

Epilepsy & Seizures
Ambiguous paroxysmal events
Mar. 22, 2024
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Laughter is a behavior that occurs in both emotional and nonemotional contexts. Emotional laughter occurs in the context of mirth (amusement), whereas nonemotional laughter is voluntary and used for social communication, usually in conversation. These two types of laughter are produced by two distinct brain networks. International classifications of epilepsy, seizures, and epilepsy syndromes have been updated in recent years. When laughter is the first feature in a focal seizure, the seizure is termed a “focal emotional seizure with laughing.” This seizure type provides unique insight into the neurobiology of laughter. In this article, the authors review the updated classification of this seizure type, associated etiologies (particularly hypothalamic hamartoma), clinical presentations, differential diagnoses, and current treatments. Newer, noninvasive surgical treatments for seizures due to hypothalamic hamartomas are also reviewed.
• Focal emotional seizures with laughing are characterized by unprovoked and stereotyped laughing or giggling that is present from seizure onset, with or without the accompanying emotion of mirth (amusement). | |
• The differential diagnosis for focal emotional seizures with laughing includes focal seizures in which laughing occurs during the evolution of the seizure (rather than at onset), focal motor seizures causing facial expression change or asymmetry, pseudobulbar affect laughter, gelastic cataplexy, gelastic syncope, and functional neurologic symptom disorder. | |
• Focal emotional seizures with laughing are classically associated with hypothalamic hamartomas but may also be secondary to structural pathology in the temporal or frontal lobes. | |
• When associated with hypothalamic hamartomas, focal emotional seizures with laughing are typically intractable to medical therapy and may be one of the early symptoms of the condition. | |
• The most effective treatment for focal emotional seizures with laughing associated with hypothalamic hamartoma involves removal, ablation, or disconnection of the hamartoma. |
The possibility of sudden emotion as a manifestation of an epileptic seizure has been recognized since the end of the 19th century (70). These emotions were usually negative, and the emotion of fear was most often described. Seizures with laughing were first described by Trousseau (77). Gowers observed emotions "with a cheerful character" as part of a seizure (27). Daly and Mulder coined the term “gelastic epilepsy” from the Greek word gelos, meaning “laughter,” to emphasize the main feature of these seizures (14). Gascon and Lombroso subsequently suggested the following criteria for the diagnosis of "gelastic epilepsy": stereotyped recurrence; absence of external precipitants; concomitance of other manifestations generally accepted as epileptic; presence of interictal or ictal epileptiform discharges on EEG; and absence of conditions in which pathologic laughter might occur (24).
In the updated classification of the epilepsies in 2017 (64), the International League Against Epilepsy (ILAE) limited epilepsy types to four: focal, generalized, combined generalized, and focal and unknown. This rendered the term “gelastic epilepsy” obsolete. In a subsequent operational definition of seizure types in 2017, the ILAE subsequently classified seizures with laughing as the first seizure feature as one type of focal (nonmotor) seizure, a focal emotional seizure—specifically a “focal emotional seizure with laughing” (allowing the synonym of “focal emotional gelastic seizure”) (18; 19). Depending on whether awareness is preserved, the seizure might be a focal aware emotional seizure with laughing or a focal impaired awareness emotional seizure with laughing. The application of the term “emotional” only requires the appearance of having an emotion (“affective manifestation”), not that the subjective emotion is present.
When used as a seizure term, “laughing (gelastic)” is defined as “bursts of laughter or giggling, usually without an appropriate affective tone.” Laughing can occur in a seizure but not as the initial feature. When it appears after another initial seizure feature, it is not used to diagnose the seizure type but is instead a “descriptive” seizure term—a term applied to describe features seen in the seizure evolution. A patient with a seizure commencing with a fast heart rate, and laughing later, would now be considered to have a focal autonomic seizure.
The distinction of whether a symptom or sign is the first feature of a seizure (and used to define the seizure type) or a later feature is important. The first feature can more precisely reflect the location of the seizure onset in the brain, whereas later features reflect seizure spread. Past literature on “gelastic seizures” did not make this distinction, and the term was previously used to encompass seizures with laughing at any time during the seizure. Therefore, the term “focal seizure with laughing” is used when referring to data from past literature on “gelastic seizures” prior to 2017, as the literature did not define whether laughing was present from seizure onset (a focal emotional seizure with laughing) or at any time in the seizure evolution. In the future, new literature may provide more precise information on the entities that result in laughing from clinical seizure onset.
It has previously been debated whether entities with focal seizures with laughing compose an epilepsy syndrome. Arguments against this were raised because the syndrome would largely be defined based on a single seizure symptom (41). In 2022, the ILAE published a classification and definition of epilepsy syndromes, confirming that a syndrome requires a cluster of clinical and EEG features that are often supported by specific etiological findings (80). The etiology-specific epilepsy syndrome “gelastic seizures with hypothalamic hamartoma” was officially recognized (83).
The clinical entities associated with focal seizures with laughing have primarily been described in historical case series and case reports. These reports may have ascertainment bias as they primarily arise from tertiary or epilepsy surgery centers. They do not distinguish whether laughing is at seizure onset or during seizure evolution. Data from the older studies can reflect limited detection of certain types of hypothalamic hamartomas or other structural brain abnormalities due to lower resolution MRI. Some studies can reflect findings from patients who developed a secondary epileptic encephalopathy and who have wider cortical and subcortical abnormalities and epilepsy comorbidities, rather than the primary epilepsy etiology. Older studies can reflect perspectives on the localization values of certain investigations or surgical techniques that have been superseded as imaging and surgical techniques have advanced. This article presents a review of the past literature, bearing in mind these limitations.
• Focal emotional seizures with laughing (focal emotional gelastic seizures) are uncommon and may not be recognized as seizures. Diagnosis may not occur until the patient presents with a different seizure type. |
Focal emotional seizures with laughing are uncommon. The presenting symptoms, physical findings, and clinical evolution seen in patients depend on the location and type of etiology underlying this seizure type.
Gelastic seizures with hypothalamic hamartoma. The clinical presentation of this epilepsy syndrome usually occurs in the first year of life (83). Most present before the age of 5 (51), though later clinical onset (eg, age 13 years) has been reported (11). Larger hamartomas may be detected antenatally.
Focal emotional seizures with laughing (less commonly, focal emotional seizures with crying) are usually the first seizure type. These may not be recognized until another seizure type has occurred. Other seizure types usually emerge later, including focal seizures with impaired awareness, motor features, autonomic features, or focal to bilateral tonic-clonic seizures (83). Focal emotional seizures with laughing are usually frequent and individually brief but occur several times daily or even hourly (83). Focal emotional status epilepticus has occasionally been reported (48; 52). Neurologic examination is normal; signs of endocrinopathy (especially precocious puberty) or Pallister-Hall syndrome should specifically be sought. A wide clinical spectrum exists (46). Some patients have few seizures and good cognitive progress (10), and other patients have significant neurocognitive and psychiatric comorbidities. Some patients can develop a secondary epileptic encephalopathy, with generalized seizure types appearing in association with cognitive regression and worsening behavioral disturbance (83).
A few cases of seizure onset in adult patients with hypothalamic hamartoma have been described. Focal emotional seizures with laughing are less frequent in this age group, and individuals are less affected by cognitive impairment and behavioral problems (46).
Other etiologies. Focal seizures with laughing have also been described in association with nonhypothalamic structural abnormalities of diverse etiology and location in the brain (42). The presenting symptoms, physical findings, and evolution are specific to the underlying etiology. Seizure onset is usually later than in patients with hypothalamic hamartomas. The age of onset can be in adulthood (42) but rarely in the elderly (09). Focal seizures with laughing are usually not as frequent as seen with hypothalamic hamartomas, though status epilepticus has occasionally been reported (57; 43).
Gelastic seizures with hypothalamic hamartoma. Children with focal emotional seizures with laughing due to hypothalamic hamartoma can have diverse neurologic, behavioral, psychiatric, cognitive, and endocrine comorbidities.
Although focal emotional seizures with laughing are the expected main seizure type, other seizure types can develop, and the prevalence of pharmacoresistant seizures is high (10). Some cases of sudden unexpected death in epilepsy (SUDEP) have been reported (10). In pediatric patients, the primary epilepsy due to the hamartoma can be complicated by a secondary epileptic encephalopathy characterized by the emergence of other focal or generalized (epileptic spasms, atypical absence, tonic, atonic, or generalized tonic-clonic) seizures and regression. In this setting, focal seizures that originate in the frontal or temporal lobes based on clinical and EEG data have been described (02; 22; 67). Surgery of the hamartoma, with resolution of the primary hamartoma-related epilepsy, can also resolve these secondary seizure types (08; 02). Some patients with longer exposure to the secondary epileptic encephalopathy have been reported (67). Removal of the hamartoma did not resolve the secondary epileptic encephalopathy, suggesting that if it has been established for some time, it may become independent. This has led to recommendations for shorter times from epilepsy onset to surgical treatment. The two stages of secondary epileptic encephalopathy have been described as “dependent-stage” and “independent” (67; 40).
Psychiatric disorders occur in up to 60% to 80% of pediatric cases, with rage attacks seen in almost half of the cases (10). Patients can also have ADHD, anxiety, and mood disorders. Sleep disorders, including reduced slow-wave sleep, have been reported (10). Cognitive impairment can range from none to severe disability, and greater impairment is associated with earlier onset of epilepsy, higher seizure burden, greater number of antiseizure medications (10), and, in some studies, greater lesion size (20). In a study of a surgical series of 48 patients, half of the patients had executive dysfunction, and 62% had verbal memory dysfunction preoperatively (78). Improvements in cognition have been demonstrated after successful surgery (72), inferring a relationship between seizures and cognition. Early surgery is thought to minimize the overall risk to cognition, but this remains unproven (10) due to the limitations in performing such a study. Impairments can predate the epilepsy, inferring that there can be direct impacts of the hamartoma itself on cognitive and psychiatric brain networks. Around 40% to 50% of patients with hypothalamic hamartomas have precocious puberty, likely due to the inappropriate premature release of hypothalamic gonadotropin-release hormone (49; 30; 10). Other endocrine abnormalities have been reported, including hypogonadism and acromegaly (30). Following surgery of the hypothalamic hamartoma, patients can experience diabetes insidious, growth hormone deficiency, central hypothyroidism, or hypothalamic obesity syndrome (hyperphagia with weight gain) (49).
Other etiologies. The comorbidities and prognosis of focal emotional seizures with laughing due to structural abnormalities in frontal or temporal lobes, or due to other etiologies, is dependent on the underlying etiology. Rates of drug resistance, frequency of seizures, risk of epileptic encephalopathy, and risk of psychiatric comorbidity are generally lower than for focal emotional seizures with laughing due to hypothalamic hamartoma. One literature review of patients with surgically amenable, extrahypothalamic causes of focal seizures with laughing identified 40 patients (20 temporal, 20 frontal pathologies) (33). Those with temporal surgery had a mean age of seizure onset of 14.8 +/- 8.7 years (1.4–31 years); 78% had an Engel class 1 seizure outcome following surgery. Those with frontal pathology had a mean age of seizure onset of 13.6 +/- 13.3 years (0.8–43 years), and 100% had an Engel class 1 seizure outcome following surgery.
A 37-year-old right-handed man with mild intellectual impairment was evaluated for intractable epilepsy. He was born at 34 weeks with no perinatal complications. He had normal or near normal development until the age of 6 months, when he developed infantile spasms with hypsarrhythmia. Following ACTH treatment, he became seizure-free until the age of 8, when initially nocturnal focal emotional seizures with laughing developed. These were characterized by an arousal with giggling. He subsequently developed multiple seizure types, including focal aware autonomic seizures, focal impaired awareness seizures with unilateral automatisms, and focal to bilateral motor seizures. The latter two seizure types occasionally included laughter. Seizures were refractory to over eight different antiseizure medications. Interictal EEG demonstrated mild to moderate background slowing, frequent generalized 2 to 3 Hz sharp-and-wave and spike-and-wave discharges, and left anterior temporal (F7-T3) spikes. Ictal EEG during focal emotional seizures with laughing was characterized by a 1- to 2-second burst of generalized spike-and-wave activity followed by a few seconds of diffuse attenuation. MRI demonstrated a non-enhancing 1 cm lesion in or abutting the left hypothalamus, protruding into third ventricle consistent with a hypothalamic hamartoma. The patient underwent endoscopic resection of the hypothalamic hamartoma via a right frontal horn approach. Seizures became less frequent (decreased from daily to monthly) but persisted for 3 years before abating completely. He was then seizure-free for at least 2 years. Surgery was complicated by memory impairment (initially severe) and transient hyperphagia with significant weight gain.
The biological basis of laughter. Laughter is a motor behavior that occurs in humans in both emotional and nonemotional contexts. Emotional laughter occurs in the context of happiness and amusement (and may not always be voluntarily suppressible), whereas nonemotional laughter can be produced voluntarily and is used in the context of social communication, often in conversation. Voluntary nonemotional laughter has also been termed “pyramidal” laughter. A number of studies have helped define the neural networks involved in these two types of laughter, including electrical stimulation studies and neuroimaging studies. Research on genuine emotional laughter is challenging, due to the spontaneous nature of its production in normal life.
Tractography studies support the finding that emotional laughter is produced by a network involving the pregenual anterior cingulate cortex, the ventral temporal pole, and the ventral striatum/nucleus accumbens, which are interconnected through the anterior cingulate bundle, the accumbofrontal tract, and the uncinate fasciculus and reach the brainstem through the mamillotegmental tract (25). Nonemotional laughter involves the frontal operculum and primary motor cortex and projects to the brainstem motor nuclei through the internal capsule. The two networks interact through the presupplementary motor area, which connects to both the pregenual anterior cingulate cortex and frontal operculum.
In keeping with neuroimaging studies, electrocortical stimulation of the pregenual anterior cingulate cortex (74; 07), the ventral temporal pole (82; 07), the frontal operculum (07), and the presupplementary motor cortex (66) have been able to generate laughter. In some but not all cases, laughter was accompanied by a positive emotion. Stimulation of the ventral striatum/nucleus accumbent (the target of deep brain stimulation for some psychiatric disorders) can produce both smiling and laughing as well as positive emotion (26).
Patients with focal emotional seizures with laughing provide further support for the neural networks involved in laughing. The most well-known pathology associated with focal emotional seizures with laughing is the hypothalamic hamartoma. Stimulation of the hypothalamus itself has never been reported to cause laughing, but stimulation of the hamartoma can produce laughing (47; 39; 34). Stereo-EEG techniques, which allow for intracranial recordings directly from the hypothalamic hamartoma, neighboring hypothalamic structures, and other bilateral cortical areas, have shown that ictal discharges localized in the hamartoma produce seizures with laughing (39; 34; 79). Therefore, it appears that the hamartoma itself, with its intrinsic epileptogenic activity, stimulates laughing via its connections, namely the mamillary bodies and, specifically, the mamillotegmental tract. Further support for this is that hamartomas can be connected via the mamillary bodies or tuber cinereum. Posterior (intra-) hypothalamic hamartomas at the level of the mamillary bodies produce focal emotional seizures with laughing, whereas anterior (para-) hypothalamic hamartomas in the region of the tuber cinereum are usually only associated with central precocious puberty and not epilepsy (39; 30; 10).
Etiologies of focal seizures with laughing. Focal seizures with laughing (whether at onset or during seizure evolution) can be seen in multiple etiologies. Hypothalamic hamartomas are the most well-known etiology; however, diverse structural etiologies in other brain areas can produce laughing during a seizure. In a review of 35 patients with focal seizures with laughing (including giggling) across five tertiary adult and pediatric epilepsy centers in Portugal, 8 (35%) had hypothalamic hamartoma, 16 had other structural abnormalities, and 9 had normal imaging (42). The range of nonhypothalamic hamartoma etiologies included dysplasias, low-grade gliomas, gliosis (eg, due to trauma, metabolic disorders), and Rasmussen syndrome. Nonhamartoma pathologies were more frequently in the temporal lobe, though abnormalities were also located in other lobes or were hemispheric. Although one patient had Niemann-Pick type C, no ictal EEG was acquired, so it is uncertain whether this patient’s events were epileptic as this disorder can also cause gelastic cataplexy. A review of all adult patients in a single tertiary center over 5 years identified 19 patients with focal seizures with laughing (0.8%), of whom one third had hypothalamic hamartomas followed by temporal lobe pathologies (38). Focal seizures with laughing have also been reported in some genetic epilepsies (16; 42) and in patients with normal imaging (42).
Pathogenesis of hypothalamic hamartomas. The cells of origin of the hypothalamic hamartoma, a congenital malformation arising in the region of the ventral hypothalamus during uterine brain development, are unknown. The sonic hedgehog (SHH) pathway in embryonic cells supports cell differentiation. The SHH protein is transcribed, secreted from the cell, and then binds to its target cell via the Patched-1 (PTCH) receptor. This results in inhibition-release activation of Smoothened (SMO) and subsequent activation of GLI transcription factors that control the transcription of SHH target genes (01). Germline (de novo) variants in the SHH transcription factor GLI3 were first found in patients with Pallister-Hall syndrome (36). Subsequently, with analysis of brain tissue from surgeries, lesion-specific somatic pathogenic variants in GLI3, but also OFD1, were found in patients with non-Pallister-Hall syndrome hypothalamic hamartomas (31; 62). OFD1 encodes a protein found in the base of primary cilia (31). With advances in genetic research, plausible molecular genetic causes have been found in more than 50% of patients (28), including single pathogenic variants or bi-allelic (one germline de novo and one somatic) variants. New cilia genes have been implicated (including DYNC2I1, IFT140, SMO). A family with SMO mutations and hypothalamic hamartoma with polydactyly has been reported (61), and the in utero development of hypothalamic hamartomas has now been conceptualized as the consequence of a ciliopathy (28), whereby abnormal primary cilia are unable to process and respond to SHH signaling as usual (01). Histologically, hamartomas have a simple structure that typically consists of abnormally distributed, but cytologically normal, neurons and glia, including fibrillary astrocytes and oligodendrocytes (12; 37).
Gelastic seizures with hypothalamic hamartoma. Mature small neurons are the most abundant cells in hypothalamic hamartomas (12; 37). These largely exist in clusters (nodules) with limited projections. They have an interneuron-like phenotype, with GABA expression and an intrinsic, pacemaker-like, firing activity, but limited interaction outside of their clusters. In contrast, larger neurons, though far fewer in number, often have pyramidal cell bodies with extensive dendritic arborization. It is hypothesized that large numbers of spontaneously firing small interneurons result in paradoxical excitement of large pyramidal neurons, contributing to the final discharges that are propagated, thereby producing the epileptic seizures (37).
Focal emotional seizures with laughter are rare, and determination of their incidence and the frequency of associated etiologies is difficult. Past literature is difficult to interpret as the term “gelastic seizure” was applied to both seizures with laughing at seizure onset and those with laughing in the evolution of the seizure. In 2007, Shahar and colleagues identified 10 cases of patients with focal seizures with laughing in a review of 2400 children referred to any of the pediatric neurology centers in Israel for the evaluation of a potential seizure disorder (0.4% of referred patients) (69). Based on this data, they estimated the prevalence of focal seizures with laughing among all Israeli children to be 0.4 in 100,000. The number of patients who had focal seizures with laughing specifically at seizure onset is likely less. Similarly, Kovac and colleagues estimated the prevalence of focal seizures with laughing to be between 0.4% and 0.8% (38).
In a Swedish study that reviewed only patients with the syndrome of gelastic seizures with hypothalamic hamartoma, the estimated prevalence of this syndrome among Swedish children was 0.5 in 100,000 (05).
There is no known strategy to prevent focal seizures with laughing or their underlying etiologies. In patients with hypothalamic hamartoma, early removal, disconnection, or ablation of the hypothalamic hamartoma is the best way to prevent the development of intractable epilepsy, epileptic encephalopathy, and cognitive and neuropsychiatric deterioration.
The differential diagnosis of a focal emotional seizure with laughing includes other focal seizure types and conditions that are not epileptic.
Focal seizures with laughing. The current use of the term “gelastic seizure” is now reserved for a focal emotional seizure with laughing (synonym: focal emotional gelastic seizure), whereby laughter or giggling is present at seizure onset. Focal seizures in which laughter is present during seizure evolution may have different etiologies. Such seizures are classified by the onset feature (eg, focal autonomic seizure, focal cognitive seizure), and the term “laughing” is then applied as a descriptor of the seizure feature seen during evolution.
Focal seizures with facial motor (expression) change. The act of laughing is a motor activity that engages specific muscles of the face, larynx, and chest (including diaphragm) in a specific stereotyped fashion, with or without the accompaniment of mirth. Focal seizures from diverse brain regions can be accompanied by a change in facial expression that is distinct from laughing and giggling. Seizures in which there is asymmetric tonic (or tonic-clonic) elevation of the angle of the mouth or “ictal smile,” a facial expression that is similar to the patient’s normal smile (45), are not considered focal emotional seizures with laughing. Similarly, focal seizures with “ictal pouting” or “chapeau de gendarme” (73) are considered distinct from focal emotional seizures with crying.
Pseudobulbar affect. Emotional disinhibition in patients with pseudobulbar affect can lead to pathologic laughter or crying. As opposed to focal emotional seizures with laughing in which laughter is unprovoked and unpredictable, pseudobulbar affect laughter usually occurs in response to an external trigger and is exaggerated and mood-congruent, but inappropriate, in its intensity. Pseudobulbar affect can be seen following strokes and in association with many neurodegenerative conditions, such as multiple sclerosis, amyotrophic lateral sclerosis, and Alzheimer disease (65).
Gelastic cataplexy. Strong emotions and laughter can trigger atonic events in patients with cataplexy, and these laughter-induced events can be mistaken for seizures. Gelastic cataplexy (cataplexy induced by laughter or emotions associated with laughter) is a relatively common symptom early in the course of late-infantile and juvenile Niemann-Pick C (54; 53; 55).
Gelastic syncope. Vigorous but appropriate laughter has been noted to induce vasovagal syncope in a handful of patients and is a rare subtype of Valsalva-induced syncope (04; 63).
Functional neurologic symptom disorder. Functional neurologic symptom disorder can present with a wide range of symptoms, including events that mimic epilepsy (known as nonepileptic seizures). Patients with recurring bouts of laughing initially diagnosed and treated as epileptic have been reported (44). Nonepileptic seizures can have a psychogenic basis. A notable epidemic of psychogenic nonepileptic laughing occurred as a mass psychogenic illness in Tanzania in 1962 (58). It began at a mission-run boarding school for girls and affected 95 of 159 pupils aged 12 to 18 years, before spreading more widely to affect thousands of individuals. Symptoms lasted a few hours to 16 days, and the epidemic died out 6 to 18 months after it started.
Pallister-Hall syndrome is a rare disorder with autosomal dominant inheritance and complete penetrance (Biesecker and Graham 1996). Most affected individuals have a hypothalamic hamartoma, polydactyly, and an asymptomatic bifid epiglottis. More severely affected individuals may have dysplastic nails, hypopituitarism, growth hormone deficiency, imperforate anus, or genital hypoplasia.
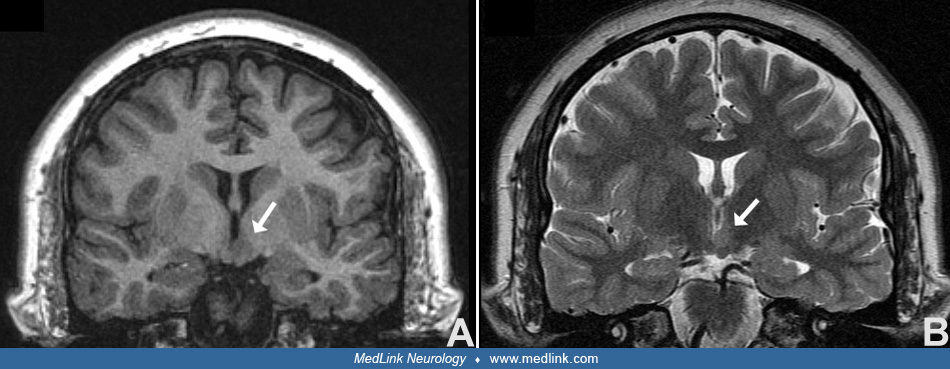
Gelastic seizures with hypothalamic hamartoma. The diagnosis of hypothalamic hamartoma is made with high-resolution MRI, which should include coronal T2 fast-spin echo sequences with thin slices and no inter-slice gap through the hypothalamus so that any hamartoma and its attachment can be visualized. Imaging characteristics of hypothalamic hamartomas include a pedunculated or sessile mass of grey matter signal intensity in the region of the hypothalamus, which is attached to the mammillary body or the tuber cinereum. If large, the mass may distort or incorporate the mammillary bodies, displace the columns of the fornix antero-laterally, and have variable extension below the third ventricle or project into the suprasellar cistern. In general, hypothalamic hamartomas are the only developmental abnormality seen on imaging (02).
Video-EEG should also be considered in patients with paroxysmal laughter. This can help confirm the diagnosis of focal emotional seizures with laughing based on ictal semiology seen on video as well as help screen for other seizure types. It is important to keep in mind, however, that due to the depth of the lesion in patients with a hypothalamic hamartoma, scalp EEG recordings may be normal or may only show slowing or discharges that do not discretely localize the seizure onset. Even ictal EEG may not show any change or may show only subtle attenuation or rhythmic slowing. In a study of 133 patients with hypothalamic hamartomas, 56% of patients had no ictal EEG change during focal seizures with laughing (76). This same study also demonstrated that video-EEG monitoring in patients with hypothalamic hamartomas did not improve surgical outcome. In the past, the use of EEG for localization could possibly lead to unnecessary frontal and temporal lobe resections, without control of the epilepsy (08). Therefore, current recommendations are that ictal and interictal EEG be used to confirm the diagnosis of focal seizures with laughing, however, not with the expectation of ictal localization for surgery when there is imaging confirmation of a hypothalamic hamartoma (10). When the epilepsy has evolved to a secondary epileptic encephalopathy, EEG may show interictal slowing and abundant interictal epileptiform abnormalities, in addition to capturing generalized seizure types.
In the past, dipole analyses of scalp EEG, MEG, diffusion tensor imaging, interictal and ictal SPECT, PET, fMRI, and intracranial EEG have been explored for their ictal localization value as part of the surgical work-up for hypothalamic hamartomas. However, the 2014 Florence consensus of the Paediatric Epilepsy Surgery Task Force of the ILAE recommends ictal/interictal EEG and MRI and specifies that PET and SPECT are not recommended given false results (32). In general, EEG and MRI are currently the main tools for the presurgical evaluation of gelastic seizures with hypothalamic hamartoma (10).
Patients with confirmed hypothalamic hamartomas should also undergo a complete endocrinological workup for precocious puberty. This includes measurement of height, weight, and height velocity as well as pubertal staging and bone age. Gonadotropin assays may also be indicated.
Other etiologies. High-resolution MRI with thin slice volumetric T1-weighted images, axial and coronal T2-weighted images, and FLAIR images are important in the diagnostic evaluation of patients with focal seizures with laughing who do not have a hypothalamic hamartoma. A structural pathology is most commonly found in the temporal or frontal lobes but can also be in any lobe or be hemispheric (42). Focal seizures with laughing have also been reported in some genetic epilepsies; therefore, genetic evaluation may be considered if MR imaging is negative (16; 42).
Gelastic seizures with hypothalamic hamartoma. Gelastic seizures associated with hypothalamic hamartoma are typically refractory to antiseizure medications (05; 10). Several surgical techniques have been used to treat the epilepsy, depending on the location and size of the hamartoma and local expertise. Initially, open or endoscopic surgeries were used. Minimally invasive techniques have since been developed. The Delalande classification of hamartomas can inform the surgical technique and approach, such as an approach from above or below the third ventricle (15). The horizontal attachment is inferior to the floor of the third ventricle in type I. The vertical attachment to the wall of the third ventricle is above the floor in type II. Type III combines types I and II, and type IV consists of giant hamartomas with a volume of 8 cm3 or larger. Seizure control can be achieved with removal, ablation, or disconnection of the hamartoma.
A transcallosal anterior interforniceal approach was initially utilized for open surgical resection of hamartomas (60). Subsequently, using frameless stereotaxy, sessile tumors less than 1.5 cm in diameter were endoscopically resected via a transcortical transventricular route through the foramen of Monro (21). Both of these surgical procedures were associated with good outcomes when performed by experienced surgeons in selected patients. Case series from the Barrow Neurological Institute reported a substantial (more than 90%) seizure reduction in 70% to 88% of cases for endoscopic and transcallosal resection, respectively (49; 50). However, a significant complication of these procedures was transient or permanent memory loss due to injury to the fornices, mamillary bodies, or their connections. In the Barrow Neurological Institute series, 8% of patients had permanent memory impairment after surgery (49; 50). Other serious complications included third nerve palsies, strokes, hyperphagia, and endocrine abnormalities, particularly diabetes insipidus.
Large series reporting endoscopic disconnection have also been published (56; 17). A large series of more than 100 children with stereotactic endoscopic disconnection by Ferrand-Sorbets and colleagues reported seizure-free rates of 57% (17). The rate was 49% in a previous report by Procaccini and colleagues (56). Both authors reported that endoscopic disconnection was more effective for patients with vertically inserted hamartomas above the floor of the third ventricle (Delalante type II); this subgroup had a 69% seizure-free outcome in the Ferrand-Sorbets study. A shorter duration of epilepsy was associated with better surgical outcome. The overall rate of permanent complication was 8% in the Ferrand-Sorbets study and included memory deficits and hyperphagia.
Calisto and colleagues compared endoscopic disconnection by monopolar coagulation to disconnection by thulium laser in a small group of patients (06). At 1-year follow-up, 42% of each group achieved Engel class I, Ia, or Ib (5 of 12 with coagulation disconnection, 3 of 7 with laser disconnection). Acute postoperative complications included oculomotor deficits, electrolyte alterations, memory deficits, and mutism in the monopolar coagulation disconnection cases. Among the laser disconnection cases, one patient had drowsiness and a possible subcortical infarct on CT, and another patient had memory deficits. The authors reported that there were no persistent complications by 1-year follow-up.
Radiosurgery is associated with a low risk of complications; however, it typically takes 1 to 2 years to reach full effect and is, therefore, best reserved for smaller hamartomas and patients without epileptic encephalopathy. In a large series of gamma-knife surgeries, rates of Engel class I outcomes were around 40% (59; 29). Interstitial radiotherapy has been associated with similar Engel class I outcome rates, but some patients can develop edema due to radiation-induced capillary leakage (68).
Minimally invasive techniques, including MRI-guided stereotactic radiofrequency thermocoagulation (SRT), laser interstitial thermal therapy (LITT), and focused ultrasound (FUS), have been introduced. Although SRT, LITT, and FUS are less invasive, some patients may need to undergo repeated treatments to achieve the best outcome. Kameyama and colleagues reported excellent seizure outcomes (86% of patients were free of focal seizures with laughing, 71% were free of all seizures) in a study of 140 SRT procedures in 100 patients with hypothalamic hamartoma (15 of 100 were giant hamartomas) (35). The majority of patients had improved cognition and resolution of behavioral problems after surgery. Shirozu and colleagues reported the outcomes of 206 SRT procedures in 150 patients with hypothalamic hamartoma (71). Forty-three patients (28.7%) underwent repeat SRTs: 33 (22.0%) patients had two SRTs; 7 (4.7%) had three SRTs; and 3 (2.0%) had four SRTs. Overall, 73% of patients became seizure free for all seizure types, and 90% were free of focal seizures with laughing. Freedom from other seizure types was achieved in 90 of 122 (73.8%) patients; however, the authors noted that this was achieved at the first SRT in all but one patient and that re-SRT may not be additionally effective for other seizure types. Complications of SRT included hyponatremia, hyperphagia, hyperthermia, memory disturbance, and disturbance of consciousness. Transient hyponatremia was more frequent after the first SRT. Persistent complications were more frequently seen after repeat SRTs, including memory disturbance, hypopituitarism, hemiparesis, and diabetes insipidus.
In contrast to SRT, LITT allows real-time MR visualization of the thermal destruction target and dose adjustment, as necessary. Curry and colleagues reported the outcomes of a large series of 71 pediatric patients who had LITT; 25% had failed other surgical and radiosurgical techniques (13). Of these, 93% were free of focal seizures with laughing at 1 year. Fourteen of the 71 patients had a second procedure, and 2 of 71 had three ablations. The authors noted that 21 of the 71 patients had other seizure types, and these were lessened by ablation and controlled with medication. Complications were uncommon and included worsening of diabetes insipidus, permanent short-term memory deficit, and transient worsening of seizures. Gadgil and colleagues retrospectively reviewed 58 pediatric cases who had LITT; 18 (31%) had prior surgery, including open resection and gamma knife (23). Repeat LITT was performed in 16 of 58 patients (12 had two LITT ablations, 4 had three ablations). Transient complications included abnormal sodium levels (n = 4), short-term memory dysfunction (n = 4), and seizure worsening (n=4). Only one patient had permanent memory deficits. The majority (81%) of patients were free of focal seizures with laughing at 6 months or more of follow-up.
MR-guided FUS thermoablation is a newer, minimally invasive technique with the potential for use in hypothalamic hamartoma surgery. However, its use has only been described in single cases or small case series to date (81; 75).
Other etiologies. The treatment approach to nonhypothalamic hamartoma focal seizures with laughing depends on the underlying etiology. Initial treatment is usually with antiseizure medication. Patients with tumors and patients who fail to achieve seizure control with medication who have a surgically amenable structural cause may be considered for surgery (38).
Early surgery can mitigate the intractable epilepsy and cognitive and neuropsychiatric comorbidities in patients with gelastic seizures with hypothalamic hamartoma. The development of minimally invasive surgical techniques has provided an alternative to open surgery. Treatment-related complications are mostly transient and tolerable. Seizure freedom, including from all seizure types, is achievable in the majority of patients.
For patients with other etiologies, outcome is dependent on the etiology and location. Seizures may remit, or seizure freedom may be possible with medications, for some patients. For others, epilepsy surgery may be an alternative, with potential for good outcome.
Women with focal seizures with laughing can carry healthy pregnancies. The management of women of reproductive age with this seizure type should be similar to that of women with other types of epilepsy. Antiepileptic drugs with a relatively low risk of teratogenesis should be used as first-line drugs, and folic acid (0.4 to 4 mg daily) should be recommended. Epilepsy surgery should be considered well in advance of attempts to conceive in women with intractable focal seizures with laughing.
Focal seizures with laughing require no specific precautions for anesthesia.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Kate Riney MB BCh BAO PhD
Dr. Kate Riney of Queensland Children's Hospital received honorariums for educational symposia, advisory boards, and consultancy work from Eisai, LivaNova, Novartis, and UCB Australia Ltd. Dr. Riney is a Zogenix shareholder.
See ProfileMatthew Lynch MD
Dr. Lynch of Queensland Children’s Hospital has no relevant financial relationships to disclose.
See Profile
Solomon L Moshé MD
Dr. Moshé of Albert Einstein College of Medicine has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Epilepsy & Seizures
Mar. 22, 2024
Epilepsy & Seizures
Mar. 20, 2024
Epilepsy & Seizures
Mar. 07, 2024
Epilepsy & Seizures
Mar. 06, 2024
Epilepsy & Seizures
Mar. 05, 2024
Epilepsy & Seizures
Mar. 05, 2024
Epilepsy & Seizures
Mar. 04, 2024
Epilepsy & Seizures
Feb. 29, 2024