




Behavioral & Cognitive Disorders
Academic underachievement
Apr. 18, 2024
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
The author explains the clinical presentation, etiology, differential diagnosis, and diagnostic workup of progressive subcortical gliosis, a chromosome-17-linked dementia with unique pathologic features. Microscopically, the major pathologic change is a marked fibrillary astrocytosis, particularly in the area of the short cortical association tracts at the junction of cortical lamina VI and the subcortical white matter and in the subpial cerebral cortex. Overall, the survival of patients with progressive subcortical gliosis averages about 10 years and is similar to that of other types of frontotemporal dementia.
• Progressive subcortical gliosis is a rare dementing disorder resembling Pick disease but with distinctive neuropathologic features. The clinical manifestations are those of a frontotemporal dementia and overlap with those of other frontotemporal dementias. | |
• Progressive subcortical gliosis has an insidious onset, generally in the fifth or sixth decade. The course is progressive, generally over 5 to 15 years, but both fulminant and protracted courses occur. | |
• Common initial manifestations include personality and emotional changes, lack of judgment and insight, deterioration in social behavior, delusions, paranoia, auditory and visual hallucinations, and depression. | |
• Microscopically, the major pathologic change is a marked fibrillary astrocytosis, particularly in the area of the short cortical association tracts at the junction of cortical lamina VI and the subcortical white matter and in the subpial cerebral cortex. | |
• Most cases have been sporadic, but some hereditary forms are recognized, with linkage demonstrated in one kindred to a region on the long arm of chromosome 17 (17q21-22). | |
• Overall, the survival of patients with progressive subcortical gliosis averages about 10 years and is similar to that of other types of frontotemporal dementia. |
Progressive subcortical gliosis is a rare dementing disorder clinically and pathologically resembling Pick disease but with distinctive neuropathologic features. In 1949, Neumann described three cases under the appellation of "Pick disease, type II" (47). In 1967, Neumann and Cohn reported four additional cases and suggested the designations "primary subcortical gliosis" and "progressive subcortical gliosis" (48); although neither of these terms is ideal, the latter has become established in the literature (10).
Although rare reports of familial cases were reported beginning in the 1940s, it was not until the late 1980s and 1990s that it was recognized that progressive subcortical gliosis could be transmitted as an autosomal dominant trait (16; 36). Progressive subcortical gliosis was linked to a tau mutation in the microtubule-associated protein tau (MAPT) gene on the long arm of chromosome 17 (54; 24) and, thus, it was included in the group of "frontotemporal dementias and Parkinsonism linked to chromosome 17" group (FTDP-17) (21; 10). More than 60 MAPT mutations have since been identified, usually causing behavioral variant frontotemporal dementia (FTD) or parkinsonism clinically (59).
MAPT-related frontotemporal dementia (MAPT-FTD) is a progressive neurodegenerative disorder characterized by frontotemporal dementia, often with parkinsonian manifestations. Most affected individuals have behavioral changes consistent with a diagnosis of behavioral variant FTD (bvFTD) or a parkinsonian syndrome, but there is considerable phenotypic heterogeneity, even between and within families with the same MAPT variant (56). End-stage disease is characterized by severe dementia often with muteness and profound functional motor impairment (often becoming chairbound or bedbound) (56). Mean disease duration is about 9 years but can be more than 30 years (56).
The concept of "frontotemporal dementias and Parkinsonism linked to chromosome 17" has since been challenged (19). Review of known cases suggested that distinct MAPT mutations result in particular tau subtypes linked to frontotemporal lobar degeneration (FTLD). As such, Forrest and colleagues argued FTLD-tau cases with MAPT mutations should be considered familial forms of FTLD-tau subtypes rather than a separate FTDP-17 category.
|
• Progressive subcortical gliosis presents as a frontotemporal dementia similar to Pick disease. | |
|
• Progressive subcortical gliosis has an insidious onset, generally in the fifth or sixth decade. | |
|
• Common initial manifestations include personality and emotional changes, lack of judgment and insight, deterioration in social behavior, delusions, paranoia, auditory and visual hallucinations, and depression. | |
|
• Terminal manifestations included profound dementia, akinetic mutism, incontinence, severe dysphagia, and extrapyramidal signs. |
Progressive subcortical gliosis has an insidious onset, generally in the fifth or sixth decade, although individuals have developed the disease as early as 30 years and as late as 79 years (36; 38). The distribution of age of onset is similar for familial and sporadic cases. There is no apparent gender predilection.
Common initial manifestations include personality and emotional changes, lack of judgment and insight, deterioration in social behavior, delusions, paranoia, auditory and visual hallucinations, and depression (36; 38; 18). Subsequently, affected individuals develop severe memory difficulty, speech output generally declines, and word-finding problems appear. Some patients develop verbal stereotypy, echolalia, or manifestations of the human Klüver-Bucy syndrome (36). Dysarthria and myoclonus have also been reported (18).
Several cases had clinical evidence of relatively localized dysfunction involving visual, motor, or cognitive domains (47; 57; 02; 05; 04; 06); in some, the changes developed rapidly over weeks, prompting a diagnosis of cerebral infarction or hemorrhage (47; 02). Isolated sporadic cases have also been reported with posterior cerebral dysfunction or atrophy (05; 04; 55; 08; 53), or the clinical manifestations of the Steele-Richardson-Olszewski syndrome (77; 01). Terminal manifestations included profound dementia, akinetic mutism, incontinence, severe dysphagia, and extrapyramidal signs (36).
The course is progressive, generally over 5 to 15 years, but both fulminant (3 months to 2 years) and protracted (spanning decades) courses occur. The duration of illness is generally longer in familial cases than in sporadic cases. The most common causes of death are pneumonia, other respiratory infections, and pulmonary emboli.
Overall, the survival of patients with progressive subcortical gliosis is similar to that of other types of frontotemporal dementia (13).
In his early 40s, the patient developed personality change, progressive memory impairment, and decreased activity. He retired from his accounting work because of forgetfulness, difficulty making decisions, and frequent errors in performing simple calculations. In his late 40s, he had poor insight and judgment, impaired memory, mild word-finding problems, and difficulty performing serial subtractions. A masked face, slightly increased appendicular and axial tone, and a fine postural tremor were present, but there were no gaze abnormalities. Complete blood count, serum chemistries, thyroid function studies, and serum rapid plasma reagin were normal. Head CT showed mild cerebral atrophy with ventriculomegaly. Radioisotope cisternography was normal. EEG showed well-modulated 10-Hz alpha activity posteriorly, but frequent, rhythmic, low-amplitude delta transients were observed in the right anterior and right midfrontal areas. Cerebrospinal fluid examination showed an opening pressure of 180 mm H2O, a protein level of 27 mg/dL, a glucose level of 55 mg/dL, and no cells.
At age 50, the patient had decreased verbal fluency, mild comprehension impairment, and word-finding difficulty with semantic paraphasic errors. Over the next several years, he became more compulsive, regimented, and perseverative. His affect became progressively blunted. He lost interest in sexual intercourse, was incontinent, and began eating excessively. He stopped speaking spontaneously but had prominent echolalia. Later he developed severe dysphagia and became bed-bound and emaciated. He died at age 58 years from aspiration.
|
• Most reported cases of progressive subcortical gliosis have been sporadic, but there are several reports of familial progressive subcortical gliosis. | |
|
• In two large kindreds, progressive subcortical gliosis was transmitted as an autosomal dominant trait. | |
|
• Linkage has been demonstrated in one kindred to a region on the long arm of chromosome 17 (17q21-22), a site previously associated with the disinhibition-dementia-parkinsonism-amyotrophy complex. | |
|
• Overproduction of 4-repeat tau is probably the primary effect of the progressive subcortical gliosis mutation. In a kindred with progressive subcortical gliosis, a tau mutation (at position +16 of the intron after exon 10) leads to an overrepresentation of the soluble 4-repeat tau isoforms that assemble into wide, twisted, ribbon-like filaments. | |
|
• Microscopically, the major pathologic change is a marked fibrillary astrocytosis, particularly in the area of the short cortical association tracts at the junction of cortical lamina VI and the subcortical white matter and in the subpial cerebral cortex. | |
|
• Neuronal cytoskeletal inclusions (Pick bodies, neurofibrillary tangles, Lewy bodies) are absent in progressive subcortical gliosis. |
Most reported cases of progressive subcortical gliosis have been sporadic, but there are several reports of familial progressive subcortical gliosis (26; 48; 31; 16; 37; 36; 68). In several of the initial reports of familial progressive subcortical gliosis, family history information is sketchy, and detailed clinical information and pathological confirmation are available only for the index cases (26; 48; 31). In a preliminary report in 1986, Currier and colleagues first demonstrated that progressive subcortical gliosis could be transmitted as an autosomal dominant trait (16). In a large kindred of 90 individuals spanning six generations, 21 of 65 members in four consecutive generations may have been affected, and five members from two generations were proved to have been affected on the basis of autopsy (36).
Lanska and colleagues subsequently confirmed these findings in a second kindred of 80 individuals spanning four generations.
Ten of 30 members in three consecutive generations may have been affected by the disease, and two siblings were proved to have been affected on the basis of autopsy (37; 36). In 1995, Petersen and colleagues suggested that familial progressive subcortical gliosis may be a prion disease (54), but this conclusion was later retracted (22).
Linkage has been demonstrated in one of the kindreds reported by Lanska and colleagues (36) to a region on the long arm of chromosome 17 (17q21-22) (54; 20; 21; 76), a site previously associated with the disinhibition-dementia-parkinsonism-amyotrophy complex (40; 75; 76). As early as 1995, it was recognized that candidate genes in the region include microtubule-associated protein tau (MAPT) (54). Subsequently, a tau mutation at position +16 of the intron after exon 10 has been identified (24) in one kindred with progressive subcortical gliosis (36; 54). The mutation destabilizes a predicted stem-loop structure and leads to an overrepresentation of the soluble 4-repeat tau isoforms that assemble into wide, twisted, ribbon-like filaments. Overproduction of 4-repeat tau is probably the primary effect of the progressive subcortical gliosis mutation. It may lead to an excess of 4-repeat tau over available binding sites on microtubules, resulting in the hyperphosphorylation of tau and its assembly into filaments (35).
Some other conditions included in the "frontotemporal dementias and Parkinsonism linked to chromosome 17" group (21), but with clinicopathologic differences from progressive subcortical gliosis (21; 41), have mutations in the tau gene (60; 63; 74; 50; 52). Several studies have identified various missense mutations, silent mutations, and amino acid deletions in the tau gene in patients with frontotemporal dementias (64; 65; 66; 39). Mutations have also been described at positions +3, +12, +13, +14, and +16 of the intron after exon 10 of the tau gene, and at the intron 10+11-splice site (61; 14; 39). Detailed tau pathology has been reported for the kindred with the +3 mutation, also known as “familial multiple system tauopathy with presenile dementia” (62; 61); affected individuals have neuronal and glial tau pathology similar to that of progressive subcortical gliosis. It is also characterized by the presence of wide, twisted ribbons made of 4-repeat tau isoforms and an overproduction of soluble 4-repeat tau, indicating that the different mutations in the intron after exon 10 cause disease through the same mechanism. These findings firmly establish that the overproduction of 4-repeat tau alone is sufficient to lead to filamentous tau pathology and a dementing disease (64).
How changes in the ratio of 3-repeat tau to 4-repeat tau produce neuronal and glial dysfunction and cell death is unknown. 3-repeat tau and 4-repeat tau may bind to distinct microtubule sites, and an altered ratio of these isoforms may disrupt microtubule function (64; 39). Also, overproduction of 4-repeat tau may simply produce an excess of free cytosolic tau, which then becomes hyperphosphorylated and is assembled into pathologic filaments (39). Other mutations alter the binding of tau to microtubules and interfere with its ability to promote microtubule assembly (64; 39).
The existence of tau mutations with distinct pathogenetic mechanisms may explain the phenotypic heterogeneity of atypical dementias that previously led to their classification into separate disease entities.
Another kindred with autosomal dominant subcortical gliosis presenting as a frontotemporal dementia, but without tau- or ubiquitin-containing inclusions and without ubiquitin or TDP-43 staining, has been reported by Swerdlow and colleagues (68). Symptom onset was generally in the fifth or sixth decade, but some individuals did not become symptomatic until their eighth decade. The TDP-43 type C form of frontotemporal lobar degeneration is characterized by the presence of immunoreactive TDP-43 dystrophic neurites, neuronal cytoplasmic inclusions, neuronal loss, and gliosis, and the absence of neuronal intranuclear inclusions (29).
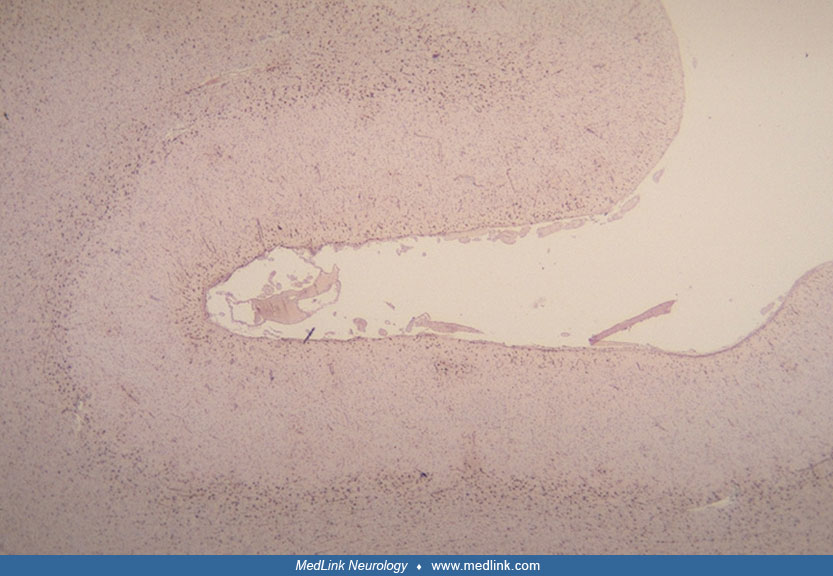
The brains of progressive subcortical gliosis cases are moderately to severely atrophic, usually with preferential involvement of the white matter of the frontal and temporal lobes (36), but several cases affected mainly the frontal lobes, and one sporadic case affected predominantly the parietal and occipital lobes (05; 04). Most often, the atrophy is symmetric (36), but in several cases, one side was more affected than the other (47; 78; 57; 72).
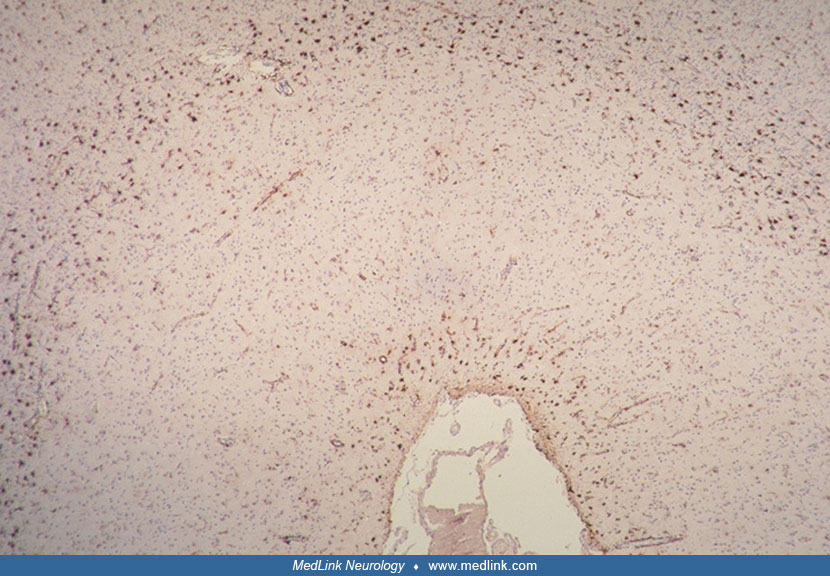
Microscopically, the major pathologic change is a marked fibrillary astrocytosis, particularly in the area of the short cortical association tracts at the junction of cortical lamina VI and the subcortical white matter and in the subpial cerebral cortex (48; 57; 42; 58; 72; 36).
This gliosis is most pronounced in the frontal and temporal lobes, occurs in the absence of identifiable myelin loss, and is out of proportion to the mild neuronal loss in the cortex (72; 36). Lesser degrees of gliosis are observed in the deep white matter, basal ganglia, thalamus, brainstem tegmentum, cerebellum, and central gray matter of the upper cervical spinal cord (36). In addition, variable protoplasmic astrocytic proliferation is evident in all layers of the cerebral cortex (57; 42; 58; 06; 36).
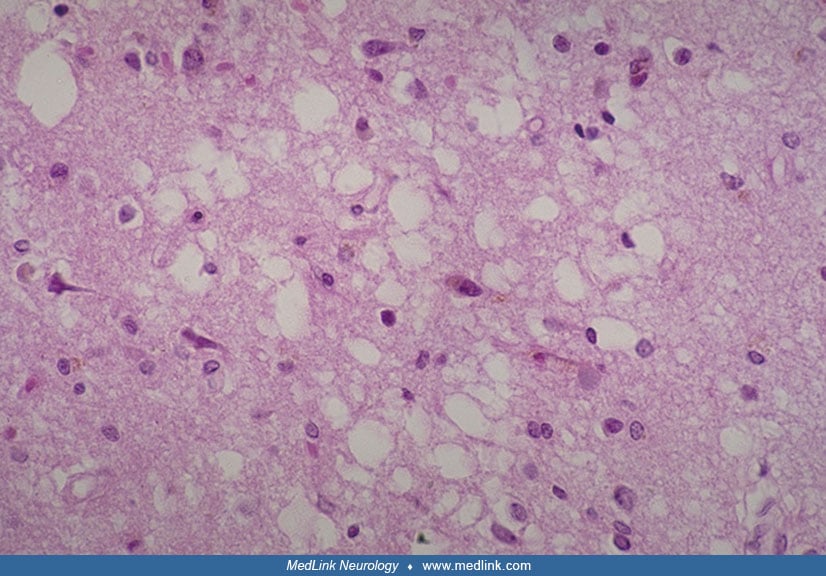
Laminar spongiosis has been variably reported in laminae II and III of the cerebral cortex in cases of progressive subcortical gliosis (48; 57; 42; 58; 72; 36).
The spongiosis may be focal and probably occurs late in the disease. It generally spares the deeper layers of the cortex as well as the basal ganglia, thalamus, brainstem, and cerebellum (36). Ultrastructural studies have shown areas of clarification and frequent vacuoles in large postsynaptic dendrites surrounded by glycogen and mitochondria (42). This spongiosis has been attributed variously to neuronal loss, loss of dendritic arborizations of neurons that expand in the second layer, and pathologic alteration and disintegration of astrocytes (42; 58; 72). The spongiosis of progressive subcortical gliosis is similar to that in other neurodegenerative diseases (eg, Pick disease, Alzheimer disease), but it differs from the spongiform change of Creutzfeldt-Jakob disease, which is pancortical, most prominent in the deeper layers, and frequently involves the thalamus, basal ganglia, brainstem, and cerebellum (43; 36).
Neuronal cytoskeletal inclusions (Pick bodies, neurofibrillary tangles, Lewy bodies) are absent in progressive subcortical gliosis (36). Several cases had occasional balloon neurons in the cerebral cortex (47; 78); however, most authors did not identify such cells. Although considered one of the histopathologic hallmarks of Pick disease (51), balloon neurons are nonspecific and do occur in several other neurodegenerative disorders.
In several cases, the pars compacta of the substantia nigra exhibited moderate to marked degenerative changes (36). There was no apparent neuronal loss or gliosis in the nucleus basalis of Meynert in the cases wherein this was examined (36).
Studies in a transgenic mouse model of frontotemporal dementia and parkinsonism linked to chromosome 17 indicate that the L-dopa-resistant parkinsonism in this tauopathy results from early loss of dopaminoreceptive neurons in the striatum (12). Accumulation of mutant tau leads to neurofibril degeneration and apoptosis through caspase-3 activation (12). In the late stages, dopaminergic neurons and dopaminoreceptive neurons decreased equally (12).
Alterations in pro-nerve growth factor and brain-derived neurotrophic factor have been identified in non-Alzheimer disease tauopathies (03). Neurotrophin dysregulation as a result of tau pathology may represent a mechanism by which tau produces neurotoxicity in progressive subcortical gliosis and other non-Alzheimer dementias (03).
Even though the hippocampus is not the primary pathologic site in progressive subcortical gliosis and other tauopathies, there are nevertheless disease-specific regional selective neuronal vulnerabilities that can be demonstrated in the hippocampus for these disorders (45).
|
• Although rare, progressive subcortical gliosis has been identified in as many as 4% to 7% of cases in autopsy series of demented individuals. |
The frequency of progressive subcortical gliosis in the general population is unknown. It is thought to be a rare disorder, but limited data from autopsy series and brain banks suggest that progressive subcortical gliosis is more common than generally recognized. In autopsy series of demented individuals, progressive subcortical gliosis has been identified in as many as 4% to 7% of cases (67; 07; 23). Cases with the disorder are present in most established brain banks; in general, the proportion of cases with progressive subcortical gliosis in established brain banks is similar to the proportions with progressive supranuclear palsy, Pick disease, or Creutzfeldt-Jakob disease. Nevertheless, the relative frequency of progressive subcortical gliosis may be overestimated by such data because cases with early onset and unusual features are perhaps more likely to be autopsied.
The clinical manifestations of progressive subcortical gliosis are those of a frontotemporal dementia and overlap with those of Pick disease and other frontotemporal dementias, including Alzheimer disease, corticobasal ganglionic degeneration, Binswanger disease, and similar conditions (33; 73; 11; 69).
Although in many cases the clinical manifestations of progressive subcortical gliosis are similar to those of Pick disease and Alzheimer disease, progressive subcortical gliosis can be distinguished on pathologic grounds (47; 48; 36; 38; 33). Progressive subcortical gliosis shares with Pick disease and other frontotemporal dementias a predilection for the frontal and temporal poles, whereas in Alzheimer disease, the posterior temporal and parietal lobes are preferentially involved. The atrophic process in progressive subcortical gliosis is not sharply demarcated, whereas in Pick disease, there are typically circumscribed areas of severe atrophy with an abrupt transition between severely affected and less affected areas.
In progressive subcortical gliosis, cortical neuron loss is mild or inconspicuous, and a characteristically distributed astrogliosis is most prominent subcortically and in the deep cortical laminae. In both Pick disease and Alzheimer disease, cerebral cortical neuron loss is prominent and is associated with a commensurate degree of cortical gliosis and little subcortical gliosis. Cerebrocortical neuronal inclusions are uniformly absent in progressive subcortical gliosis but are pathologic hallmarks of Alzheimer disease and Pick disease, seen even in early or incipient cases (44).
Despite these distinctions, the differentiation of progressive subcortical gliosis from Pick disease may be difficult, and some authors likely lump progressive subcortical gliosis with putative subtypes of Pick disease without Pick bodies (eg, Pick disease "type C" of Constantinidis and colleagues) (15; 70; 28).
Compared with other forms of frontotemporal dementia (27; 30), progressive subcortical gliosis appears to have a distinctive clinicopathologic profile (36; 21; 41), but several specific forms of frontotemporal dementia, including progressive subcortical gliosis, have now been linked to chromosome 17 (21; 14).
Thalamic degenerations involve similar neural structures and have histopathologic similarities to progressive subcortical gliosis (49; 06; 36). Primary thalamic degenerations are frequently accompanied by secondary gliosis in the cerebral cortex, subcortical white matter, basal ganglia, and various brainstem nuclei. Thalamic gliosis and neuron loss occur in progressive subcortical gliosis (78; 48; 06); however, they apparently develop independently of the other pathologic changes because (1) there is prominent gliosis of the short ipsilateral cortical association fibers that have no direct thalamic connections; (2) there is neither demyelination nor axon loss in the thalamic radiations; (3) gliosis in the cerebral cortex generally spares laminae III and IV, where the majority of thalamocortical projection fibers terminate (36).
Some of the "atypical" dementias have features in common with progressive subcortical gliosis. Although progressive subcortical gliosis and "frontal lobe degeneration of non-Alzheimer type" are clinically similar (25), pathologic differences exist between the two conditions (09; Englund and 09; 36). In frontal lobe degeneration, little atrophy occurs, and brain weights are generally greater in frontal lobe degeneration than in progressive subcortical gliosis. In addition, the pathologic changes of frontal lobe degeneration are more evident in the cerebral cortex rather than in the subcortical areas, with prominent cortical neuron loss and mild gliosis involving mainly the outer cortex. Finally, areas typically involved in progressive subcortical gliosis (basal ganglia, thalamus, substantia nigra, inferior olivary nucleus) are either not affected or are much less affected in frontal lobe degeneration. Several other reported cases of familial (32; 46; 34) and sporadic (71; 34) atypical non-Alzheimer dementia had clinical and pathologic similarities to progressive subcortical gliosis, but in all of these cases, there was prominent cortical neuron loss in addition to variable degrees of gliosis.
|
• Cerebral imaging is normal early in the course, but later shows generalized cerebral atrophy, sometimes preferentially affecting the frontal and opercular areas. |
Cerebral imaging is normal early in the course but later shows generalized cerebral atrophy, sometimes preferentially affecting the frontal and opercular areas.
Brain MRI may show hyperintensities in the subcortical white matter on T2-weighted images (18). Electroencephalograms remain normal except for mild slowing late in the illness. Screening for tau gene mutations in patients with frontotemporal dementia is still largely a research tool and is unlikely to identify pathogenic mutations in sporadic cases (66).
|
• Management is supportive. | |
|
• Although antipsychotic medications have been used to control hallucinations and paranoia, such agents may increase the extrapyramidal dysfunction that is commonly present in the later stages of the disorder. |
Management is supportive. Although antipsychotic medications have been used to control hallucinations and paranoia, such agents may increase the extrapyramidal dysfunction that is commonly present in the later stages of the disorder.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Douglas J Lanska MD FAAN MS MSPH
Dr. Lanska of the University of Wisconsin School of Medicine and Public Health and the Medical College of Wisconsin has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Behavioral & Cognitive Disorders
Apr. 18, 2024
Behavioral & Cognitive Disorders
Apr. 17, 2024
Behavioral & Cognitive Disorders
Mar. 29, 2024
Neuroimmunology
Mar. 24, 2024
Infectious Disorders
Mar. 24, 2024
Stroke & Vascular Disorders
Mar. 21, 2024
Behavioral & Cognitive Disorders
Mar. 07, 2024
Behavioral & Cognitive Disorders
Mar. 07, 2024