






Epilepsy & Seizures
Ambiguous paroxysmal events
Mar. 22, 2024
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Progressive myoclonic epilepsy type 1 (EPM1) is a progressive myoclonus epilepsy caused by pathogenic, autosomal recessive variants in the cystatin B (CSTB) gene mapped to chromosome 21q22.3. The disease is regarded as the “purest” progressive myoclonus epilepsy due to disabling stimulus-sensitive and action myoclonus and generalized tonic-clonic seizures whereas cognitive deterioration and cerebellar signs are mild and slowly progressive. Age at onset ranged between 6 and 13 years (mean, 10 years) and a relatively high frequency is observed on the shores of the Baltic and Mediterranean Seas, probably due to founder effects. Widening the knowledge over the molecular basis of this disease may also expand our chance of effective treatment, hence, the field opened by neuroinflammation, which is increasingly becoming a suitable target for precision medicine-based approaches also in this disease.
|
• EPM1 is an autosomal recessive progressive myoclonus epilepsy. | |
|
• It manifests with action and stimulus-sensitive myoclonus, generalized tonic-clonic seizures, mild and slowly progressive cerebellar ataxia, and cognitive deficits. Myoclonic seizures are severe and debilitate teenagers throughout adolescence and early adulthood. | |
|
• It is mainly caused by a homozygous expansion of a dodecamer repeat near the promoter of the cystatin B (CSTB) gene mapped to chromosome 21q22.3. Thus, the diagnosis can be confirmed by appropriate molecular genetic testing. | |
|
• Valproate, levetiracetam, zonisamide, perampanel, benzodiazepines, and topiramate are first-choice antiseizure drugs for treatment. Conversely, carbamazepine and derivatives, phenytoin, gabapentin, pregabalin, and vigabatrin are contraindicated. |
Progressive myoclonic epilepsy type 1 (EPM1) is also known as Unverricht-Lundborg disease. Heinrich Unverricht was a German physician, born on September 18, 1853, in Breslau; he died on April 22, 1912, in Magdeburg.
He was Professor of Medicine in Dorpat, now Tartu, Estonia. In 1891 he described a family of Baltic German origin in which several siblings suffered from a progressive neurologic disease with prominent myoclonus and generalized seizures. Tremor and rigidity have not been described in his patients (90). He also published a volume named Die Myoclonie (106).
Lundborg, from 1898 to 1910, compiled information on 17 patients afflicted by what is now known as progressive myoclonic epilepsy type 1 (EPM1 disease). They all belonged to one extensive kindred, the "Lister" family in Listerlandet of southern Sweden. He and Blekinge traced the affected family back to the 1700s (70; 71; 72). Lundborg's thesis on “Die progressive Myoklonus-Epilepsie (Unverricht's Myoklonie)” was one of the earliest descriptions of recessive inheritance (71). He had published the names of those affected, and he had concluded that the family had degenerated because of “unwise marriages.” Subsequently, a marriage of relatives was carefully avoided in the group and no more cases occurred. According to Puschmann, Lundborg distinguished three stages of the disorder: Stage 1 began in childhood or early adolescence and consisted of nocturnal attacks of involuntary symmetric muscle twitches, which often were painful, caused anxiety, and reminded Lundborg of clonic, tonic-clonic, or tetanic seizures (90). Patients were awake and conscious during the attacks. Stage 2 appeared a few years later with diurnal tremor, myokymia, and myoclonic or dystonic muscle contractions. Typically, the contractions initially affected the upper extremities symmetrically and subsequently involved lower extremities, head, neck, and finally, all voluntary muscles. Tactile and auditory stimuli elicited and stress aggravated these involuntary muscle contractions. Symptom severity fluctuated markedly from day to day. Gradually, increased muscle tone was noted in the interval between attacks. Stage 3 appeared several years to some decades later with the disappearance of the nocturnal attacks whereas daytime muscle contractions became more and more pronounced. Regularly, marked generalized rigidity developed, leaving some patients utterly stiffened in certain poses, incapable of any voluntary movements. Lundborg noted that the clinical picture of some of his patients in the terminal phase reminded him of Parkinson disease.
Herman Bernhard Lundborg was a Swedish physician, born on April 7, 1868, in Vase, Varmlands lan, and he died in 1943. His radical racist ideas and his influence of Nazi ideology and acts overshadow his contribution to medicine. He was extremely negative to Jewish people and strongly involved in the ideology of racial hygiene. He was also responsible for the massive mapping, The Racial Character of the Swedish Nation, from 1926. He characterized 15% of patients in his work as morally or socially inferior (alcoholic, criminal, wanton, of bad character) and another 9.5% as psychiatrically inferior. He had served as a Professor for Racial Hygiene and he advocated the ideology of race biology. His influence contributed to the implementation of forced sterilization programs in Sweden and his view was that "The future belongs to the racially fine people" (73).
Over the next 80 years, there was a great deal of discussion and misunderstanding of the nature and classification of the progressive myoclonus epilepsies (08; 15; 31; 99; 69; 87; 07; 49; 90), of which EPM1 is the most common. Advances in molecular biology have identified EPM1 disease as a specific genetic disorder encountered in many populations and parts of the globe. It has been described under different names, including Baltic myoclonus or Mediterranean myoclonus, before their identification by genetic studies as the same disorder as EPM1. Similarly, Ramsay Hunt syndrome or dyssynergia cerebellaris myoclonica is no longer considered to be a separate disease (02; 36; 78).
An excellent source of information is an online review of EPM1 by Lehesjoki and Kalviainen (64) and other updates (50).
Patients with progressive myoclonic epilepsy type 1 (EPM1) are initially normal. Myoclonic jerks make their appearance between the ages of 6 and 18 years, with a peak at 9 to 13 years (86% of patients). The myoclonic jerks are action activated and stimulus sensitive.
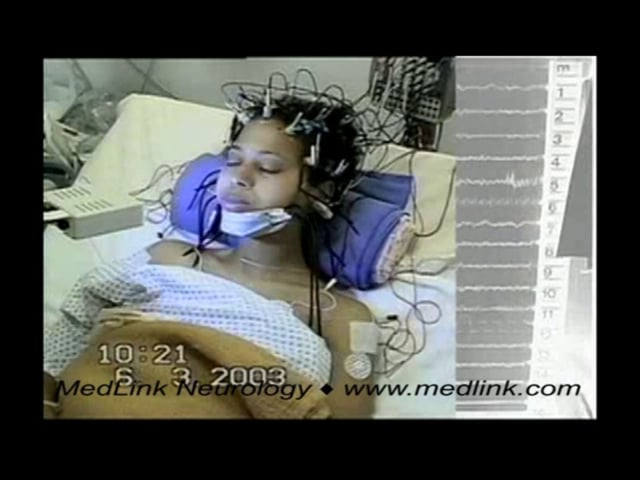
This 18-year-old girl has progressive myoclonic epilepsy type 1 (EPM1) disease that began at 12 years of age with action myoclonus and generalized tonic-clonic seizures. Despite major antimyoclonic therapy, including valproate,...
They may be provoked by light, physical exertion, and stress. Stimulus-sensitive myoclonus is the first symptom in at least 50% of patients and is an essential feature for diagnosis. They may be focal or multifocal and affect predominantly the proximal muscles of the extremities. They are small and mild, but they may also become so violent as to cause frequent falls. In a study of nine patients with genetically proven EPM1, myoclonus was activated by action but was virtually absent at rest and poorly induced by stimuli. Positive myoclonus was multifocal, often rhythmic and of brief duration, with top-down pyramidal temporospatial propagation (34). Compared with normal subjects, patients with EPM1 demonstrated decreased saccadic gain, increased gain dispersion, and a higher frequency of hypermetric saccades associated with decreased peak velocity (34).
Generalized convulsions, sometimes nocturnal, are initially infrequent and may not occur in all patients. These are at times clonic-tonic-clonic convulsions.
Myoclonus and GTCS progressively worsen over the 3 to 8 years following the onset of the disease. Severe action, reflex, and spontaneous myoclonus predominates and becomes the most incapacitating symptom. It takes the form of continuous shivering-like, small amplitude myoclonus affecting every muscle of the body, including the tongue and mouth. Violent myoclonic jerks erupt out of this “background myoclonic noise,” causing falls and injuries.
Progression to ataxia, incoordination, intention tremor, and dysarthria with mental deterioration are typical late features. However, the severity of the symptoms varies significantly even within affected members of the same family.
There is a slowly progressive mental deterioration, evaluated as a loss of one IQ point per year (59; 60), though most authors consider that this is usually mild and does not reach the levels of dementia (31). However, three neuropsychological studies demonstrated that patients with EPM1 show significant impairment of abstract reasoning, attention, planning, word fluency, constructive praxis, and visuospatial memory and learning in comparison with controls (32). Moreover, patients have lower scores on all short-term memory and executive function tasks, which are associated with the duration of disease and severity of myoclonus (25; 01).
Overall, the disease tends to stabilize in adulthood but with a severe disability.
Notably, patients who are compound heterozygous for the dodecamer repeat expansion and the c.202C>T mutations have a more severe clinical form of EPM1 than patients homozygous for the expansion mutation. They have an earlier age at onset of symptoms, more severe myoclonus, drug-resistant tonic-clonic seizures, moderate to severe cognitive impairment, and, sometimes, psychiatric symptoms (57; 13). Conversely, atypical presentations with milder, not full-blown, symptomatology and late onset have also been described. Nasri and colleagues described the case of a 50-year-old patient exhibiting progressive myoclonus and cerebellar ataxia, together with anxiety disorders (ie, specific phobia and agoraphobia) due to recurrent initial falls (81). High intra-familial phenotypic variability was also described among those relatives who tested positive for Unverricht-Lundborg disease. Hence, the authors underlined the importance of clinical suspicion in the case of suggestive family history also in older patients.
Patients with Unverricht-Lundborg disease often have comorbidities. Trauma and mental health risks should be especially followed and acted upon (102). Nevertheless, the outcome seems related to the severity of the myoclonus, tonic-clonic seizures, and ataxia.
The ataxia is greatly increased by the use of some antiepileptic drugs, notably phenytoin, prescribed to many of these patients in the past. Withdrawal of phenytoin has led to a marked improvement (23).
There is some disagreement as to whether intelligence remains normal (29) or may deteriorate even when optimal antiepileptic and antimyoclonic treatments are used (60; 16). See the Clinical manifestations section for further discussion.
Life expectancy may not be affected. However, though the long-term functional outcome and survival have improved, the overall efficacy of antimyoclonic drugs remains unsatisfactory (16).
One study showed that EPM1 progresses only over a limited period and stabilizes thereafter (74). However, in a multicenter study of the circumstances of death amongst 19 patients with EPM1, six patients died of sudden unexpected death in epilepsy, four patients died of a stroke, drowning, complications of chronic alcoholism, and Wernicke encephalopathy, two committed suicide, and one died in a car accident (52). The authors concluded that although the prognosis of EPM1 has improved, there are still severe forms and high risk of early death, including sudden unexpected death in epilepsy (52).
Earlier age at onset for EPM1 and longer disease duration are associated with more severe action myoclonus, lower performance IQ, increased motor threshold, and prolonged silent period (41).
Canafoglia and associates explored the course of EPM1 and identified the risk factors for severity detectable near disease onset (12). They retrospectively evaluated 59 Italian patients carrying the CSTB expansion mutation and coded the information every 5 years after the disease onset to describe the cumulative time-dependent probability of reaching disabling myoclonus, relevant cognitive impairment, and inability to work, and evaluated the influence of early factors using the log-rank test. The risk factors were included in a Cox multivariate proportional hazards regression model. Disabling myoclonus occurred an average of 32 years after disease onset, whereas cognitive impairment occurred a little later. Age at onset of fewer than 12 years, the severity of myoclonus at the time of first assessment, and seizure persistence more than 10 years after onset affected the timing of disabling myoclonus and cognitive decline. Most patients became unable to work years before the appearance of disabling myoclonus or cognitive decline. The authors concluded that a younger age at onset, early severe myoclonus, and seizure persistence are predictors of a more severe outcome. All of these factors may be genetically determined, but the greater hyperexcitability underlying more severe seizures and myoclonus at onset may also play a role by increasing cell damage due to reduced cystatin B activity (12).
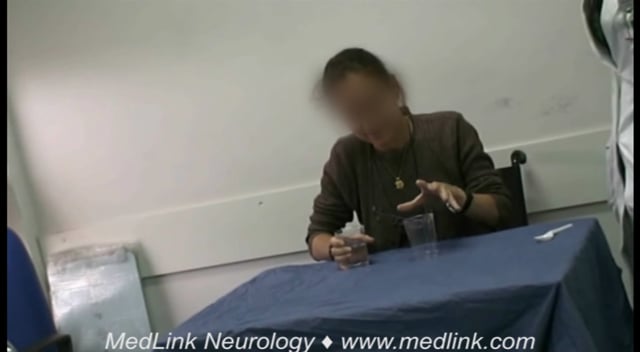
An 18-year-old girl had EPM1 that began at 12 years of age with action myoclonus and generalized tonic-clonic seizures. Despite major antimyoclonic therapy, including valproate, phenobarbital, benzodiazepines, and piracetam at 27 g/day, she was markedly handicapped by major myoclonic jerks elicited by action, intention of action, or even any cutaneous stimuli. There was also a pseudocerebellar syndrome with the finger-to-nose maneuver and marked photosensitivity (104).
This 18-year-old girl has progressive myoclonic epilepsy type 1 (EPM1) disease that began at 12 years of age with action myoclonus and generalized tonic-clonic seizures. Despite major antimyoclonic therapy, including valproate,...
Progressive myoclonic epilepsy type 1 (EPM1) is inherited as an autosomal recessive trait.
Mutations of the cystatin B (CSTB) gene (formal name = EPM1) are responsible for the disease. The CSTB gene has been mapped to chromosome 21q22.3 and encodes a protein that attains several biological roles from functioning as a lysosomal cysteine protease inhibitor to regulating glial activation, oxidative stress, serotonergic neurotransmission, and hyperexcitability (see Table 1) (67; 66; 63; 65; 68; 38; 99; 69; 27; 43; 44).
Linkage to chromosome 21q22.3 has been confirmed in families from several different ethnic groups, suggesting that nonallelic genetic heterogeneity is unlikely. Six-point mutations have been found, two of which are seen in numerous unrelated patients (69).
In almost all affected individuals, EPM1 is caused by an increase in the size of the CSTB gene. One region of the CSTB gene has a particular repeating sequence of 12 DNA building blocks (nucleotides). This sequence is normally repeated two or three times within the gene and is called a dodecamer repeat. The main CSTB mutation in EPM1, even among patients of different ethnic origins, is an expansion of a dodecamer repeat (ie, CCCCGCCCCGCG) in the 5' flanking area of CSTB (62; 61). Most normal alleles contain either two or three repeats, whereas rarer normal alleles that are highly unstable contain between 12 and 17 repeats. Mutant expanded alleles have been reported to contain between 30 and 80 copies and are also highly unstable, particularly via parental transmission. Mutant repeat length and disease phenotype are not correlated. Although the repeat expansion is outside the CSTB transcriptional unit, it results in a marked decrease in CSTB expression, at least in certain cell types in vitro. In a study of 66 patients (33 men and 33 women) with genetically confirmed EPM1 and homozygous for the CSTB expansion mutation, the number of dodecamer repeats in CSTB alleles varied between 38 and 77 (41). On average, the size of the longer expanded alleles of patients was independently associated with motor threshold, but exerted only a modulating effect on age at onset, myoclonus severity, and silent period.
CSTB homozygous knockout mice show some parallels to the phenotype of human EPM1 including myoclonic seizures, development of ataxia, and neuropathological changes associated with cell loss via apoptosis. Loss of CSTB function due to mutations is consistent with the observed neurodegenerative pathology and phenotype, but the functional link to the epileptic phenotype of EPM1 remains largely unknown (61).
Though the genetic background of EPM1 is a homozygous dodecamer repeat extension mutation in the CSTB gene, mutations occurring in a compound heterozygous form with the expansion mutation have also been reported (20; 57; 13; 04; 10). Heterozygous compound mutations are described to induce a more severe phenotype than that of homozygous dodecameric repetition. However, female patients usually present with less severe symptoms than male patients (04).
|
Gene |
Chromosome locus |
Protein |
|
CSTB |
21q22.3 |
Cystatin B |
|
OMIM entries |
254800 Myoclonic epilepsy of Unverricht and Lundborg | |
|
601145 Cystatin B (CSTB) | ||
|
| ||
Another clinical and molecular form of EPM1 localized by homozygosity mapping has been described by Berkovic and colleagues (09). This was in an in-bred Arab family with a clinical pattern compatible with EPM1, but mutations in the CSTB gene were absent. The family was mapped to chromosome 12. A similar Arab family with the gene mapping also to the pericentromeric region of chromosome 12 has also been described by El-Shanti and colleagues (22). Chromosome 12 does not contain genes for any other form of progressive myoclonus epilepsy, nor does it have genes known to be related to cystatin B.
The pathogenesis of EPM1 is still unknown. The increased number of dodecamer repeats in the CSTB gene seems to interfere with the production of the cystatin B protein. Levels of cystatin B in affected individuals are only 5% to 10% of normal, and cathepsin levels are significantly increased. It is presumed that with mutations in cystatin B and loss of its inhibition of cysteine proteases, apoptosis proceeds abnormally and neurodegeneration results (99). A cystatin B multiprotein complex may have a specific cerebellar function.
Another hypothesis is that in at least a subset of patients with EPM1, cystatin B protein may aggregate in the cell (14). Protein aggregation can be secondary to external insults, aging, or inherited forms of neurodegenerative diseases. Protein aggregates lead to increased production of the reactive oxygen species, dysfunction of the ubiquitin/proteasome system, mitochondrial dysfunction, and dysfunctional Ca2+ signaling. Moreover, RNA extracted from the brains of Cstb knockdown mice was analyzed by Joensuu and colleagues using microarray techniques to identify the transcriptomic signature of EPM1 (45). Of note, the upregulation of genes encoding complement proteins (C1qa, C1qb, C1qc, C4b, and C3ar1), major immunohistocompatibility complex class I (MHC-I), b2-microglobulin (B2m), glial fibrillary acid protein (Gfap), chemokines (Cxcl13 and Ccl6), immunoglobulin receptors (Fcgr3 and Fcgr1g), and other immune and defense response genes was found. Hence, early-onset neuroinflammation and the related glial activation was believed to contribute to the pathogenesis of the disease, promoting recurrent seizures and neuronal degeneration (45). Additionally, the CNS’s proinflammatory state is accompanied by peripheral inflammation demonstrated by high chemokines and cytokines serum concentrations (particularly, Cxcl13 has also been proposed as a marker of the disease) (85). Presently, it still sounds difficult to rule out the exact “order of events” in the neuroinflammatory process, although the possibility to identify disease-specific biomarkers (as Cxcl13) will be of undoubted interest (97; 24).
Buzzi and associates provided direct evidence that loss of cortical GABA input leads to a condition of latent hyperexcitability that favors myoclonus and seizures (11). They tested the hypothesis that EPM1 is accompanied by a loss of cortical GABA inhibition in a murine model (the CSTB knockout mouse) and a human case. Cortical GABA signaling has been investigated by measuring VGAT immunohistochemistry (a histological marker of the density of GABA terminals), GABA release from synaptosomes, and paired-pulse stimulation. In CSTB knockout mice, a progressive decrease in neocortex thickness was found and was associated with a prevalent loss of GABA interneurons. A marked reduction in VGAT labeling was found in the cortex of both CSTB knockout mice and a patient with EPM1. This implicates a reduction in GABA synaptic transmission, which was confirmed in the mouse model as a reduction in GABA release from isolated nerve terminals and as loss of electrophysiologically measured GABA inhibition. The alterations in VGAT immunolabeling progressed in time, paralleling the worsening of myoclonus (11).
Danner and associates aimed to characterize the relationship between the excitability and anatomy of the motor cortex and their association with the severity of the clinical symptoms. Seventy genetically verified patients were compared with 40 healthy controls (19). The symptoms were evaluated with the Unified Myoclonus Rating Scale. Navigated transcranial magnetic stimulation (TMS) was applied to characterize the excitability of the primary motor cortex by determining the motor thresholds and cortical silent periods. Also, the induced cortical electric fields were estimated using individual scalp-to-cortex distances measured from MRIs. A cortical thickness analysis was performed to elucidate possible disease-related anatomical alterations. The motor thresholds, cortical electric fields, and silent periods were significantly increased in the patients (P < 0.01). The silent periods correlated with the myoclonus scores (r = 0.48 to r = 0.49, P < 0.001). The scalp-to-cortex distance increased significantly with disease duration (r = 0.56, P < 0.001) and correlated inversely with cortical thickness. The authors concluded that their results may reflect the refractory nature of the myoclonus and indicate a possible reactive cortical inhibitory mechanism to the underlying disease process (19).
Similarly, Julkunen and colleagues studied seven genetically verified patients with EPM1 and six healthy control subjects (47). They utilized transcranial magnetic stimulation to induce cortical responses measured with EEG in order to observe prevailing cortical excitability/inhibition changes, as well as power and coherence of the cortical oscillations in EPM1. Navigated TMS was focused on the left primary motor cortex at the representation area of the right thumb. TMS-EEG responses were measured at 90% of the resting motor threshold intensity in 110 to 150 trials. It was found that P30 waveform following the TMS was significantly (p < 0.05) increased in patients with EPM1, suggesting increased cortico-cortical excitability, whereas the later N100/P180 waveform was significantly (p < 0.05) decreased, indicating reduced inhibition. In the event-related spectral perturbation, alpha, beta, and gamma band oscillations following the TMS were significantly lower in power in the patients with EPM1 compared to controls. In the alpha and beta bands, the intertrial coherence (ITC) representing the degree of synchronization was also decreased in EPM1. These results suggest abnormal reactivity in EPM1, and may indicate impaired cortico-cortical inhibition and attenuation of subsequent cortical circuits or the thalamic or subcortical nuclei (47). The same group of researchers studied the neurophysiological mechanisms behind the impaired thalamocortical system in patients with EPM1 through short-term adaptation of the motor cortex to transcranial magnetic stimulation via repetition suppression phenomenon (46). Their results suggested that neural processing of external stimuli is progressively impaired in EPM1, possibly due to anatomically impaired thalamocortical system or inhibitory tonus preventing sufficient adaptive reactiveness to stimuli (46).
Sensory hyperexcitability in EPM1 as revealed by giant somatosensory evoked potentials (SEP) has been studied (107; 53). In one study of 25 patients with EPM1 compared with 20 controls, SEPs were evaluated by applying median nerve stimuli and the SEP recovery function (SEP-R) using paired stimuli at interstimulus intervals of between 20 and 600 ms (107). The SEPs were considered "giant" if the N20P25 and P25N33 amplitudes exceeded normal mean values by +3SD. It was found that during the paired-stimulus protocol, the SEPs elicited by the second stimulus (S2) were detectable at all interstimulus intervals but consistently suppressed in the 13 patients with giant SEPs, reflecting a significantly delayed SEP-R. Maximal suppression roughly corresponded to the plateau of a broad middle latency (> 100 ms) wave pertaining to the S1 response. The authors concluded that cortical processing dysfunction generating giant SEPs in patients with EPM1 consistently combines with a long-lasting suppression of hyperexcitability that leads to a delayed giant SEP-R without obstructing the response to incoming stimuli. The delayed SEP-R is not due to true inhibition but the suppression of aberrant hyper-synchronization sustaining giant SEPs. A broad middle latency SEP component adds a significantly suppressive effect. This suggests that cortico-subcortical circuitries contribute to both the gigantism and the delayed SEP-R (107). In the other study, SEPs to median nerve stimulation were repeatedly examined in seven genetically diagnosed Japanese patients with a mean interval of 11.9 years (53). The degree of temporal changes in the amplitude of three early cortical components, N20, P25, and N35, to the age was analyzed and compared with that of healthy subjects. It was found that the clinical course of the patients was almost stable during the follow-up period, namely cessation of generalized tonic-clonic seizures and little or no progression of myoclonus. SEP amplitudes of P25 and N35 were enlarged in all patients and were gradually decreased with aging in EPM1 on average. The degree of temporal changes of P25 and N35 in patients was similar or even lower than that of healthy subjects.
Manninen and colleagues performed a translational diffusion-tensor imaging study in patients with EPM1 and cystatin B-deficient mice (77). Diffusion-tensor imaging and tract-based spatial statistics (TBSS) were used to compare fractional anisotropic (FA) results and axial, radial, and mean diffusion among patients with EPM1 (n = 19) and control subjects (n = 18). Ex vivo diffusion-tensor imaging and TBSS were used to compare CSTB-/- mice (n = 9) with wild controls (n = 4). Areas of FA decrease in mice were characterized by means of immunohistochemical analysis and transmission electron microscopy. Student t test statistics were applied to report significant findings (threshold-free cluster enhancement, P < .05). It was found that patients with EPM1 showed significantly (P < .05) reduced FA and increased radial and mean diffusion in all major white matter tracts compared with those of control subjects, shown as global FA decrease along the TBSS skeleton (0.41 +/- 0.03 vs. 0.45 +/- 0.02, respectively; P < 5 x 10(-6)). CSTB-/- mice exhibited significantly reduced FA (P < .05) and antimyelin basic protein staining. Transmission electron microscopy revealed degenerating axons in CSTB-/- mice versus controls (979 axons counted, 51 degenerating axons; 2.09 +/- 0.29 per field vs. 1072 axons counted, 9 degenerating axons; 0.48 +/- 0.19 per field; P = .002). These results indicate that EPM1 is characterized by widespread alterations in subcortical white matter, the thalamocortical system, and the cerebellum, which result in axonal degeneration and white matter loss. Motor disturbances and other symptoms in patients with EPM1 involve not only the cortical system but also the thalamocortical system and cerebellum (77).
Another study estimated the appropriateness of generalized partial directed coherence (GPDC) in detecting myoclonus-related EEG-EMG connectivity pattern and the information flow between sensorimotor cortex and muscles in patients with typical cortical myoclonus due to EPM1 (88). In 13 patients with cortical myoclonus, the authors analyzed the EEG and EMG signals recorded during simple voluntary motor activities using GPDC, a frequency domain linear index of connectivity estimated from a multivariate autoregressive model. The results were compared with those obtained in 12 healthy controls. The GPDC revealed a peculiar pattern characterizing patients with cortical myoclonus with respect to healthy subjects. Patients consistently had a significantly more robust outflow toward activated muscle originating from cortical areas exceeding the motor one. Moreover, they also had a more robust EMG outflow directed toward a wider cortical area contralateral to the activated hand and sometimes also toward the ipsilateral central region. These results indicate the recruitment of extensive cortical network in afferent and efferent EEG-EMG relationships. Given that robust cortical outflow can be considered as the pathogenic mechanism sustaining myoclonus, the perturbation from the EMG outflow could lead to the involvement of the large cortical area implied in sensorimotor integration and become capable of generating and maintaining the jerk recurrence.
Kononen and associates aimed to elucidate possible alterations in cortical activation related to motor performance in EPM1 (54). Fifteen patients with EPM1 and 15 healthy volunteers matched for age and sex underwent motor functional MRI. Group differences in activations were evaluated in the primary and supplementary motor cortices and sensory cortical areas. Furthermore, in patients with EPM1, the quantitative fMRI parameters were correlated with the severity of the motor symptoms. It was found that the patients with EPM1 exhibited decreased activation in the left inferior frontal junction during right hand voluntary motor task when compared with controls. In the quantitative analysis, patients with EPM1 had significantly weaker activation than controls in the hand knob and supplementary motor areas. The volume of activation in M1 decreased with age and duration of disease in the patient group, whereas the volume increased with age in controls. Negative correlations were observed between fMRI parameters of supplementary motor areas and disease duration or age in patients but not in controls. The authors concluded that the weaker motor fMRI activation observed in patients with EPM1 parallels previous neurophysiological findings and correlates with the motor symptoms of the disease. Thus, the observed decrease in inferior frontal junction activation in patients with EPM1 may be associated with the difficulties in initiation or termination of motor execution, which is a typical clinical symptom in EPM1 (54).
Rossi Sebastiano and colleagues investigated whether the structural cortical changes observed in patients with EPM1 did correlate with the functional reorganization of the motor cortex (96). Seven adult patients with EPM1 and seven age-matched controls underwent transcranial magnetic stimulation-based cortical mapping of the motor hand areas combined with functional magnetic resonance imaging and cortical thickness analyses. Vertex-wise cortical thickness analysis revealed thinning in the precentral gyrus bilaterally and in the left supplementary motor area. Eventually, the motor-evoked potentials centers of gravity of the abductor brevis pollicis muscles and the peak of activation at functional magnetic resonance imaging were shifted toward the posterior regions, suggesting a reshaping of the sensorimotor areas. In conclusion, a transcranial magnetic stimulation-based approach to brain mapping may be used to identify the cortical reorganization of the cortical motor system possibly deriving from both plastic changes and the same pathophysiological process underlying the disease (96).
Pathology. Vacuoles may be present in eccrine sweat glands.
Their contents have not been identified. They may not be exclusive manifestations of EPM1 and have occasionally been found in other, clearly unrelated, disorders (17).
The pathological abnormalities of EPM1 first described by Haltia and colleagues involved mainly the cerebellum with Purkinje cell loss (35). There has been a great deal of variation in the description of the pathology, and many of the additional changes described most likely represented artifacts (33).
The disorder is most common in Finland, which has an incidence of 1/20,000 births and a prevalence of 1/25,000 (64; 101). The increased prevalence there most likely represents a founder effect with a heterozygote frequency estimated at 1/70 (83). Finland is a country in which road transportation and communications have been difficult for many centuries. Increased incidence of this disorder is also present in North Africa, mainly in Algeria and Tunisia (93), but affected families with the common homozygous dodecameric repeat expansion have also been reported from Egypt (39). Moreover, cases have been described from Japan, Italy, France, Turkey, Sweden, the United States, and other countries. Serbian patients with EPM1 share the same common ancestor with patients from Baltic countries and North Africa (51). The main aggregates in addition to Finland are found in areas with high consanguinity (100).
Recognition of the autosomal recessive inheritance of this disorder may lead to a reduction in multiplex siblings. Amniocentesis with molecular analysis may now be feasible. However, because the onset of symptoms is relatively late, birth of affected siblings cannot be entirely prevented.
Progressive myoclonic epilepsy type 1 (EPM1) may be underdiagnosed in patients with severe myoclonus epilepsy (21).
The most common forms of progressive myoclonus epilepsy are EPM1, Lafora disease, sialidosis, neuronal ceroid lipofuscinoses, and myoclonus epilepsy with ragged-red fibers (MERRF).
A more recently described syndrome of myoclonus epilepsy and ataxia due to recurrent KCNC1 p.R320H potassium channel mutation resembles EPM1, despite genetic and biological differences (86). Symptoms begin at between 3 and 15 years of age, with progressively severe myoclonus and rare tonic-clonic seizures. Ataxia presents early, but quickly becomes overshadowed by myoclonus. Mild cognitive decline occurs in half. Electroencephalogram shows generalized spike and polyspike wave discharges, with documented photosensitivity in most. Polygraphic EEG-electromyographic studies demonstrated a cortical origin for myoclonus and striking coactivation of agonist and antagonist muscles. Magnetic resonance imaging reveals symmetrical cerebellar atrophy, which appears progressive, and a prominent corpus callosum. Transient clinical improvement with fever is often noted in most patients. Myoclonus epilepsy and ataxia due to potassium channel mutation causing loss of the channel functioning (ie, reduced or absent potassium currents) have a relatively homogeneous presentation, resembling EPM1, despite the genetic and biological basis being quite different (86; 108).
Lafora body disease, a form of progressive myoclonus epilepsy starting between the ages of 11 and 18 and progressing relentlessly to dementia with an average age at death of 24 years, must be considered. Sialidosis, or cherry-red spot myoclonus syndrome, is a form of progressive myoclonus epilepsy not associated with mental deterioration. The eye findings are characteristic, and oligosaccharides are found in the urine. The juvenile form of neuronal ceroid lipofuscinosis, Spielmeyer-Vogt-Sjögren or Batten disease, begins between the ages of 5 and 8 years of age with progressive visual impairment, followed in the early teens by myoclonus, other types of seizure, deterioration of cognitive skills and speech, ataxia or clumsiness, and Parkinsonian-like extrapyramidal symptoms. As the disease progresses, myoclonus and other symptoms rapidly deteriorate and the patient loses all skills of communication, mobility, and self-help. The CLN3 gene in chromosome 16 accounts for 73% of all cases of juvenile neuronal ceroid lipofuscinosis. The diagnosis of neuronal ceroid lipofuscinosis is based on clinical findings, electron microscopy of tissue biopsy, and, in some instances, an assay of enzyme activity and molecular genetic testing.
Myoclonus epilepsy with ragged-red fibers often presents in this age group. There may be evidence for peripheral nerve dysfunction, and muscle involvement may be demonstrated by the finding of ragged-red fibers on biopsy. This maternally inherited disorder may be associated with features of MELAS syndrome. Short stature, optic atrophy, and bilateral lipomas may be found. Uncommon forms of progressive myoclonus epilepsy include juvenile neuroaxonal dystrophy, the neuronopathic form of Gaucher disease, atypical inclusion-body disease, and renal failure myoclonus syndrome (05).
The diagnosis of EPM1 is made based on family history, age at onset, geographical and ethnic context, and on the typical features of myoclonus and epilepsy, in the absence of severe cognitive and sensory deficits (29).
Exclusion of other forms of progressive myoclonus epilepsy in the presence of the characteristic clinical features makes the diagnosis possible. EPM1 should be suspected when there are significant periods of worsening and improvement as well as a substantially beneficial effect of alcohol on myoclonus.
Detection of CSTB gene mutations confirms the clinical suspicion of EPM1. Genetic testing for heterozygotes and even prenatal diagnosis are both possible (64). All other tests are not specific for this disease. The changes of mitochondrial disease or of Lafora body disease are absent from muscle and skin biopsy material in EPM1. Vacuoles in eccrine sweat glands may be present, but there is no storage material. Biochemical abnormalities have not been demonstrated.
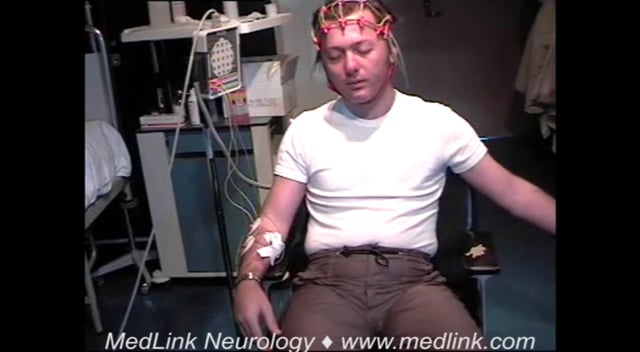
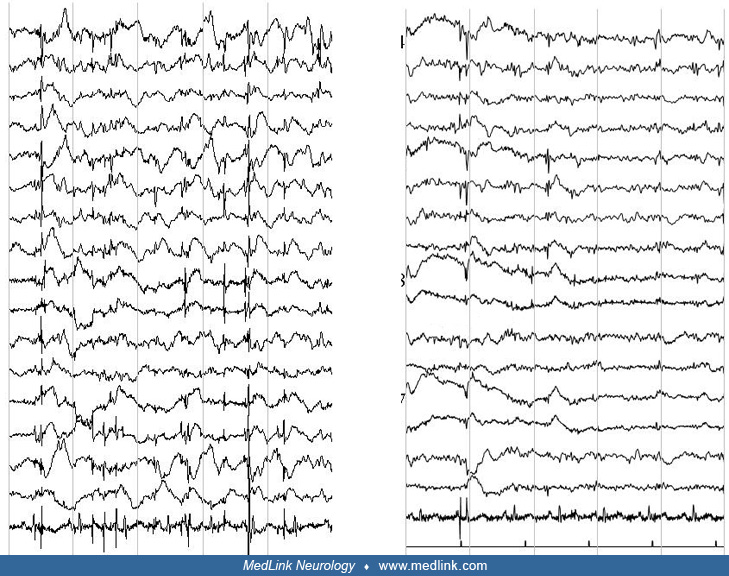
The interictal EEG is initially normal but abnormalities always appear as the disease progresses. Myoclonic jerks may or may not be associated with EEG changes.
Photosensitivity is common. In a long-term EEG evaluation of patients with EPM1, alpha activity was normal in 68% of patients at the beginning of the disease and kept stable over the years (26). Generalized spike-wave discharges were present in 92% of patients at the onset of the disease and gradually disappeared during the follow-up with a significant difference (p< 0.001) after the 15th year of the disease. Photoparoxysmal responses were present in 88% of patients at the disease onset and gradually disappeared with a significant difference (p< 0.001) after the 10th year of the disease. A gradual reduction of generalized spike-wave discharges and a progressive disappearance of physiological sleep patterns were observed in sleep EEGs. Gargouri-Berrechid and associates studied the long-term evolution of EEG in EPM1 and concluded that the progressive disappearance of EEG abnormalities is rather due to antiepileptic treatment than a gradual spontaneous tendency to decrease over time (28).
Specific neurophysiological testing as per any other cortical myoclonus is abnormal (87; 103; 34). Cortical neurophysiology reveals a transient wave preceding myoclonus on EEG jerk-locked back-averaging, enlarged somatosensory evoked potentials, and positive long-loop C-reflexes at rest.
Brain MRI is normal at the initial stages of disease onset. An MRI morphometric analysis of 10 patients demonstrated a significant loss of bulk of the basis pontis, medulla, and cerebellar hemispheres compared to controls (79). T1-weighted, 3D images analyzed with voxel-based morphometry revealed atrophy in the bilateral primary, premotor, and supplementary motor cortex (56). The thalamus and precuneus were also bilaterally affected, but no infratentorial changes were detected in the group analysis (56). Patients with EPM1 have significantly more T2-weighted high-intensity signals on MRI than control subjects due to the increased prevalence of these signals in the basal ganglia (55). Koskenkorva and associates evaluated possible changes in CTH of patients with EPM1 compared with healthy controls and correlated those changes with clinical parameters (58). Fifty-three genetically verified patients with EPM1 and 70 healthy volunteers matched for age and sex underwent 1.5T MR imaging. T1-weighted 3D images were analyzed with CTH analysis to detect alterations. The patients were clinically evaluated for myoclonus severity by using the UMRS. Higher UMRS scores indicate more severe myoclonus. CTH analysis revealed significant thinning of the sensorimotor and visual and auditory cortices of patients with EPM1 compared with healthy controls. CTH was reduced with increasing age in both groups, but in the patients the changes were confined specifically to the aforementioned areas whereas in controls the changes were more diffuse. Duration of the disease and the severity of myoclonus correlated negatively with CTH. The authors concluded that cortical thinning in the sensorimotor areas in EPM1 correlated significantly with the degree of the severity of the myoclonus and is most likely related to the widespread stimulus sensitivity in EPM1.
Nigri and associates assessed the structural and functional involvement of the cerebellum in 13 patients with mild to moderate myoclonus of EPM1 (82). They used voxel-based morphometry and spatially unbiased infratentorial template analyses of structural magnetic resonance imaging scans and functional MRI scans during block and event-related go/no-go motor tasks. The results were compared with those obtained in 12 age-matched healthy controls and in 12 patients with hereditary spinocerebellar ataxia. Structural analyses revealed different patterns of atrophic changes in the patients with EPM1 and those with hereditary spinocerebellar ataxia: in the former, they involved both cerebrum and cerebellum, but in the latter, only the cerebellum. During fMRI, block and event-related go/no-go tasks similarly activated the cerebellum and cerebrum in the patients with EPM1 and healthy controls, whereas both tasks revealed much less cerebellar activation in the patients with hereditary spinocerebellar ataxia than in the other two groups. Volumetric evaluation of the patients with EPM1 showed that the cerebellum seemed to be marginally involved in a widespread atrophic process, and fMRI showed that it was not functionally impaired during motor tasks (82).
This includes prophylactic drug treatment, rehabilitation, and psychosocial support. Valproate, clonazepam, piracetam, and more recently levetiracetam and perampanel are the main antiepileptic agents (18). Zonisamide may also be effective in some cases (37; 42). Generalized tonic-clonic convulsions are relatively easily controlled but the myoclonus has usually been the limiting factor. Unlike most forms of epilepsy, progressive myoclonus epilepsy is best treated by polytherapy (84).
An important aspect of management is to avoid the use of contraindicated antiepileptic drugs such as phenytoin, carbamazepine, gabapentin, or vigabatrin (31). Lamotrigine is also contraindicated. A study found dose-related exacerbation of myoclonus, putative late-onset aggravation, or lack of improvement in five patients treated with adjunctive lamotrigine (29). Because of a promyoclonic effect in focal epilepsies (40), pregabalin is also probably contraindicated in EPM1. Removal of phenytoin may lead to striking improvement and a return of the ability to walk (23).
Magaudda and colleagues studied the efficacy of levetiracetam in 13 patients with EPM1 about seizure frequency and myoclonus by using a simplified myoclonus rating score and comparing the patients' status before levetiracetam and at the last follow-up (75). Levetiracetam was given at 2000 to 4000 mg/day for 0.5 to 26 months (mean, 13.8 months). One patient stopped levetiracetam within 2 weeks because of side effects and lack of efficacy. None of the other 12 patients reported side effects. The average myoclonus score significantly changed from 3.1 to 2.4 (p = 0.01), but only eight patients had a measurable improvement. The best effects were noted in the younger patients. In patients previously treated with high-dose piracetam, discontinuation of piracetam was not always well tolerated, and a combination of piracetam at lower doses and levetiracetam appeared to be a practical solution. The authors concluded that levetiracetam should probably be considered as a major treatment option early in the course of EPM1 (75). Similarly, Papacostas and colleagues reported three patients with genetically confirmed EPM1 who were treated with levetiracetam as adjunctive therapy for their myoclonus (89). All cases responded with a decrease of their myoclonus and improvement of their quality of life. Two were able to return to or continue their employment. Patients tolerated the drug well without reported side effects.
Italiano and associates carried out a pilot, open-label trial of add-on zonisamide in 12 patients with EPM1 (42). Oral zonisamide was gradually titrated until the target dose of 6 mg/Kg per day. Unified Myoclonus Rating Scale was obtained in each subject before and after zonisamide add-on. A significant reduction of myoclonus severity was reached after zonisamide introduction. Two patients withdrew due to adverse effects.
In a retrospective study of 20 patients with EPM1 at Helsinki University Hospital, the majority of the patients had severe functional disabilities (94). In the year preceding the last hospital visit, all patients were receiving polytherapy, and 14 patients had been free of tonic-clonic seizures. During follow-up, improvement in myoclonia had been recorded for the majority of patients with either add-on piracetam, topiramate, or levetiracetam, but valproate was still in use by all patients. Treatment with lamotrigine had been started and retained less often relative to other antiepileptic drugs. Add-on antiepileptic drug treatment was often associated with significant adverse effects.
Brivaracetam, an antiepileptic agent that is chemically related to levetiracetam, has been granted orphan medicinal designation in Europe and the United States for the treatment of symptomatic myoclonus and progressive myoclonic epilepsies (76; 49; 98). Kalviainen and colleagues assessed the results of brivaracetam in two randomized, double-blind, placebo-controlled studies of EPM1 (48). The effect of brivaracetam on action myoclonus was not statistically significant. However, (1) action myoclonus score showed wide intrapatient variability and may not have been the optimal tool to measure the severity of myoclonus in EPM1, and (2) both studies had very high completion rates (95.3% overall) and a high percentage of patients (88.7% overall) entered long-term follow-up; both likely to be influenced by good tolerability. Thus, the authors concluded that these studies demonstrate the feasibility of rigorous trials in progressive myoclonic epilepsy (48). Patients completing these studies and expected to benefit from brivaracetam treatment were then eligible for the long-term open-label extension study (N01125; NCT00175916) (06). Ninety-four patients with EPM1 (11.0% of the entire cohort) were enrolled into this phase 3, single-arm, open-label extension trial with the primary objective to evaluate long-term safety and tolerability of adjunctive brivaracetam (mean, 100 mg/day in the progressive myoclonic epilepsy type 1 subgroup but up to 200 mg/day). Maintenance of efficacy over the time was a secondary endpoint. As a result, brivaracetam was overall well-tolerated with treatment-emergent adverse events reported by 84.4% of all patients and is primarily related to the CNS, particularly headache and somnolence in the EPM1 subgroup.
In a study of 12 patients with this disease, with perampanel, 10 patients had a clear clinical response of myoclonus, and five were able to reduce concomitant therapy (18). Improvement was noted sometimes as soon as with 2 mg/day. Epileptic seizures stopped on perampanel in the six patients who still had experienced generalized tonic-clonic or myoclonic seizures (100%). Some abatement of efficacy on myoclonus was seen in two patients who still retained some benefit. Weight gain was reported in six patients. Psychological and behavioral side-effects were observed in six patients and led to the withdrawal of perampanel in three cases and dose reduction in three, with abatement of the problems. The authors concluded that: (1) perampanel may show marked efficacy even in severe cases, particularly against myoclonus, but also against seizures; (2) perampanel should be tried in patients with EPM1 whose seizures are not satisfactorily controlled; and (3) its use is limited because of psychological and behavioral side effects, with higher doses of approximately 6 mg/day or greater likely risk factors (18). A systematic review explored the outcomes with perampanel in 59 patients with progressive myoclonic epilepsies in two non-randomized interventional studies (Trinka and Lattanzi 2021). Reductions in myoclonic seizures frequency and severity were seen across study types and different etiologies, including Lafora disease and Unverricht–Lundborg disease; reductions in primary generalized tonic-clonic seizures were also often reported. Considering the uncontrolled nature of these studies and different populations and settings, results are reasonably consistent with meaningful responses in 50% to 80% of patients.
Repetitive transcranial magnetic stimulation can be safely used in patients with EPM1, improves action myoclonus, and partially restores deficient cortical inhibition (95). In one study, nine patients with EPM1 underwent two series of 500 stimuli at 0.3 Hz through a round coil twice a day for five consecutive days (95). Clinical and neurophysiological examinations were performed 2 hours before starting the first session and 2 hours after the end of the last session. Eight patients completed the protocol; one discontinued because of a transient increase in spontaneous jerks. The unified myoclonus rating scale indicated a 25% reduction in posttreatment myoclonus, with an action score associated with an increase in the cortical motor threshold and lengthening of the cortical silent period. The decrease in the myoclonus with action scores correlated with the prolongation of cortical silent period.
An etiologically oriented treatment has also been tested: cell-permeable peptide TAT-protein transduction domains (PTD) fused to cystatin as a potential protein therapy of EPM1 is problematic because TAT-PTD-CSTB does not penetrate the cells despite initial evidence of time- and concentration-dependent transduction. Therefore, it cannot be used as a form of replacement of the intracytoplasmic protein missing in EPM1 (03; 92).
As abnormalities of CSTB mRNA are common effects of various mutations in EPM1, Matos and colleagues developed an antisense oligonucleotide in a Portuguese patient with the homozygous c.66G>A CSTB mutation, which activates a cryptic splice site downstream in CSTB intron 1 (80). A specific locked nucleic acid antisense oligonucleotide was designed to complementary target the cryptic donor splice site in intron 1 of the CSTB and redirect splicing. This highlights the feasibility of antisense oligonucleotides therapies and the relevance of personalized treatment for patients (80; 92; 97).
Lastly, as one of the pathological determinants of EPM1 is reactive oxygen species and the oxidative stress, high doses of N-acetyl-cysteine have been tested in some patients showing a marked improvement in seizures, ataxia, and a blockade of symptoms progression (97). This suggests the need to further dissect the specific altered inflammatory pathways in this condition to that to use specific antiinflammatory compounds (92).
In conclusion, the progress in the understanding of the biological bases of EPM1 will provide the bases for precision medicine-based approaches, aiming at targeting the specific mutations and possibly striking the neurodegenerative process underlying this disease. Moreover, the development of new, wearable devices to objectively quantify myoclonus in real life will undoubtedly prove valuable to evaluate treatment effects (91).
The outcome seems related to the severity of the myoclonus, generalized seizures, and ataxia.
The ataxia is greatly increased by the use of some antiepileptic drugs, notably phenytoin, prescribed to many of these patients in the past. Withdrawal of phenytoin has led to a marked improvement (23).
There is some disagreement as to whether intelligence remains normal (29) or may deteriorate even when optimal antiepileptic and antimyoclonic treatments are used (60; 16).
Life expectancy may not be affected. However, though the long-term functional outcome and survival have improved, the overall efficacy of antimyoclonic drugs remains unsatisfactory (16).
One study showed that EPM1 progresses only over a limited period and stabilizes thereafter (74). However, in a multicenter study of the circumstances of death amongst 19 patients with EPM1, six patients died of sudden unexpected death in epilepsy, four patients died of stroke, drowning, complications of chronic alcoholism, and Wernicke encephalopathy, two committed suicide, and one died in a car accident (52). The authors concluded that although the prognosis of EPM1 has improved, there are still severe forms and high risk of early death, including sudden unexpected death in epilepsy (52).
There is no record of people with this disorder bearing children, nor is there information on the effect of pregnancy on the disorder.
There is no record of particular sensitivity to anesthetic agents.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Pasquale Striano MD PhD
Dr. Striano of the University of Genova, Istituto Gaslini, has no relevant financial relationships to disclose.
See ProfileAntonella Riva MD
Dr. Riva of the University of Genoa has no relevant financial relationships to disclose.
See Profile
Solomon L Moshé MD
Dr. Moshé of Albert Einstein College of Medicine has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Epilepsy & Seizures
Mar. 22, 2024
Epilepsy & Seizures
Mar. 20, 2024
Epilepsy & Seizures
Mar. 07, 2024
Epilepsy & Seizures
Mar. 06, 2024
Epilepsy & Seizures
Mar. 05, 2024
Epilepsy & Seizures
Mar. 05, 2024
Epilepsy & Seizures
Mar. 04, 2024
Epilepsy & Seizures
Feb. 29, 2024