Developmental Malformations
Walker-Warburg syndrome
Apr. 14, 2024
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Although first recorded by Wagner in 1863 (150), polymyositis became a recognized clinical entity 75 years later when Walton and Adams published a remarkable monograph titled Polymyositis (151). These authors defined the term “polymyositis” as an all-inclusive designation of a group of myopathies characterized by a single basic disease process related to "other collagen" diseases or arising as the result of "hypersensitivity response to allergic inflammation." Today, described by neurologists as a neuromuscular disorder, polymyositis is cared for not only by neurologists but also by rheumatologists, who approach the illness as in a setting of other rheumatic diseases. Polymyositis is a rare disorder that, by all accounts, is the most overdiagnosed acquired myopathy and, although now unclear whether it exists as a distinct entity, it is discussed not only for historical reasons but also because it is still diagnosed by various practitioners. Although this author believes that it does not exist as a single entity, the evolution and now almost extinction of what was considered as polymyositis 3 to 4 decades ago, has taught us a lot about muscle immunopathology. It is discussed here in the context of two similar but now distinct and more frequent entities, which have evolved in the last two decades, namely necrotizing autoimmune myositis and overlap myositis, while emphasizing its distinction from inclusion body myositis.
The study of polymyositis requires a scholarly review of the neurologic examination, muscle histopathology, immunopathology, and biochemistry to ensure that toxic, metabolic, or mitochondrial muscle diseases are not missed and that the two more common entities, the inclusion body myositis and the necrotizing autoimmune myositis, are not overlooked (12; 73; 29; 30; 36; 37; 39; 46; 47; 49; 132; 40; 114; 46; 46).
Polymyositis, as traditionally had been viewed and with the reservations discussed above, has no unique clinical features, and its diagnosis is one of exclusion (29; 30; 36; 37; 39; 49; 132; 40; 46). Polymyositis is best defined as an inflammatory myopathy of subacute onset (weeks to months) and steady progression occurring in adults not presenting the following symptoms: (1) rash; (2) involvement of eye and facial muscles; (3) family history of a neuromuscular disease; (4) endocrinopathy; (5) history of exposure to myotoxic drugs or toxins; and (6) neurogenic or dystrophic disorder, metabolic myopathy, or inclusion body myositis, or necrotizing autoimmune myositis, as determined by muscle enzyme histochemistry and biochemistry. Unlike dermatomyositis, in which the rash secures early recognition, the actual onset of polymyositis cannot be easily determined, and the disease may exist for several months before the patient seeks medical advice.
Patients with polymyositis commonly present with a myopathy characterized by proximal and often symmetric muscle weakness manifested as difficulty performing tasks requiring the use of proximal muscles. Symptoms develop relatively slowly (weeks to months), occasionally insidiously, but rarely acutely (30; 36; 37; 39; 49; 46). Symptoms include difficulty getting up from a chair, climbing steps, stepping onto a curb, lifting objects, and combing hair. In contrast, fine-motor movements that depend on the strength of distal muscles, such as buttoning a shirt, sewing, knitting, or writing, are affected late in the disease. If these muscles are affected early and the patient complains of frequent falling, then the diagnosis of inclusion body myositis is more likely (30; 36; 37; 39; 43; 46; 47; 49; 40; 46). Ocular muscles remain normal even in advanced untreated cases, and if these muscles are affected, then the diagnosis of inflammatory myopathy should be in doubt. Facial muscles also remain normal except for rare advanced cases; in contrast, facial muscles are often affected in patients with inclusion body myositis. The pharyngeal and neck-extensor muscles can be involved, causing dysphagia or premature fatigue and difficulty in holding up the head. In advanced cases and rarely in acute cases, as in necrotizing autoimmune myositis, respiratory muscles may also be affected. Severe weakness is almost always associated with muscular wasting. Sensation remains normal. The tendon reflexes are preserved, but may be absent in severely weakened or atrophied muscles. Myalgia and muscle tenderness may occur in some patients, usually early in the disease and especially when polymyositis occurs in the setting of a connective tissue disorder. When the fascia is affected, as in fasciitis, there is diffuse muscle induration and tenderness but minimal muscle weakness.
Primary cardiac abnormalities due to myocarditis may be present in a small number of patients, mainly manifested as atrioventricular conduction defects, tachyarrhythmias, low-ejection fraction, dilated cardiomyopathy, or congestive heart failure. Cardiac abnormalities, however, appear overall to be secondary to hypertension associated with long-term steroid use or to pulmonary hypertension related to interstitial lung disease (30; 38; 49; 40). Interstitial lung disease may occur in up to 10% to 40% of these patients, who do not have polymyositis but the antisynthetase syndrome with anti-Jo-1 antibodies or antibodies to various ribonucleoproteins (29; 30; 38; 49; 55; 40; 01; 144). The prevalence of interstitial lung disease, best detected with high-resolution CT, is up to 70% among patients with anti-Jo-1 antibodies. Interstitial lung disease, however, may rarely be iatrogenic due to methotrexate use, a known but very rare cause of pneumonitis (“methotrexate-pneumonitis”). General systemic disturbances, such as fever, malaise, weight loss, arthralgia, and Raynaud phenomenon denote antisynthetase syndrome or overlap myositis but may occur when polymyositis is associated with a connective tissue disorder.
Polymyositis is a rare disease seen exclusively in adults. The disease is extremely rare in childhood, and if a diagnosis is made in patients below the age of 16 years, then a careful review is needed to exclude another disease, especially certain dystrophies that may be associated with endomysial inflammation (40; 49). Most patients still diagnosed as having polymyositis actually have another disease—either inclusions body myositis (described in a separate article), necrotizing autoimmune myositis (described below), or antisynthetase syndrome (most often anti-Jo-1 syndrome; also described below). The rarity of polymyositis has now become a reality; it is not included, and correctly so, in the newest classifications of inflammatory myopathies. A previously reported cohort of 255 patients from the United Kingdom, 37 of whom were classified some years ago as having polymyositis based on Eular/ACR criteria, highlights the rarity of the disease (109). A re-review of those patients did not show any convincing cases of true polymyositis, with the authors concluding “is there anything left?” Accordingly, as polymyositis is now correctly being questioned as a distinct clinicopathologic entity, a view we have been advocating for years, the inflammatory myopathies can be safely classified into four major subgroups: necrotizing autoimmune myositis, antisynthetase syndrome overlap myositis, dermatomyositis, and inclusion body myositis (144).
The natural history of what had been considered as polymyositis is unknown because of the almost universal administration of early steroid treatment and the disease rarity. The mortality rates reported 30 to 40 years ago are outdated (30; 132). Several cases of probable polymyositis, referred to our institution as definite polymyositis, have been diagnosed as necrotizing autoimmune myositis, inclusion body myositis, toxic myopathy, antisynthetase syndrome, or inflammatory dystrophy on careful clinical examination and repeated muscle biopsy. In general, older age, interstitial lung disease, and frequent pneumonias due to esophageal dysfunction are factors associated with poor prognosis. Some patients still do not adequately respond to therapies and remain disabled; whether in these circumstances the disease was a bona fide polymyositis or another disorder erroneously diagnosed as polymyositis is unclear.
For years we have been advocating that when treatment of polymyositis is unsuccessful, the patient should be reevaluated and the muscle-biopsy specimen reexamined. A second biopsy might be considered to affirm the correct diagnosis. As mentioned, disorders most commonly mistaken for polymyositis include: inclusion body myositis; sporadic limb-girdle muscular dystrophy, which is suspected when the disease has a slow onset and progression, and the muscle biopsy specimen does not show primary inflammatory features; metabolic myopathy (eg, phosphorylase deficiency); endocrinopathy; and neurogenic muscular atrophies.
In cases of eosinophilic myositis associated with eosinophilic infiltration of an organ (heart, lung), the prognosis usually depends on the severity of cardiac or pulmonary involvement. Such cases may not respond to immunosuppression and usually carry an unfavorable prognosis (105). These patients are also predisposed to develop hematological malignancies (75; 76). Otherwise, uncomplicated cases of eosinophilic myositis usually respond to nonsteroidal anti-inflammatory or steroid medications, either with resolution or marked improvement. About one third of these patients, however, develop recurrences when these medications are discontinued (99) and need prolonged immunosuppression. Macrophagic myofasciitis also responds to steroids.
Polymyositis appears to be a syndrome of diverse causes that often occurs in association with systemic autoimmune diseases, viral infections, or connective tissue disorders. As a stand-alone clinical entity, polymyositis is a very uncommon disorder, if it exists. Other than D-penicillamine, tacrolimus (149), and immune checkpoint inhibitors, the other potential myotoxic drugs such as zidovudine, emetine, chloroquine, or ipecac do not cause polymyositis. Instead, myotoxic drugs elicit a toxic noninflammatory myopathy that is histologically different (or histologically distinct) from polymyositis and do not require immunosuppressive therapy (45). Certain cholesterol-lowering drugs may rarely exert an immune-modulating effect and have been thought to possibly trigger histologic features of autoimmune myositis responding to immunotherapies (45). As discussed below, however, statins can rarely cause acute rhabdomyolysis or myalgia within the first six weeks of therapy initiation and, although they have been implicated in the cause of necrotizing autoimmune myopathies, their causative role in patients who develop such myositis after a chronic statin use remains still uncertain. Several animal parasites, such as protozoa (Toxoplasma, Trypanosoma), cestodes (cysticerci), and nematodes (Trichinae), may produce a focal or diffuse inflammatory myopathy known as “parasitic polymyositis” (12). In the tropics, a suppurative myositis known as “tropical polymyositis” or “pyomyositis” may be produced by Staphylococcus aureus, Yersinia, Streptococcus, or other anaerobes. Pyomyositis, previously rare in the West, had been seen in patients with AIDS early in the epidemic when no treatment was available (78; 50). Certain bacteria, such as Borrelia burgdorferi of Lyme disease and Legionella pneumophila of Legionnaire disease (11; 152), may infrequently be the cause of polymyositis.
Polymyositis can be seen in patients with common connective tissue disorders such as systemic lupus erythematosus, Sjögren syndrome, or rheumatoid arthritis. In contrast, systemic sclerosis and mixed connective tissue diseases are more often associated with dermatomyositis than with polymyositis (30; 38; 39; 41; 49; 132). Patients with systemic sclerosis and mixed connective tissue disease may develop a rare but distinct myositis subtype identified as brachio-cervical inflammatory myopathy, characterized by weakness of proximal muscles of the upper limbs and cervical flexor and extensor muscles (128; 77).
Polymyositis can also be rarely manifested during the course of another systemic autoimmune disease, such as Crohn disease, vasculitis, sarcoidosis, primary biliary cirrhosis, adult celiac disease, chronic graft-versus-host disease, discoid lupus, ankylosing spondylitis, Behcet disease, myasthenia gravis, acne fulminans, dermatitis herpetiformis, psoriasis, Hashimoto disease, granulomatous diseases, agammaglobulinemia, hypereosinophilic syndrome, Lyme disease, Kawasaki disease, autoimmune thrombocytopenia, hypergammaglobulinemic purpura, hereditary complement deficiency, IgA deficiency, and AIDS (12; 29; 30; 50).
Among viruses, HIV and HTLV-I are the retroviruses more clearly associated with polymyositis and inclusion body myositis (62; 32; 63). Claims that other viruses, such as enteroviruses, can be causally connected with polymyositis are unproven (30; 41; 106). In some cases, however, polymyositis or necrotizing autoimmune myositis (NAM) has followed an acute viral illness. The same applies with SARS-Cov-19 during the present pandemic. A number of cases have been reported in connection with COVID-19, but few have been fully substantiated (55). Autopsy studies from patients hospitalized with severe pulmonary and systemic complications have shown some nonspecific endomysial inflammatory features mostly related to cytokine release and the strong inflammatory state triggered by the coronavirus; however, no convincing signs of SARS-Cov-19 have been detected in the muscle tissue and, like the other viruses, there is no evidence that SARS-Cov can infect the muscle fibers (55; 56; 57; 58).
Prior claims that cancer is considered to have increased association with polymyositis (139) or plays a role in its pathogenesis have not been substantiated (30). Cancer is, however, more frequently associated with dermatomyositis and necrotizing autoimmune myositis (04). Some reports suggesting patients with increased risk of cancer for all inflammatory myopathies have not taken into account the subtype of myopathy and other independent cancer-associated risk factors, such as long-term immunotherapy and increasing age (21; 85).
Eosinophilic myositis. This is a rare form of myositis characterized by eosinophilia in the peripheral blood and eosinophilic infiltrations in the endomysial tissue. The term “eosinophilic myositis” was coined by Layzer and colleagues in 1977 to describe cases in which eosinophils were the most prominent inflammatory cells within the endomysial infiltrate (105). Some of these patients may have involvement of other organs (heart, lungs, bone marrow, or skin) at some point in the course of their disease. Eosinophilic myositis can be seen in the context of parasitic infections, vasculitis (especially Churg-Strauss syndrome), mixed connective tissue disease, L-tryptophan-induced eosinophilia-myalgia syndrome (84; 92), toxic oil syndrome, or idiopathic hypereosinophilic syndrome (137). When the pathology is predominant in the fascia, the disease presents with skin induration and pain and is often referred to as “eosinophilic fasciitis” (Shulman syndrome) (138). At times, the skin is spared and the pathology predominates in the perimysium; such cases are referred to as “eosinophilic perimyositis” (91; 74; 104; 137; 147). Accordingly, an eosinophilic inflammatory muscle disease can present either as polymyositis with proximal muscle weakness or, most often, as fasciitis clinically manifesting as focal or generalized myalgia, muscle induration, tenderness, and cramps, with various involvements of the skin and the subcutaneous tissue. Eosinophilic myositis may overlap with hypereosinophilic syndrome, eosinophilic fasciitis, and eosinophilic perimyositis, implying a continuum of inflammatory involvement that extends from the fascia into the perimysium and endomysium. Several cases of eosinophilic myositis and fasciitis have been associated with drugs, such as tranilast (an antiasthmatic), phenobarbitol (101; 09), or contaminated L-tryptophan (84; 136; 92). Mutations in the calpain gene have also been associated with eosinophilic myositis, which often presents in young adults as hyperCKemia and minimal muscle weakness (102).
Macrophagic myofasciitis. This type of fasciitis seems to be a distinctive disorder identified years ago in French patients presenting with myalgias, early fatigue, and mild muscle weakness (80; 51). Muscle biopsy revealed pronounced infiltration of the connective tissue around the muscle (epimysium, perimysium, and perifascicular endomysium) by sheets of periodic acid-Schiff-positive macrophages and occasional CD8+ T cells. Creatine kinase or erythrocyte sedimentation may be at times elevated. Most patients respond to glucocorticoid therapy, and the overall prognosis is favorable. The pathology was almost always seen at the sites of previous vaccinations, even several months later, and had been linked almost exclusively in France to a type of aluminum component used as a substrate for preparation of their vaccines. Some evidence suggests that examination of the fascia along with the muscle may enhance the diagnostic yield for an overlapping myositis (108).
Necrotizing autoimmune myositis (NAM), also referred to as immune-mediated necrotizing myopathy (IMNM). Necrotizing autoimmune myositis is turning out to be the commonest subset among all the inflammatory muscle diseases, if inclusion-body myositis is excluded, based on the number of cases identified each year in busy referred clinics and reported in large series; accordingly, it should not be missed because it is potentially treatable if identified early (49; 154). NAM still, however, remains an overlooked entity, often misdiagnosed as polymyositis. The patients present with high CK, in the thousands; moderate to severe muscle weakness of acute or subacute onset; and histological features of muscle fiber necrosis mediated by macrophages as the main effector cell. There are no T-cell infiltrates or ubiquitous MHC-I expression as seen in polymyositis and inclusion body myositis, but the MHC-I expression is spotty (46; 47; 48; 49). In a number of patients, the muscle biopsies show deposition of complement on necrotic fibers and at times the blood vessels (83; 25; 01). Up to 65% of these patients have antibodies against signal recognition particles (SRP) or against a 100-kd autoantigen identified as 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR) (83; 25; 111).
The cause of NAM is multifactorial. A higher incidence of cancer has been observed in several cases (04) whereas others developed NAM during therapy with checkpoint inhibitors for advanced malignancies (96; 53; 93); some patients have an active viral infection (ie, HIV) and others may have a smoldering underlying autoimmune process, but still others have no other disease or apparent exposure to exogenous agents (48; 49). Some cases, however, have a statin exposure, which is implicated as a cause based on the assumption that statins, in addition to rarely causing a self-limited toxic myopathy, can also induce an autoimmune necrotizing myositis that persists after statin withdrawal (47; 111). Although the statin connection is widely accepted, a statin-triggered muscle autoimmunity remains still dubious among some critics (49; 31; 52; 53; 54; 05). NAM is probably an antibody-mediated disease, as implied by the presence of specific antibodies and complement with endomysial macrophages, which have been interpreted to represent an antibody-dependent cell-mediated cytotoxicity (ADCC) process (25; 01). This hypothesis, however, as discussed, lacks specificity because these antibodies are not pathogenic, whereas complement deposition and MHC-I expression are always associated with muscle fiber necrosis due to any cause, being ubiquitous in necrotic muscle fibers in all patients with nonimmune myopathies, such as muscular dystrophies (71; 97; 52; 53; 54; 55; 56). It has been proposed that NAM may differ from nonimmune-mediated toxic or dystrophic necrotizing myopathies by a strong expression of type 1 helper T cell (T1)/classically activated macrophage M1 and elevated level of interferon-γ, tumor necrosis factor-α, IL-12, and STAT1 levels in the muscle tissue (129).
The connection of necrotizing autoimmune myositis with statins has been based on a retrospective–over many years–historical chart review and the observation that anti-HMGCR antibodies were present in a number of these patients (111); because statins upregulate the expression of HMGCR in cultured cells, and HMGCR is the target of action of statins, this is a logical assumption. In this retrospective chart review, anti-HMGCR antibodies were detected in 45 of 750 patients (6%) with all types of myopathies, but among patients older than 50 years of age, 92.3% had at some point taken statins (111). This assumption did not, however, establish a causative role of statins and did not take into account that 25% of Americans above 40 years take statins. Subsequent studies from many centers throughout the world have now shown that anti-HMGCR autoantibodies are more often seen in statin-naïve patients with autoimmune necrotizing myositis (03; 69; 120; 02; 05; 98; 153), challenging a strict statin connection. Considering that NAM is now one of the commonest inflammatory myopathies and more than 25% of Americans above 40 years take statins, the association between statins and NAM needs to be objectively viewed as to whether it represents a chance phenomenon (53; 54; 55; 56). Some authors have proposed that the term “statin myopathy” should not be used (05) because only a minority of NAM patients had statin exposure and, most importantly, they develop NAM many years after statin initiation that makes their causative role dubious. These reservations are of general practical significance because in the author’s experience many patients with preexisting myopathic weakness, even genetic myopathies, or even just hyperCKemias, are deprived of statins by their primary care physicians fearing worsening of muscle weakness.
Because statins have immunomodulatory potential in autoimmune diseases, an interesting controlled study showed that simvastatin 20 mg/kg in an experimental mouse model of autoimmune myositis decreased the severity of the disease and significantly improved muscle strength and histopathology compared to placebo-treated mice (110). A trend towards higher serum Th17 percentage population was also noted in statin-treated and improved mice. These authors concluded that simvastatin can be a candidate as a disease-modifying agent in inflammatory myopathies.
“Antisynthetase syndrome” or “overlap myositis” or “anti-Jo-1 syndrome”. Anti-Jo-1 antibodies, the commonest among the anti-antisynthetases, directed against the histidyl-transfer RNA synthetase, define the now distinct antisynthetase or “overlap myositis” syndrome, characterized by: (a) myositis with prominent pathologic changes at the periphery of the fascicles and the perimysial connective tissue in the form of necrotizing perimysial and perifascicular myositis with actin myonuclear inclusions (115; 140); (b) interstitial lung disease; (c) arthritis; (d) Raynaud phenomenon; (e) fever; and (f) mechanic’s hands. The association of this clinicopathological phenotype with anti-Jo-1 antibodies appears strong in defining this distinct myositis subset, even if the Jo-1 antibodies are not pathogenic.
Presence of autoantibodies. Various autoantibodies against nuclear (antinuclear antibodies) or cytoplasmic antigens are found in as many as 20% of patients with inflammatory myopathies. The antibodies to cytoplasmic antigens are directed against ribonucleoproteins that are involved in translation and protein synthesis and include various synthetases and translation factors, including Jo-1, PL-7, PL-12, OJ, EJ, PMS1, and PMS2 (145; 94; 82; 02). Of these, anti-Jo-1, directed against the histidyl-transfer RNA synthetase, accounts for 75% of all the antisynthetases and is clinically useful in defining the distinct subset mentioned above. Overall, however, these antibodies may not be muscle-specific or pathogenic because: (1) they are directed against ubiquitous intracellular targets; (2) their exact myopathogenicity has not been defined; (3) they are almost always associated with interstitial lung disease or seen in patients with interstitial lung disease who do not have active myositis; and (4) they also can occur in inclusion body myositis and dermatomyositis, despite the clinical and immunopathologic differences of these disorders. Other autoantibodies are seen when myositis is associated with connective tissue disease and are the hallmarks of the other disease, such as Ro/SSA, La/SSB, Ki/SL in Sjögren; or tRNA, ANA, LE, or a 56 kDanRNP in lupus. The autoantibodies associated with scleroderma and mixed connective tissue concern only patients with dermatomyositis, the only form of inflammatory myopathy that overlaps with these two conditions (as discussed in the related article). Because these antibodies recognize ubiquitously expressed molecules and seem to be associated with some distinct clinical phenotypes, it has been suggested that the targeted tissue itself might regulate the phenotype-specific immune response owing to altered structure of tissue autoantigens during periods of cell stress, apoptosis, or acquisition of adjunct proteins (141). This hypothesis remains to be tested. As mentioned above, two autoantibodies, anti-SRP and anti-HMGCR, appear to be good markers of necrotizing myositis, and they are of diagnostic value because they are detected in up to 65% of patients with NAM (25), regardless of statin exposure. Claims that these antibodies may be pathogenic because they can cause muscle fiber atrophy and affect regeneration in vitro are not of direct relevance to the mechanism of the disease because the main causative characteristic of NAM is a macrophage-mediated muscle fiber necrosis with devastating muscle destruction and not muscle fiber atrophy (10; 53; 54; 55; 56).
Immunopathology. In polymyositis, original studies provided evidence suggesting a T cell-mediated cytotoxic process directed against unidentified muscle antigens in a pattern identical to the one described and studied in inclusion body myositis. Considering that polymyositis is very rare, it is conceivable that some of the studied specimens might have been from patients with inclusion body myositis, which is often erroneously diagnosed as polymyositis. The immunology and immunopathology have been, however, of extraordinary scientific value in understanding the role of cytotoxic T cells and, even if polymyositis is now rare, the information is applicable to all forms of autoimmune myopathies, hence, the need to be reviewed.
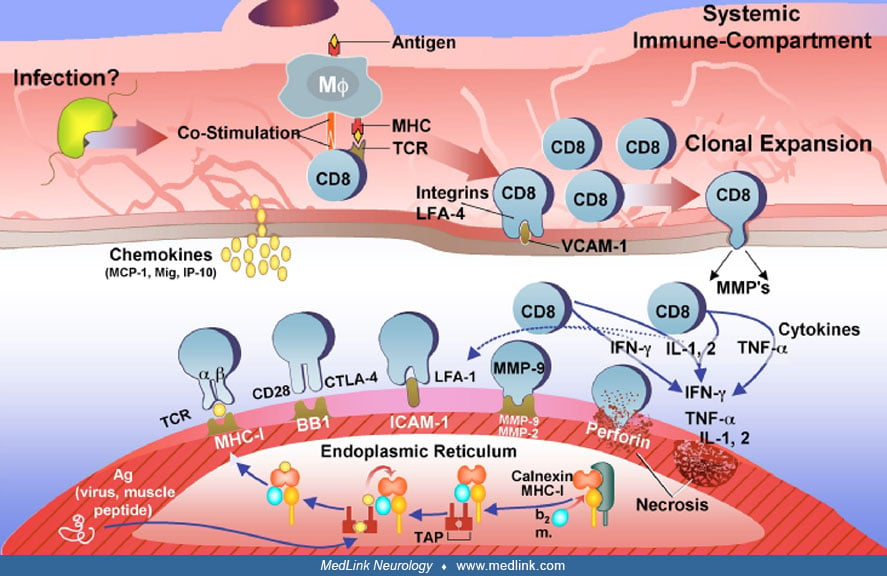
The T cell-mediated process is supported by the presence of CD8+ cells that, along with macrophages, initially surround healthy, nonnecrotic muscle fibers and eventually invade and destroy them (07; 08; 72; 70; 88). Muscle fibers, either next to or remote from the areas of inflammation, strongly express the class I major histocompatibility complex antigen, which is absent from the sarcolemma of normal muscle fibers (97). Cytotoxic T cells recognize antigenic targets in association with class I major histocompatibility complex antigen; these findings indicate that in polymyositis the primary immunopathologic mechanism is T cell-mediated and class I major histocompatibility complex antigen-restricted process. In one case, gamma/delta cytotoxic T cells were responsible for the muscle fiber injury (89); in vitro studies have further shown that this patients' circulating T lymphocytes are sensitized and exert cytotoxic effect to their homologous myotubes (87).
The specificity of the T cells has been examined by studying the gene rearrangement of the T cell receptors of the autoinvasive T cells, with similar findings in both polymyositis and inclusion body myositis. In patients with polymyositis, as well as inclusion body myositis, only certain T cells of specific T cell receptor alpha and T cell receptor beta families are recruited to the muscle from the circulation (112; 124; 15). Cloning and sequencing the amplified endomysial or autoinvasive T cell receptor gene families has demonstrated a restricted use of the J-beta gene with conserved amino acid sequence in the CDR3 region, indicating that CD8+ cells are specifically selected and clonally expanded in situ by muscle-specific autoantigens (112; 124; 15). Studies combining immunocytochemistry with polymerase chain reaction and sequencing of the most prominent T cell receptor families have shown that only the autoinvasive, not the perivascular, endomysial CD8+ cells are clonally expanded (15). Comparison of the T cell receptor repertoire between polymyositis and dermatomyositis with spectral type has confirmed that perturbations of the T cell receptor families occur among the peripheral blood lymphocytes, and they are specific for polymyositis and inclusion body myositis but not dermatomyositis (16). Further, among the circulating T cells, clonal expansion occurs only of the cytotoxic CD8+ cells that express genes for perforin and infiltrate the major histocompatibility complex-I-expressing muscle fibers (122). The clonal restriction of the autoinvasive T cells was confirmed with laser microdissection studies followed by sequencing of the T cell receptor gene families (86).
In order for antigen presentation and recognition by the T cells to occur, the muscle fibers and the autoinvasive CD8+ T cells need to coexpress the costimulatory molecules (B7-1, B7-2, BB1, CD40, or ICOS-L) and the respective counter-receptors [CD28, CTLA-4 (cytotoxic T lymphocyte antigen 4), CD40L or ICOS]. Several studies have now confirmed that the MHC-I-positive muscle fibers express BB1 (CD80) and make cell-to-cell contact with their CD28 or CTLA-4 ligands on the autoinvasive CD8+ T cells (14; 35). The CD40 molecule is also upregulated in muscle fibers, and the CD40 ligand is expressed in the infiltrating T cells (142). Further, there are ICOS/ICOS-L interactions between the autoinvasive CD8+ cytotoxic T cells and the MHC-I-expressing muscle fibers (157; 158; 156; 134).
Finally, cytokines, chemokines, and metalloproteinases (fundamental molecules for T cell activation, trafficking, antigen recognition, and T cell attachment) are upregulated in the muscle tissue of polymyositis patients. The mRNA of IL-1, IL-2, tumor necrosis factor-alpha and its receptor, tumor necrosis factor and its receptor, INF-gamma, T cell growth factor beta, granulocyte-macrophage colony-stimulating factor, IL-6, and IL-10 have been amplified from the muscle of most polymyositis and inclusion body myositis patients (146; 34; 67). Some of these cytokines, such as INF-gamma, ILI-1 beta, and tumor necrosis factor-alpha exert a direct cytotoxic effect on the muscle fibers (95; 40; 49; 50). Chemokines, a class of small cytokines participating in the leukocyte recruitment, trafficking, and activation, are also upregulated in polymyositis and inclusion body myositis. Among them, the chemokines interleukin-8, RANTES (regulated on activation, normal T cell expressed and secreted), monocyte chemoattractant protein 1, macrophage inflammatory protein 1a (MIP-1a), and IP-10 and its receptors are expressed in the endomysial inflammatory cells and in the neighboring extracellular matrix (26; 68; 66; 131). Adhesion of lymphocytes to muscle may be facilitated by metalloproteinases, a family of calcium-dependent zinc endopeptidases involved in the remodeling of the extracellular matrix. Among metalloproteinases, the metalloproteinase-9 and metalloproteinase-2 are upregulated in the nonnecrotic and major histocompatibility complex-I-expressing muscle fibers of patients with polymyositis and inclusion body myositis (24; 100). The metalloproteinase-2 is expressed on the autoinvasive CD8+ T cells that make cell-to-cell contact with the muscle fibers.
The release of cytokines and chemokines upregulates the expression of vascular cell adhesion molecule-I and intracellular adhesion molecule-I in the endothelial cells (32). These molecules serve as ligands for the integrins VLA-4 and lymphocyte function associated antigen-I that are expressed in T cells and facilitate their exit through the blood vessel wall into the perimysial and endomysial spaces.
Plasma cells and myeloid dendritic cells, which are potent antigen-presenting cells, are also seen among the endomysial infiltrates of patients with polymyositis, as well as inclusion body myositis and dermatomyositis and may play a significant role (81). Myeloid dendritic cells can be candidate cells for antigen presentation to surrounding T cells. Based on their immunoglobulin gene isotype, the endomysial plasma cells appear to mature and expand in situ implying an antigen-driven response (20). Because the same cells also have been found in inclusion body myositis and dermatomyositis, they may reflect a peculiarity of the inflammatory response within the closed muscle microenvironment. Similar clusters have been seen in the target organs of other autoimmune diseases, such as the synovium in rheumatoid arthritis or the brains of patients with multiple sclerosis.
Because in polymyositis, similarly to inclusion body myositis, the MHC-I is expressed in all fibers, regardless of if they are invaded by T cells, and the MHC-I remains strong throughout the disease; it was proposed that the MHC-I upregulation may exert a stress effect to the endoplasmic reticulum of the myofiber independent of T cell-mediated cytotoxicity (121). The assembly and folding of MHC-I occurs in the endoplasmic reticulum and matures only when binds to an antigenic peptide synthesized in the cytosol. A system of chaperone proteins, including calnexin, calreticulin, GRP94, GRP78, and ERP72, that form the MHC-loading complex ensure the proper maturation of MHC for antigen processing (42). If the “MHC-class-I loading complex,” does not bind to suitable antigens, the heavy chain glycoprotein is misfolded and removed from the endoplasmic reticulum to the cytosol for degradation. In polymyositis as well as inclusion body myositis, the muscle fibers are overloaded by MHC molecules, and the antigenic peptides cannot undergo proper conformational change to bind to MHC-I complex leading to endoplasmic reticulum stress. This is supported by upregulation of the aforementioned chaperone proteins and the activation of NF-kB, a means by which the cells protect themselves from endoplasmic reticulum stress. Such stressor effects are also seen in MHC-I transgenic mice, suggesting that overexpression of MHC-I alone may be sufficient to induce endoplasmic reticulum stress (121). This hypothesis is reasonable in explaining the continuous MHC-expression and chronic inflammation as seen in polymyositis and inclusion body myositis (42), but needs to be tested further.
The factors triggering the T cell-mediated process in polymyositis remain unclear. Viruses, especially the retroviruses HIV and HTLV-I, have been etiologically connected with the disease in infected individuals (62; 118; 32; 41; 92; 107), but these viruses are visible only on some infiltrating macrophages and not within the muscle fibers (44). Viral-specific T cells have been seen among the endomysial infiltrates in polymyositis and inclusion body myositis (63). The most interesting triggering factor is now becoming the immune-check-point inhibitors, which are directed against: (1) CTLA-4 (ipilimumab); (2) PD-1 (pembrolizumab and nivolumab); and (3) PD-L1 (atezollizumab, avelumab, and durvalumab). These drugs are used for advanced malignancy and prevent the CTLA-4 or PD-1 from binding to their respective receptors CD80/86 and PDL-1 and, by doing so, inhibit the “inhibitory” costimulatory interactions between T cells and tumor cells, resulting in positive costimulation and strong cell activation, like taking the “brakes off” the immune system (53). This blockade allows T cells to kill tumor cells, but the resulting enhanced costimulation causes an uncontrolled T cell activation that disrupts immune tolerance, resulting in immune-related events against muscle, mainly expressed as polymyositis and NAM, which at times occurs concurrently with myasthenia gravis (96; 53). Eosinophilic fasciitis and orbital myositis have been also seen.
New work on anti-synthetase syndrome myositis patients using proteomic analysis on peripheral blood cells concluded that a B-cell-driven pathology with persistence of a specific subtype of plasma cells is involved in self-perpetuating chronicity of the myositis process (130). Although the endomysial cells were not studied, the information indirectly suggests that B cells and plasma cells may be considered as therapeutic targets.
The triggering factors in eosinophilic myositis are also unclear, but trauma, drugs, or viral infections have been implicated. The cytokine interleukin-5 may play a role in inducing eosinophilia (148). Activated eosinophils infiltrate tissues and degranulate, releasing cytotoxic factors such as cytotoxic granule protein, major basic protein, and eosinophil cationic protein (155; 92; 99; 148). Eosinophil granule proteins are known to be toxic to cultured cardiac muscle (143) and may induce a similar effect in the skeletal muscle. Eosinophilic infiltration of skeletal muscle, however, does not account for all the parenchymal destruction because in many cases, in spite of peripheral eosinophilia, the eosinophilic infiltrates have been rare or transient within the muscle. Perimysial deposition of major basic protein has been demonstrated in some cases and is thought to contribute to tissue damage (92; 99). The rarity of these syndromes has precluded a systematic study of their immunopathogenesis. In macrophagic myofasciitis, the triggering factors have been thought to be aluminum-containing vaccines (13).
The exact incidence of polymyositis (even if exists as a stand-alone entity, as discussed earlier) remains unknown. It has been so uncommon and often overdiagnosed that its existence has been correctly challenged even 20 years ago (06). Nevertheless, along with overlap myositis, necrotizing autoimmune myositis, dermatomyositis, and inclusion body myositis, seem to occur in approximately 1 in 100,000 adults (40; 49; 50). The emergence of autoimmune necrotizing myositis seems now more common, and it is likely that several cases of NAM have been misdiagnosed over the years as polymyositis (48; 49; 55).
Polymyositis is diagnosed by exclusion (29; 30; 36; 37; 39; 43; 49; 132; 36; 40). Therefore, all diseases that cause an acquired myopathy should be considered before the diagnosis of polymyositis is established and the disease treated. The following groups of diseases should be excluded: (1) hereditary neuromuscular diseases, especially muscular dystrophies wherein endomysial inflammation can occur, such as dysferlinopathies, anoctaminopathies, fascioscapulohumeral dystrophy, or Becker muscular dystrophy; these dystrophies are more common in patients below the age of 18 years, whereas polymyositis is rare; (2) metabolic myopathies, endocrinopathies, electrolyte disturbances, mitochondriopathies; (3) any systemic medical illness, including malabsorption syndromes, alcoholism, cancer, vasculitis, systemic viral or bacterial infections, various autoimmunities, sarcoidosis, granulomatous disease, or treatment with various known myotoxic drugs or a combination of unknown, but potentially myotoxic, drugs, or toxins; (4) neurogenic muscular atrophies or neurogenic conditions; (5) biochemical muscle diseases (enzyme deficiencies) and inclusion body myositis excluded by muscle enzyme histochemistry or biochemical analysis; and (6) necrotizing autoimmune myositis, which is characterized by acute or subacute onset of muscle weakness and macrophage-mediated muscle fiber-necrosis without T-cell infiltrates or ubiquitous expression of MHC-I class antigen but with anti-SRP or anti-HMGCR antibodies in 65% of patients.
The clinical suspicion of polymyositis, overlap myositis, or necrotizing autoimmune myositis is bolstered by evaluation of serum muscle enzymes and by electromyography and is confirmed by a diagnostic muscle biopsy.
Serum muscle enzymes. The most sensitive enzyme is creatine kinase, which in polymyositis can be as much as 50 times higher than normal whereas in NAM can be more than 50 times the upper limit of normal, often in the thousands. Although creatine kinase usually parallels disease activity, it can be normal in chronic polymyositis and chronic NAM. Creatine kinase may also be normal in patients with polymyositis and in a setting of a connective tissue disease (“overlap myositis”), reflecting the preference of the pathologic process for the intramuscular vessels and the perimysium. Along with creatine kinase, serum SGOT and SGPT, and LDH may also be elevated. The presence of high SGOT, SGPT, and LDH levels in a patient with early disease and minimal weakness often suggests the diagnosis of liver disease leading to an unnecessary liver biopsy if the creatine kinase level is not concurrently checked. If SGOT is higher than SGPT, a myogenic cause should be suspected; when SGPT is higher than SGOT and the gamma-GT is also elevated, liver disease is more likely.
Electromyography. Needle electromyography shows myopathic potentials characterized by short-duration, low-amplitude polyphasic units on voluntary activation and increased spontaneous activity with fibrillations, complex repetitive discharges, and positive sharp waves. This electromyographic pattern occurs in a variety of acute, toxic, and active myopathic processes and should not be considered diagnostic for the inflammatory myopathies. In fact, no pattern is characteristic of polymyositis. Mixed myopathic and neurogenic potentials (polyphasic units of short and long duration) are more often seen in inclusion body myositis, but they can be seen in both polymyositis and dermatomyositis due to muscle fiber regeneration and the chronic nature of the disease. Electromyographic studies are generally useful to exclude neurogenic disorders and to assess whether the myopathy is active or inactive
Muscle biopsy. Muscle biopsy is the definitive test not only to establish the diagnosis of polymyositis but also to exclude other neuromuscular diseases. In what is referred to as polymyositis, the presence of inflammation is the histologic hallmark of the disease. The endomysial infiltrates are mostly in foci within the fascicles (endomysially) initially surrounding healthy muscle fibers but eventually resulting in phagocytosis and muscle fiber necrosis. The inflammatory infiltrates may be so small and multifocal that they are missed in a small-size muscle biopsy specimen. Occasionally, inflammation can be better seen in longitudinal sections. In polymyositis, as in inclusion body myositis, the inflammation is primary, a term used to indicate that CD8+ cells invade histologically healthy fibers that express MHC-I antigen. We refer to this lesion as the “CD8/MHC-I complex” (40) and have proposed it to be a specific lesion for polymyositis that secures the histologic diagnosis. Eosinophils are rare, but, if abundant, the diagnosis of eosinophilic myositis should be considered; in cases of suspected fasciitis or perimyositis, a biopsy of skin, fascia, or muscle in a wedge-like fashion is most informative. The perifascicular atrophy and prominent perivascular infiltrates seen in dermatomyositis are not present in polymyositis, and the blood vessels are normal. When the disease is chronic, the connective tissue is increased. In polymyositis, there should be no vacuolated fibers with cytoplasmic inclusions, typical of inclusion body myositis. Magnetic resonance imaging (MRI) is helpful for diagnosis when muscle edema is present or myofasciitis is suspected. MRI can be also helpful in identifying the most affected muscles and guide the diagnostic biopsy (49).
In necrotizing autoimmune myositis, there are abundant necrotic fibers invaded or surrounded by macrophages. Lymphocytic infiltrates are sparse, and MHC class 1 upregulation is spotty but sometimes prominent beyond the necrotic fibers. Necrotizing autoimmune myositis seems mediated, in the majority of the patients, by specific antibodies against SRP or HMGCR, which are seen in up to 65% of these patients; although not pathogenic, they are very good markers. Complement deposits are frequent but not unexpected because based on classic immunopathology studies, all necrotic fibers in nonimmune myopathies, such as muscular dystrophies, unambiguously activate complement, which in turn stimulates cellular infiltrates and macrophages (71).
Primary intramuscular inflammatory response is an invariable feature of polymyositis, and absence of inflammation early in the illness should raise a critical concern about the diagnosis. In the old diagnostic criteria introduced by Bohan and Peter (17; 18), the main diagnostic features of proximal muscle weakness, myopathic findings on the electromyogram, elevated creatine kinase levels, and inflammation in the muscle biopsy had equal diagnostic weight. Further, the diagnosis of polymyositis was acceptable even without the presence of muscle biopsy findings specific for the disease. Consequently, inclusion body myositis was overlooked, and various noninflammatory myopathies were erroneously diagnosed as polymyositis, as repeatedly emphasized (30; 36; 40). This has prompted the introduction of diagnostic criteria (30; 49; 40). The diagnosis of polymyositis is definite when a patient has: (1) acquired, subacute myopathy fulfilling the exclusion criteria described earlier and lacking the distribution of weakness typically seen in inclusion body myositis; (2) elevated creatine kinase; (3) a muscle biopsy with the histologic features of polymyositis, including the MHC-I/CD8 lesion (40); and (4) exclusion of inflammatory dystrophies and NAM. The diagnosis is probable polymyositis if the muscle biopsy shows nonspecific myopathic features but widespread MHC-I expression without apparent T cell infiltrates, macrophages, or vacuoles and the patient does not have the clinical phenotype of inclusion-body myositis (see relevant MedLink Neurology article for Inclusion-body myositis). A repeat muscle biopsy from another site may prove informative in these cases and might be considered to secure a definitive diagnosis.
The most common erroneous practice that impacts investigative and therapeutic decisions is the failure to distinguish PM from IBM, NAM, and inflammatory dystrophies (ie, Duchenne muscular dystrophy, dysferlinopathy, calpainopathy, merosin deficient sarcoglycanopathy), owing to erroneous pathologic interpretation of the biopsy and the failure to correlate the histology with the clinical phenotype. The muscle fiber necrosis in PM is due to invasion of seemingly intact muscle fibers by cytotoxic lymphocytes; in contrast, the invasion of muscle fibers in dystrophies is mostly by macrophages. However, in cases where endomysial infiltration is associated with lymphocytes, these cells lack the MHC/CD8 complex that is typical of PM and IBM as noted above. Some observations have confirmed that up to 15% of biopsies from patients with typical clinical features of IBM demonstrate only inflammation, like that seen in PM, but without the classic vacuoles (22). These patients labeled as PM/IBM or probable IBM need to be distinguished from PM on the basis of the typical clinical phenotype and the large number of COX-negative and “ragged-blue” fibers seen in the biopsy. In NAM the predominant cell invading muscle fibers are macrophages but, in contrast with dystrophies where the disease manifests slowly over years, NAM evolves acutely or within weeks and is often associated with antibodies. Errors can be avoided by a combined evaluation of the clinical with the histologic and immunopathologic findings.
The treatment of polymyositis, NAM, and anti-synthetase syndrome remains empirical, and separate large-scale, prospective, controlled clinical studies have not been performed (30; 36; 37; 39; 46; 49; 36; 40; 114). In most old series, patients had not been screened to exclude inclusion body myositis, which appears to resist all therapies.
The main goal of therapy in all three myositis groups is to improve function in activities of daily living by improving muscle strength. Although improvement in strength is usually accompanied by a fall in serum creatine kinase, decreases of serum creatine kinase alone need to be interpreted with caution because most immunosuppressive therapies lower serum muscle enzyme levels without necessarily improving muscle strength. Unfortunately, this has been misinterpreted as "chemical improvement" and has formed the basis for the common habit of "chasing" or "treating" the creatine kinase level instead of monitoring muscle strength, a practice that has led to prolonged use of unnecessary immunosuppressive drugs and erroneous assessment of their efficacy (33; 46). The most commonly used drugs are steroids and nonsteroidal immunosuppressive agents.
Corticosteroids. Prednisone is the first-line drug of this empirical treatment.
Response to prednisone determines whether or not stronger immunosuppressive drugs will be needed; therefore, one may prefer an aggressive approach with high-dose prednisone beginning early in the disease (33; 46; 2015a). A high, single-daily, morning dose of 60 to 80 mg for an initial period of 3 to 4 weeks is preferable. Prednisone is then tapered over a 6 to 8-week period to an 60 to 80 mg single dose every other day by gradually reducing the "off-day" dose by 10 mg per week or faster, if side effects occur (though this carries a greater risk of breakthrough of disease). If there is evidence of efficacy and no serious side effects, then the dosage is reduced gradually until the lowest possible dose that controls the disease is reached. If by the time the dosage has been reduced to every other day and no objective benefit (defined as increased muscle strength) has been observed, the patient may be considered unresponsive to prednisone; tapering may be then accelerated while a second-line therapy is considered (28).
The merits of a single-dose, alternate-day program in minimizing side effects (cushingoid appearance, diabetes, obesity, high blood pressure, osteoporosis, avascular necrosis of the hip, retarded growth in children) while adequately controlling the underlying disease have been previously discussed (28; 30).
Sometimes the long-term use of prednisone may cause increased weakness with a normal or unchanged creatine kinase level, a situation often referred to as "steroid myopathy." This condition is rather uncommon with prednisone but more often seen with dexamethasone. In a patient who has previously responded to high doses of prednisone, the development of increased weakness may be related to steroid-induced myopathy or to disease activity that either will respond to a higher dose of steroids or has become resistant to steroids. It may be difficult to distinguish one situation from the other because the two can coexist or be complicated by other factors, such as decreased mobility, infection, or associated systemic illnesses. In these circumstances, the decision to raise or lower the prednisone dosage may be influenced by reviewing changes in the patient’s muscle strength during the preceding two months in connection with mobility, serum creatine kinase levels, and changes in medications. If none of these are informative, then prednisone dosage can be adjusted (increased or decreased) arbitrarily. The cause of the weakness may be evident in the ensuing 2 to 8 weeks, based on the changes in the patient's strength.
Prednisone failures and nonsteroidal therapies. Almost all patients with polymyositis, NAM, and anti-synthetase syndrome myositis respond to steroids to some degree and for some period of time. The rationale for starting another immunosuppressive drug in those patients is based on the following criteria: (1) the need for a "steroid-sparing" drug when the patient has developed significant complications despite steroid responsiveness, (2) repeated relapses when attempting to lower a high steroid dosage, (3) ineffectiveness of an adequate dose of prednisone for at least a 2- to 3-month period, and (4) rapidly progressive disease with evolving severe weakness and respiratory failure. The preference for selecting the next-in-line immunosuppressive therapy is, however, empirical. The choice is typically based on a physician's own bias, personal experience with each drug, and assessment of the relative efficacy to safety ratio. The following immunomodulating or immunosuppressive therapies are used in the treatment of these patients.
Azathioprine. Although low doses (1.5 to 2 mg/kg/day) are commonly used, one may prefer higher doses up to 3 mg/kg for more effective immunosuppression in difficult cases. This drug is well tolerated and has fewer side effects than others, but it acts slowly (effects are not seen until 3 to 6 months of treatment). It follows that patience is required before concluding that the drug is ineffective. Azathioprine, metabolized by xanthine oxidase, can be severely toxic to the liver or bone marrow if given concurrently with allopurinol; therefore, combined use should be avoided.
Mycophenolate mofetil. Mycophenolate mofetil is an immunosuppressive drug that inhibits the purine pathway on the T cells with promising results (135), although control studies have not been performed. It has the advantage of working faster than azathioprine, and it is well tolerated. Doses up to 2000 to 3000 mg per day in two divided doses are recommended.
Methotrexate. Methotrexate is preferred by some practitioners because it acts faster than azathioprine. It can be given orally, starting at 7.5 mg weekly for the first three weeks (given in a total of three doses, 2.5 mg every 12 hours), increasing it gradually by 2.5 mg/week up to a total of 15 to 25 mg weekly. An important very rare side effect is methotrexate-pneumonitis, which can be difficult to distinguish from the interstitial lung disease accompanying some of these patients, especially those with Jo-1 antibodies, as described above.
Cyclophosphamide. Preferably, cyclophosphamide is given intravenously at doses of 0.5 to 1 gm/m2 per month. Cyclophosphamide has shown promising results in some patients (19). Although in our hands it was ineffective in patients with severe disease, the drug may be helpful in a subset of patients with interstitial lung disease (27).
Cyclosporine. The advantage of cyclosporine, although used with limited success, is that it acts faster than azathioprine, and its toxicity can be monitored by measuring optimal trough serum levels (which vary between 100 and 250 ng/mL).
Plasmapheresis. Plasmapheresis was not helpful in a double-blind, placebo-controlled study that we conducted (117).
Tacrolimus. Acting as a calcineurin inhibitor, tacrolimus has shown promise in the treatment of difficult cases. However, the experience with this drug is limited.
Rituximab. Rituximab, the first anti-CD20 monoclonal antibody that causes depletion of B cells, is promising in some polymyositis, NAM, and anti-synthetase syndrome patients based on a series of anecdotal reports. However, a completed controlled study comparing rituximab-early and rituximab-late (given 8 weeks later and serving as a placebo arm) groups showed no difference between the two arms in the time to achieving the definition of improvement during the 44-week period (123). Eighty-three percent of patients in both groups, however, met the definition of improvement. Although the nature of the study design does not allow conclusions on efficacy of this drug versus placebo, the observation that patients in both groups improved suggests that rituximab may play a role in patients resistant to therapies (49).
Intravenous immunoglobulin. In a double-blind study, intravenous immunoglobulin was effective in patients with refractory dermatomyositis (32; 35) and has been effective in the majority of polymyositis and NAM patients tried (23; 35; 113; 79; 56). However, controlled data are lacking. Subcutaneous immunoglobulin was also shown to be of benefit (127; 64; 65).
Considering all the aforementioned therapies, our step-by-step approach is as follows (40; 46; 49):
Step 1: High-dose prednisone (oral or intermittent intravenous)
Step 2: Initial or subsequent optional immunosuppressant, ie, mycophenolate, azathioprine, or methotrexate, only for steroid-sparing effect
Step 3: If steps 1 and 2 fail, high-dose intravenous immunoglobulin
Step 4: If Step 3 fails, consider a trial, with guarded optimism, of rituximab. If interstitial lung disease is present, one of the following agents, chosen according to the patient’s age, degree of disability, tolerance, experience with the drug and general health, are additional options: cyclosporin, cyclophosphamide, or tacrolimus. Other agents, mostly in the form of monoclonal antibodies or fusion proteins, seem now very promising, especially anti-B cell agents like rituximab, a B-cell-depleting monoclonal antibody (42; 43; 44; 49).
New biologics. New biologics may becoming promising therapeutic options. JAK kinase inhibitors, such as tofacitinib (103; 126) and ruxolitinib (90) seem effective, probably by affecting circulating cytokine levels and regulating the activation of dendritic cells and T lymphocytes, known to be activated in the disease (124). Monoclonal antibodies directed against complement, such as eculizumab directed against C5 blocking the generation of C5a and the membrane attack complex assembly, are being tested in these patients (55; 56; 57). A double-blind study in patients with NAM and anti-synthetase syndrome is ongoing.
Polymyositis can occur in the last trimester of pregnancy or during the puerperium, but it is not known whether pregnancy triggers the disease. Pregnant women with polymyositis have been treated with steroids and have delivered normal, but small, babies (125). Yet, miscarriages and stillborn babies have been reported. In two reported cases, the newborn babies had elevated creatine kinase for a few months, but they were normal otherwise (116). The mothers responded well to steroids.
Altered response to succinylcholine (theoretical hyperkalemia) and possible sensitivity to nondepolarizing muscle relaxants. Avoid succinylcholine.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Marinos C Dalakas MD
Dr. Dalakas of the National and Kapodistrian University of Athens Medical School in Greece and Thomas Jefferson University, Philadelphia, Pennsylvania received speaker honoraria and consultancy fees from Alexion, Argenx, Grifols, CSL, Sanofi, and UCB.
See Profile
Michael K Hehir MD
Dr. Hehir of University of Vermont Medical Center received a consulting fee from Immunovant, Janssen, and UCB Pharma.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Developmental Malformations
Apr. 14, 2024
Neuroimmunology
Apr. 07, 2024
Neuroimmunology
Mar. 24, 2024
Neuroimmunology
Mar. 24, 2024
Neuromuscular Disorders
Mar. 11, 2024
Movement Disorders
Mar. 06, 2024
Neuromuscular Disorders
Mar. 04, 2024
General Neurology
Mar. 03, 2024