (35; 89). Finally, sialidosis patients should not be confused with those patients suffering from free sialic storage disease or Salla disease, both of which are the result of the defective transport of sialic acid across lysosomal membranes (04).
In an early review, Lowden and O'Brien proposed that patients with a primary neuraminidase 1 deficiency could be divided into two groups (36). They classified the more mildly affected patients, those who lacked physical changes (normosomatic), as sialidosis type I. The more severely affected patients, having obvious physical changes (dysmorphic), were designated as sialidosis type II. Sialidosis type II can present with severe dilated cardiomyopathy (19) or marked ascites (73). These broad categories have been further subdivided (52). For example, Type II sialidosis (dysmorphic form) has been classified into three subgroups based on the age of onset and the clinical severity: (1) congenital or neonatal; (2) infantile (onset 0 to 12 months); and (3) juvenile form (onset 13 months to 20 years) (73).
• Patients with a primary neuraminidase 1 deficiency are typically divided into two groups: sialidosis type I refers to more mildly affected patients, who lack Hurler-like physical changes (normosomatic), whereas sialidosis type II refers to more severely affected patients, having obvious physical changes (dysmorphic). | |
• The presenting feature of this form of the disorder is often the loss of visual acuity, the development of myoclonus, or both, usually in the second or third decade of life ("cherry-red spot-myoclonus" syndrome). | |
• Sialidosis type II usually presents at an early age and includes many Hurler-like changes, such as coarse facial features, increased head size, and short stature. |
Patients with a primary neuraminidase 1 deficiency are typically divided into two groups: sialidosis type I and type II (36; 52). Sialidosis type I refers to more mildly affected patients, who lack Hurler-like physical changes (normosomatic), whereas sialidosis type II refers to more severely affected patients who have obvious physical changes (dysmorphic).
Sialidosis type I. Sialidosis type I is distinguished from type II by both a milder clinical course and the absence of Hurler-like somatic changes (52; 01). In contrast to type II patients, these individuals usually have normal growth and intellect. The presenting feature of this form of the disorder is often the loss of visual acuity, the development of myoclonus, or both, usually in the second or third decade of life (11).
The myoclonus is usually induced by stimulation, movement, or emotion. The myoclonus may have both cortical and subcortical origins (84; 88). EMG recordings in two patients with sialidosis type I have demonstrated clear episodes of bilaterally synchronous myoclonic activity in contralateral homologous muscles with a high muscular-muscular coherence with near-zero time-lag between the right and left homologous muscles (84); a unilateral cortical source cannot fully explain this type of myoclonic activity, so there must exist a subcortical mechanism for bilateral synchronization. In another case with sialidosis type I, EEG revealed extensive paroxysmal spiky beta brush, and somatosensory evoked potentials (SEP) after stimulation of median nerves demonstrated giant SEP and C-reflex, supporting a cortical origin of myoclonus (88).
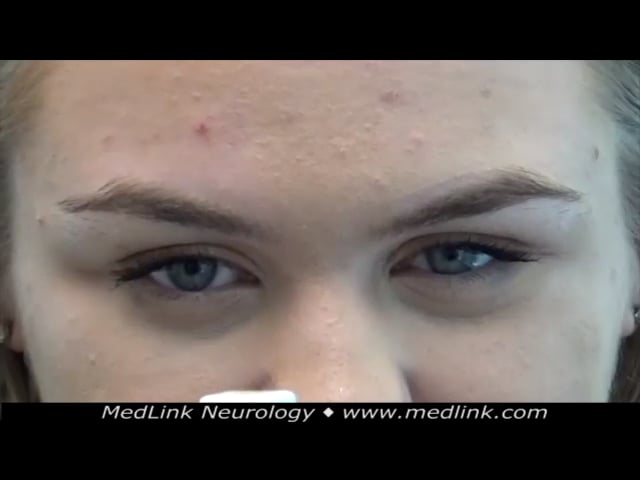
It is often at this time that cherry-red spots in the macular region of the fundus are discovered, although many patients never develop cherry-red spots (32); rarely, patients may present with isolated cherry-red spots (45). Although visual acuity commonly, but not universally, decreases during the course of the disease, most patients maintain relatively preserved visual acuity, even in adulthood; those with poor visual acuity often have optic atrophy (16). Because many patients with sialidosis type I develop ataxia, it should be considered in the differential diagnosis of late-onset ataxia and myoclonus (11; 40; 60). Rarely, the patient may have epilepsy only without visual impairment due to retinal abnormalities (43).
As the disease progresses, the myoclonus may interfere with walking, sitting, etc. Because of the prominence of the eye findings and the myoclonus, this form of the disorder has also been referred to as the "cherry-red spot-myoclonus" syndrome (18; 51; 59; 75; 60).
Sialidosis type II. In the more severely affected group of patients, sialidosis type II is characterized by the early onset of a progressive mucopolysaccharidosis-like phenotype (52; 92; 05; 10). Clinical findings, usually present at an early age, include many Hurler-like changes, including coarse facial features, increased head size, and short stature. Other "Hurleroid" findings include lens opacities, corneal clouding, visceromegaly, joint stiffness, hernias, and sensorineural hearing loss. Most patients are intellectually disabled and have macular cherry-red spots and myoclonus (68), and many have ataxia. Radiographic changes resemble those seen in the mucopolysaccharidoses, with beaking of the lumbar vertebrae, broadening of the ribs, and thickening of the skull. Rarely, deposits in the spinal roots and paravertebral ganglia can cause spinal cord compression (06).
Type II patients can be further subdivided into an infantile form of the disease, in which patients are relatively normal or only minimally affected at birth, and a more severe disorder called congenital sialidosis (33). This latter form of the disorder is characterized by neonatal ascites and hydrops fetalis and often by stillbirth or death at an early age (29). This is one of at least 10 different lysosomal disorders that have been diagnosed in infants with hydrops fetalis (71; 02; 28). A nephropathic phenotype of sialidosis type II has also been described (61). There can be significant intrafamilial clinical heterogeneity (85).
The prognosis depends on the form of sialidosis. Congenital sialidosis is characterized by neonatal ascites, hydrops fetalis (34), and often by stillbirth or death at an early age (29). Sialidosis type I is distinguished from type II by a milder clinical course with typically normal growth and intellect. As the disease progresses, however, the myoclonus may interfere with walking, sitting, etc. Visual acuity also commonly decreases during the course of the disease.
Case 1: Sialidosis type I. A 7-year-old girl with normal prior psychomotor development had worsening vision, and ophthalmological evaluation revealed bilateral cherry-red spots (60). The diagnosis of sialidosis was established after a skin biopsy showed reduced neuraminidase activity. Genetic testing for NEU1 gene mutations revealed a heterozygous pathogenic variant (c.727G> A, p.G243R) and a new missense variant c.544A>C (p.S182R). Levels of oligosaccharides were increased, although lower than expected, consistent with her milder phenotype (type I).
A few years after her initial diagnosis, she developed severe depression with suicide attempts and was treated with sertraline and escitalopram. Around age 13, she developed myoclonic jerks in her lower limbs while asleep. Her vision loss progressively worsened, with a prominent decline around age 17 years, accompanied by the onset of oscillopsia. She also had burning sensations in her hands and feet and was diagnosed with a small fiber neuropathy; neuropathic symptoms were relieved with pregabalin.
Neurologic examination around 19 years of age revealed (1) visual acuity of 20/400 in each eye; (2) prominent myoclonus of her lower extremities that was triggered by walking and caused multiple falls; (3) mild action myoclonus of the arms, left more than right; (4) action myoclonus of the legs while standing; (5) cerebellar signs (eg, overshoot with tracking a moving object with her finger); and (6) a moderately widebased gait with superimposed myoclonic jerks.
Action myoclonus was significantly improved by the light touch of a wall or a supportive person, and she also reported that this was lessened with alcohol. Treatment with trihexyphenidyl, clonazepam, levetiracetam, and miglustat did not produce significant benefit. Cranial MRI showed only small caliber optic nerves and chiasm.
Oculomotor examination revealed a complex but variable pattern of abnormal spontaneous eye movements in central gaze that may have developed in part because of her substantial central vision loss: specifically, square wave jerks in central gaze alternated with variably jerky or pendular nystagmus.
When nystagmus was present in central gaze, nystagmus could also be visualized in eccentric gaze without a null zone. Saccadic intrusions were superimposed on smooth pursuit. Saccadic hypermetria was present.
Eye movement recordings (when the patient reported being able to see the visual target during recordings) demonstrated marked difficulty with following saccadic targets, with excess erratic saccades poorly related to target jumps, saccadic hypermetria, macro square wave jerks, and saccadic smooth pursuit. Saccade velocity was preserved in both horizontal and vertical planes as demonstrated by normal main-sequence relationships between horizontal saccade amplitude and peak velocity. Later eye movement recordings demonstrated the variability of eye movements in the central position.
Case 2: Sialidosis type I. A 40yearold Ecuadorian man was diagnosed with sialidosis type I at 38 years of age (60). He was born prematurely and was jaundiced but had normal motor and cognitive development. At age 8 years, his vision began to decline. At age 14, he developed involuntary myoclonic movements, particularly affecting his hands during action, and 3 years later, his balance began to worsen. Treatment trials of valproic acid, levetiracetam, perampanel, clonazepam, and zonisamide were either ineffective or not tolerated. Because his myoclonus was very alcohol-responsive, sodium oxybate (gamma-hydroxybutyrate) was tried and clearly improved his symptoms but was discontinued due to the emergence of visual hallucinations. Because of the marked visual impairment, he required the use of a white cane to walk.
Visual acuity was 20/400 in both eyes, and cherry red maculae were identified on ophthalmological evaluations.
At age 38 years, confirmatory genetic testing revealed a homozygous missense variant c.629C>T, p.Pro210Leu, in the NEU1 gene. Sialidase activity was absent in fibroblasts whereas betagalactosidase activity was preserved.
Neurologic examination around 40 years of age revealed vocal myoclonus (with paroxysms triggered by speaking) and prominent limb myoclonus at rest and with action. Action myoclonus was exacerbated with fine action, such as copying a spiral or writing, although it improved if he performed the same activity with his eyes closed. Gait was narrowbased. Romberg sign was absent, but standing for a prolonged period provoked myoclonic volleys.
Oculomotor examination revealed subtle downbeat nystagmus on lateral gaze and subtle gaze-evoked nystagmus; saccadic smooth pursuit and saccadic hypermetria were present. Eye movement recordings were unsuccessful due to his poor visual acuity.
Case 3: Sialidosis type I. A 31yearold man of Haitian heritage was diagnosed with sialidosis type 1 at 12 years of age after developing difficulty seeing the board at school (60). There was no consanguinity in the family. Ophthalmological evaluation revealed bilateral macular cherry red spots.
Confirmatory genetic testing disclosed two variants in the NEU1 gene: c.649G>A (p.V217M) and c.644T>C (p.L215P).
Subsequently, his color perception and visual acuity progressively worsened. Around age 20 years, he developed slurred speech, mild gait ataxia, and myoclonus, the latter particularly affecting his upper limbs. A trial of clonazepam was not helpful. Levetiracetam was begun after four generalized tonicclonic seizures, with coincident improvement in myoclonus. Because his myoclonic symptoms were exquisitely alcohol-responsive, sodium oxybate (gamma-hydroxybutyrate) was begun and produced a marked improvement, with complete resolution of stimulussensitive myoclonus and with only very mild postural and action myoclonus of his hands remaining. Gait ataxia remained mild.
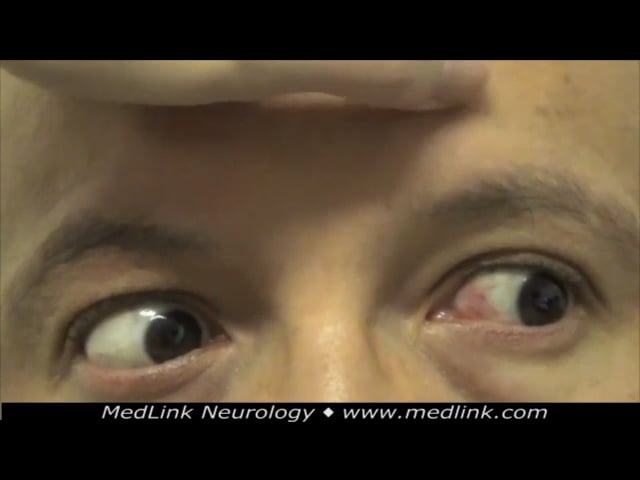
Ocular motor examination revealed saccadic pursuit (worse with horizontal than vertical smooth pursuit), intermittent horizontal and vertical saccadic hypometria, and prolonged vertical saccade latency. Horizontal optokinetic nystagmus was normal, but only slow phases were present with vertical optokinetic testing, suggesting a possible early saccadic gaze palsy. In addition, there was mild right abducens paresis and convergence insufficiency. Eye movement recordings showed saccadic pursuit, both horizontally and vertically, saccadic hypometria (undershoots), and slow horizontal and vertical saccades (supporting a saccadic gaze palsy, possibly from brainstem burst neuron involvement).
A plot of saccadic amplitude versus saccadic peak velocity shows slow horizontal saccades, particularly at larger saccadic amplitudes. (Source: Riboldi GM, Martone J, Rizzo JR, Hudson TE, Rucker JC, Frucht SJ. Looking "cherry r...
Cranial MRI showed reduced size of the cerebellar vermis. Brain magnetic resonance spectroscopy revealed elevated creatinine in the superior cerebellar vermis. EMG and nerve conduction studies were interpreted as consistent with a distal motor neuropathy. Pure tone audiometry showed high-frequency sensorineural hearing loss.
Case 4: Sialidosis type II. A 12-year-old boy was the second child of healthy, unrelated Indian parents (92). His older brother was healthy, and there was no other relevant family history. He was born at term with a birth weight of 2.1 kg. He was first investigated at 18 months of age because of short stature and delayed milestones. He first walked at 20 months and began talking at 2 years. By age 3 years, he had developed coarse facial features, so a diagnosis of a mucopolysaccharidosis was considered, although there was no hepatosplenomegaly or excess mucopolysacchariduria. Developmental assessment at that time revealed an IQ of approximately 75. He was reassessed at the age of 9 years because of poor school performance and failing vision. His coarse, “Hurleroid” face was noted. The Wechsler Intelligence Test for Children assessed his full-scale IQ at 67.
His first grand mal convulsion occurred at 11 years of age; subsequently, he had frequent myoclonic jerks, particularly at night. Repeat IQ assessment at age 11 years indicated further cognitive deterioration with a full-scale IQ of 55.
At 12 years of age, his height (118.5 cm), weight (20.5 kg), and head circumference (48 cm) all fell well below the third percentile. His face was coarse with prominent lips, large tongue, and gingival hypertrophy. There was limitation of abduction at the shoulders and of external rotation at the hips, with mild limitation of extension at both elbows and knees, although movement at other joints was normal. The liver and spleen were not enlarged. Neurologic findings included fine vertical nystagmus, ataxia with an intention tremor, mild generalized hypotonia, ankle clonus, and extensor plantar responses. Visual acuity was 6/60 in each eye with a low myopic correction. Ocular and vision findings included faint opacification of the superficial corneal stroma, extensive dot lens opacities clustered around the lens nucleus, bilateral optic atrophy, cherry-red spots, bilateral central scotomas, and grossly abnormal color perception across the spectrum. Ocular movements were full, but there were bursts of fine vertical nystagmus. The visually evoked potential to a flash stimulus showed a delayed major positive peak. Audiologic testing revealed mild bilateral conductive hearing loss. A skeletal survey showed flattening of the lumbar vertebrae with anterior tonguing and irregular end plates, a J-shaped pituitary fossa, a small irregular left femoral epiphysis, and mild pointing of the proximal end of the metacarpals.
Thin layer chromatography of urinary oligosaccharides showed a strongly staining band characteristic of sialidosis. Neuraminidase activity in cultured skin fibroblasts was very low in the proband and in the heterozygous range in both parents. This case was reported before genetic testing was available.
• Sialidosis types I and II are the result of primary neuraminidase 1 (sialidase1) deficiencies. | |
• Sialidosis types I and II are both inherited in an autosomal recessive manner. |
Sialic acids. Sialic acids are a class of alpha-keto acid sugars with a nine-carbon backbone. The name "sialic acid" (from the Greek for saliva) was introduced by Swedish biochemist Gunnar Blix (1894-1981) in 1952 (39). Sialic acids are common components of glycoproteins, glycolipids, and gangliosides, where they decorate the end of sugar chains on soluble proteins or on glycoproteins on the surface of cells.
Sialic acids are derivatives of neuraminic acid, an acidic amino sugar with a 9-carbon backbone. The name "neuraminic acid" was introduced by German biochemist Ernst Klenk (1896-1971) in 1941, in reference to the brain lipids from which it was derived as a cleavage product. Neuraminic acid and its sialic acid derivates have an open-chain form, the latter of which can be shown in Haworth projections or in chair conformations, as is the case for simple sugars (eg, glucose, mannose, etc.).
Neuraminic acid does not occur naturally, but many of its derivatives are found widely distributed in animal tissues, especially in glycoproteins and gangliosides. The N- or O-substituted derivatives of neuraminic acid are collectively known as sialic acids, the predominant form in mammalian cells being N-acetylneuraminic acid (Neu5Ac or NANA), which in its cyclic form exists as alpha and beta anomers that can be visualized in a standard stick representation, or in their chair conformations.
N-acetylneuraminic acid is found in complex glycans on mucins, glycoproteins at the cell membrane, and in glycolipids, known as gangliosides, a crucial component of neuronal membranes in the brain. Despite its general involvement in preventing infections (mucus associated with mucous membranes), N-acetylneuraminic acid is a receptor for influenza viruses, allowing virion attachment to mucous cells via hemagglutinin (an early step in influenza).
In humans, the highest sialic acid concentration is in the brain, where these acids are important for neural transmission and synaptogenesis (86).
Sialidosis. The various forms of sialidosis all apparently result from inherited deficiencies of acid neuraminidase (08; 58). The identification, isolation, and characterization of cDNA that codes for the lysosomal form of this enzyme confirm this conclusion (08; 58). The lysosomal neuraminidase gene maps to chromosome 6 at band 6p21 (specifically, 6p21.33), a region known also to contain the human leukocyte antigen locus (08; 42; 58). In a study of genomic DNA of nine patients with sialidosis, the effects of the mutations were clustered in one region of the surface of the sialidase molecule, a region likely involved in the binding of the multienzyme complex required for enzyme activity (37). More than 20 mutations have been identified in the NEU1 gene (01).
Both the early-onset, severe (type II) and later-onset, milder (type I) forms of sialidosis are the result of primary neuraminidase 1 (sialidase 1) deficiencies. These are both inherited in an autosomal recessive manner.
Neuraminidase 1 catalyzes the hydrolysis of the terminal neuraminic acid (sialic acid) from a variety of naturally occurring compounds such as glycoproteins and glycolipids. Normally, neuraminidase 1 cleaves the terminal alpha(2-3)- and alpha(2-6)- sialyl linkages of a variety of oligosaccharides and glycoproteins (14; 12; 26; 25). Because the sialic acid residues must be removed before the other carbohydrate moieties of glycoproteins and oligosaccharides can be cleaved by other enzymes, neuraminidase 1 plays an initial and key role in the lysosomal degradation of this class of compounds.
Early investigations, carried out on crude extracts of fibroblasts and other tissues, established that the enzyme is one of a large number of lysosomal, acidic hydrolyses responsible for the catabolism of large molecular compounds in a wide variety of normal cells. In subsequent investigations, the enzyme was isolated from human placenta and more fully characterized (83; 82; 26; 25).
In the lysosome, neuraminidase 1 is part of a heterotrimeric complex together with beta-galactosidase and cathepsin A (the latter also referred to as "protective protein") (20; 21; 79; 57; 77). Neuraminidase must interact with this auxiliary protein before it can segregate to mature lysosomes (80). Some sialidosis patients have mutations that cause a disruption of the lysosomal multienzyme complex (38).
Deficiency of neuraminidase 1 results in excessive amounts of urinary and tissue concentrations of compounds having N-acetylneuraminic acid (the predominant sialic acid found in human cells) at the nonreducing terminus. In most cases, N-acetylneuraminic acid is linked to galactose via either an alpha(2-3) or alpha(2-6) linkage (31; 72; 81; 47). The structures of the neuraminic acid-rich oligosaccharides that accumulate in the absence of neuraminidase are closely related and consistent with the evidence that they originate from the sugar side chains of glycoproteins.
Cultured fibroblasts from sialidosis patients, when cultured in the presence of radiolabeled acetylmannosamine, accumulate labeled neuraminic acid covalently bound to at least nine different sialyl oligosaccharides (64). Neuraminidase 1 regulates lysosomal exocytosis (91), particularly at the cell surface where it participates in formation and maturation of elastin fibers (24). Loss of NEU1 results in hypersialylation of LAMP1 at the lysosomal membrane, which makes this organelle preferentially dock at the plasma membrane and engage in exocytosis of lysosomal enzymes (17). These enzymes, particularly serine proteases, cause the bony abnormalities and other changes seen in this disease.
In the mouse model for this disease, NEU1 deficiency causes damage in cochlear marginal cells that contribute to hearing loss and defective muscle regeneration (90; 49).
The pathology varies from region to region and shows some developmental abnormalities that, in general, are not related to the extent of lysosomal storage (78).
• Sialidosis is a rare, panethnic disorder. |
Sialidosis appears to be rare, occurring with an incidence of 1 per 4.2 million live births in Australia (41). Case reports suggest that it is a panethnic disorder, although ethnic predilections cannot be excluded (07).
• Close-degree relatives can be tested for reduced levels of sialidase; the presence of reduced levels--approximately 50% of normal--is taken as evidence that a person is a disease carrier. | |
• As neuraminidase activity is present in fresh, normal, cultured amniotic fluid cells as well as in chorionic villi samples, prenatal monitoring can be offered in at-risk situations. |
As sialidosis is an autosomal recessive disorder, any effort at prevention must be directed toward identifying couples who are both carriers. Because no large public health (eg, mass screening) programs are presently directed at the identification of carriers for this disorder, almost all carriers are identified after the birth of an affected child within the immediate family. Other close-degree relatives can be tested for reduced levels of sialidase in various cells (eg, fresh, white blood cells or cultured fibroblasts); the presence of reduced levels--approximately 50% of normal--is taken as evidence that a person is a disease carrier.
As neuraminidase activity is present in fresh, normal, cultured amniotic fluid cells as well as in chorionic villi samples, prenatal monitoring can be offered in at-risk situations (eg, to carrier couples with a 25% risk of an affected offspring at each pregnancy) to establish the diagnosis of both affected and unaffected fetuses (29; 44). Preimplantation genetic diagnosis should be applicable to sialidosis.
Neuraminidase 1 deficiencies are most often confused with other inherited disorders resulting from lysosomal enzyme deficiencies.
In the more severe form of disease, sialidosis type II, the clinical findings of affected patients overlap those seen in patients with the severe forms of mucopolysaccharidosis (eg, mucopolysaccharidosis II or VII, or one of the closely related oligosaccharide storage disorders) (52). In contrast to mucopolysaccharidosis patients, sialidosis patients have normal concentrations and patterns of mucopolysaccharides in their urine. Analysis of oligosaccharides in urine should also help distinguish these patients from patients with other disorders of oligosaccharide metabolism, such as mannosidosis, fucosidosis, or aspartylglycosaminuria.
Cherry-red spots and a progressive clinical course might cause confusion with Tay-Sachs disease. Measurement of hexosaminidase A in serum or white blood cells will easily confirm or exclude this possibility.
In the milder form of this disease, sialidosis type I, cherry-red spots in combination with a late-onset and progressive course often leads to consideration of adult or late-onset Tay-Sachs disease or other lysosomal storage disorders having similar eye findings. Appropriate enzyme studies on white cells or fibroblasts will confirm or rule out these possibilities.
• The clinical workup of patients suspected of having sialidosis includes careful examination for evidence of organomegaly, an eye examination for corneal clouding and cherry-red spots, x-ray studies for evidence of abnormalities of the type seen in many lysosomal storage disorders, and close examination of white blood cells for inclusions. | |
• As the enzyme defect in sialidosis patients results in an inability to remove the terminal neuraminic acid residues, compounds rich in this complex are present in increased amounts in the urine of affected patients. | |
• Definitive diagnosis of sialidosis should be based on the determination of neuraminidase activity in fresh cells from the patient (eg, fibroblasts, white blood cells, or cultured amniotic cells). | |
• DNA diagnosis should be routinely performed using Sanger sequencing or next-generation methods. |
Sialidosis partis of a large group of inherited, lysosomal storage disorders with progressive courses (56). The clinical workup of this group of patients usually includes careful examination for evidence of organomegaly, an eye examination for corneal clouding and cherry-red spots, x-ray studies for evidence of abnormalities of the type seen in many lysosomal storage disorders, and close examination of white blood cells for inclusions.
As the enzyme defect in sialidosis patients results in an inability to remove the terminal neuraminic acid residues, compounds rich in this complex are present in increased amounts in the urine of affected patients. Screening tests based on the demonstration of abnormal patterns and concentrations of neuraminic acid-rich compounds are available (27; 66; 67). For these analyses, urine oligosaccharides are placed on thin-layer chromatography plates, separated with one of several suitable solvents, and then stained with either orcinol or resorcinol. A technique for simultaneously examining urine samples for both abnormal oligosaccharides and glycopeptides has been described (63). Urine samples from type I and type II patients (36), as well as from newborns with the congenital form of this disorder (34), yield abnormal patterns with these techniques. The concentrations of the urinary oligosaccharides are correlated with the clinical severity of the affected patients, ie, 3.5 times higher in the infantile type of sialidosis than in the milder, later onset form of the disease (72).
Although the above methods can serve as screening tests, the definitive diagnosis of sialidosis should be based on the determination of neuraminidase activity in fresh cells from the patient (eg, fibroblasts, white blood cells, or cultured amniotic cells) (46; 52). As neuraminidase is unstable, cells or tissues that have been frozen or exposed to prolonged sonication should not be used (74). DNA diagnosis should be routinely performed using Sanger sequencing or next-generation methods (45).
The neuro-ophthalmic manifestations found in sialidosis types I and II have been described in detail (76; 68). Optical coherence tomography, which shows thickened optic nerve fibers and increased reflectivity of the nerve fiber and ganglion cell layers, may be helpful in diagnosing sialidosis type I patients (30; 93; 23). Fundus autofluorescence typically reveals hyperautofluorescent areas surrounding the fovea in patients with sialidosis type I (23).
Brain MRI of a 40-year-old woman with sialidosis type I showed atrophy of the cerebellum, pontine region, cerebral hemispheres, and the corpus callosum (55). The cerebral atrophy is diffuse and progressive (65). The histological and ultrastructural lesions found in this form of the disease have been described in a postmortem study (03).
• No specific therapy has been developed for sialidosis |
Once a diagnosis is established, one should ensure that the precise genetic aspects of the disorder are clearly understood by the immediate family, ie, the parents and other close relatives and siblings who might be carriers for the disorder. The risks for future pregnancies by the same parents (25% for each pregnancy) should be reviewed in detail. Various options, including prenatal diagnosis, should be covered as part of the counseling of the family. The current status of various treatments for the affected proband should be explored. Finally, the family should be made aware of the progressive nature of this disorder to make appropriate decisions.
Enzyme replacement therapy. Preclinical experiments in a mouse model indicate that enzyme replacement therapy can attenuate nephropathy, restore neuraminidase 1 activity in multiple tissues, and reduce lysosomal storage (87; 48). Unfortunately, recombinant neuraminidase 1 is inefficiently transferred across the blood-brain barrier: indeed, even in the mouse model, enzyme replacement therapy restored neuraminidase activity in all tissues except the brain and ears (87; 48). In addition, enzyme replacement therapy in the mouse model triggered a severe immune response against the exogenous enzyme, which has concerning implications for potential future use in sialidosis patients.
Hematopoietic stem cell transplantation. The first use of hematopoietic stem cell transplantation for sialidosis type II was in a child who presented at age 1.5 months with failure to thrive, dysostosis multiplex, and dysmorphic features (48). Diagnosis was made based on urine and protein analyses prior to the onset of clear neurologic impairment. Allogenic hematopoietic stem cell transplantation at age 9 months was accomplished with successful engraftment. Asymptomatic proteinuria was noted at age 25 months; a subsequent renal biopsy showed large cytoplasmic vacuolation producing enlargement of glomerular epithelial and tubular cells. The patient subsequently developed renal failure, requiring hemodialysis at age 6 years and renal transplantation a year later. By age 11 years, the patient remained in poor condition with significant psychomotor retardation and musculoskeletal involvement. Hematopoietic stem cell transplantation did not prevent renal disease or failure.
Gene therapy. Although specific therapy has not been developed yet, the combination of a potent proteosomal inhibitor and an enhancer of the protein-folding machinery in the endoplasmic reticulum increases the level of the mutated sialidase protein in vitro (54). Gene transfer of the chaperone, protective protein/cathepsin A to a mouse model of type I sialidosis that ubiquitously expresses a NEU1 variant carrying a V54M amino acid substitution, showed increased enzyme activity and decreased storage (09). Such an approach may help future patients who have misfolded enzymes due to a missense mutation.
Expected problems include difficult airway, complicated management of associated cardiopathy, and difficult weaning from mechanical ventilation (Gonzalez Gonzalez and Lopez 2006).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Douglas J Lanska MD FAAN MS MSPH
Dr. Lanska of the University of Wisconsin School of Medicine and Public Health and the Medical College of Wisconsin has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Childhood Degenerative & Metabolic Disorders
Apr. 07, 2024
Childhood Degenerative & Metabolic Disorders
Mar. 22, 2024
Developmental Malformations
Mar. 22, 2024
Childhood Degenerative & Metabolic Disorders
Mar. 22, 2024
Childhood Degenerative & Metabolic Disorders
Mar. 15, 2024
Childhood Degenerative & Metabolic Disorders
Feb. 27, 2024
Childhood Degenerative & Metabolic Disorders
Feb. 27, 2024
Childhood Degenerative & Metabolic Disorders
Feb. 27, 2024