
Neuroimmunology
Acquired sensory neuronopathies
Apr. 07, 2024
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Autoimmune glial fibrillary acidic protein astrocytopathy is a form of monophasic or relapsing steroid-responsive meningoencephalomyelitis often accompanied by a hallmark radial periventricular brain MRI enhancement pattern and inflammatory cerebrospinal fluid with lymphocytic pleocytosis. GFAP-IgG was first described at the Mayo Clinic in the United States and subsequently confirmed by multiple international groups (33; 10).
|
• Autoimmune GFAP astrocytopathy manifests as a meningoencephalomyelitis (or limited form thereof) with dramatic steroid-responsiveness. Early relapses may occur if steroids are deescalated and discontinued quickly. | |
|
• Brain MRI may reveal a typical pattern of radial gadolinium enhancement; the spine MRI abnormalities are often hazy and longitudinally-extensive T2 hyperintensities with central cord enhancement. | |
|
• CSF testing for GFAP-IgG is more specific. Both the characteristic astrocytic, filamentous immunofluorescence pattern of CNS tissue staining (meeting all criteria) and positivity for GFAP-alpha isoform-specific-IgG are required for diagnosis. | |
|
• In most cases the etiology is unknown; in 25% of patients a neoplasm is detected after neurologic symptoms onset (most common being ovarian teratoma). |
In 1999, Uchida and colleagues identified glial fibrillary acidic protein antibodies in the serum and cerebrospinal fluid samples of two pug dogs with canine necrotizing encephalitis, a severe and rapidly progressive form of encephalitis characterized by mood alteration, ataxia and seizures, and features of necrosis and lymphocytic inflammation in pathological studies (31). Further studies confirmed the presence of GFAP-IgG in larger cohorts with similar clinical phenotype (28).
In humans, GFAP-IgG was described as a biomarker of meningoencephalomyelitis in 2016 by Fang and associates in a cohort of 16 patients who presented with subacute cognitive and behavioral impairment, inflammatory CSF, pathology consistent with lymphocytic inflammation, and robust steroid therapy-responsiveness (07). Some of those cases had been previously reported, though utilizing pathologic descriptors for nomenclature, such as “nonvasculitic autoimmune inflammatory meningoencephalitis (NAIM)” (02) or “chronic microglial encephalomyelitis” (01).
|
• Autoimmune GFAP astrocytopathy presentation is subacute (over days to a few weeks). | |
|
• Flu-like symptoms are common prior to onset of neurologic symptoms. | |
|
• Headache, neck stiffness, blurred vision, altered mental status, seizures, and psychiatric symptoms are the most common presenting manifestations. |
The most common clinical phenotype is consistent with meningoencephalitis, which may or may not be associated with myelitis. On the contrary, isolated myelitis is rarely found (05; 27). Onset is acute or subacute in the majority of patients, and prodromal flu-like symptoms with fever, cough, rhinorrhea, and sore throat are commonly reported. Subsequently, patients develop meningeal symptoms (headache, neck-stiffness, vomiting), encephalopathy, seizures, psychiatric disorders, and tremor. Ataxia and other movement disorders (including parkinsonism) are less commonly part of the clinical phenotype but may be more frequent in older patients (15). Dysautonomia occurs in 25% of patients (08). Rhombencephalitis featured prominently (65%) in a French cohort of GFAP astrocytopathy with prominent eye movement disorders, dysphagia, and cerebellar signs (10). Peripheral neuropathy occurs in up to 24% (typically as part of a multifocal syndrome) and presents usually with a lower-limb-predominant axonal or demyelinating motor neuropathy (25; 10; 04). Given the prodromal symptoms and meningeal involvement, patients are frequently diagnosed speculatively with “test negative” meningitis of infectious etiology. When spinal cord involvement occurs, it manifests with predominant bowel and bladder dysfunction and sensory symptoms, whereas motor impairment is milder in comparison to other autoimmune myelopathies (such as classical neuromyelitis optica). Ocular findings include blurred vision with optic disc edema but intracranial pressure is generally normal or only slightly elevated (03).
Median age at onset of autoimmune glial fibrillary acidic protein astrocytopathy is in the fifth decade, although patients of all ages may be affected (08; 05; 10); both sexes are affected equally. Children account for about 10% of patients. Clinical phenotypes in the pediatric and adult populations appear to be the same (05; 13; 10).
Neoplasia is detected in one third, the majority after neurologic symptom-onset (08; 10). The most common neoplasm reported is ovarian teratoma, occurring in women usually when n-methyl-D-aspartate receptor (NMDA-R)-IgG, aquaporin-4 (AQP4)-IgG, or both are in coexistence. The positive predictive value for teratoma when all three antibodies coexist is 71% (08), though approximately 50% of NMDA-R encephalitis patients in general have teratoma detected. Other diverse neoplasms have been described, including adenocarcinomas, lymphoma, renal cell carcinoma, and others. (08; 05; 10).
Although the coexistence of AQP4-IgG seems not to influence the clinical phenotype of glial fibrillary acidic protein astrocytopathy, patients with NMDAR-IgG and GFAP-IgG manifest with typical features of NMDA-R-encephalitis (08; 34). Coexisting autoantibodies less frequently reported include those specific for glutamic acid decarboxylase 65, striational, ganglionic acetylcholine receptors, Purkinje cell cytoplasmic antibody type 1 (PCA-1 or anti-Yo, seen with ovarian adenocarcinoma), antineuronal nuclear antibody type 1 (ANNA-1 or anti-Hu, seen with small-cell lung cancer), leucine-rich glioma inactivated 1, contactin-associated protein-like 2, gamma-aminobutyric acid A, myelin oligodendrocyte glycoprotein (MOG), and voltage gated calcium channels (08; 17; 21; 34).
Autoimmune GFAP astrocytopathy may be monophasic with complete or incomplete recovery after steroid therapy. A relapsing-remitting course has been described in 20% to 50% of the patients who have required long-term immunosuppression, especially if the initial steroid treatment is tapered early on (08; 05; 16). Progressive neurologic decline has been described in patients that have not received any immunotherapy (05; 17). Although limited prognostic factors have been identified to date, a pooled analysis of published cases suggests that the presence of myelitic lesions in GFAP autoimmunity is associated with a better response to initial immunotherapy, but also higher risk of subsequent relapse compared to cases without myelitis (32). Overall, the outcome is generally favorable if patients are treated with corticosteroids early, at high doses and for long periods. The outcomes are worse in patients not treated with immunosuppression (deaths have been described), with underlying malignancies or coexisting autoimmunity, as seen in the cases of NMDAR encephalitis. In contrast to reports from other countries, Chinese patients had severe disease and less immunotherapy-responsiveness. It is not apparent whether this was due to delayed initiation of therapy in erstwhile idiopathic cases, more severe disease among an Asian population, or other factors yet to be unearthed (21; 38).
Prior to the discovery of GFAP-IgG as a biomarker of autoimmune CNS disease, a 51-year-old man presented with subacute (over 2 months) cognitive impairment, tremor, and gait imbalance. He also reported a prodrome of new onset chronic daily headache, abdominal constipation, and urinary retention. On examination, he had diffuse hyperreflexia, significant action and rest tremor, and intermittent myoclonus.
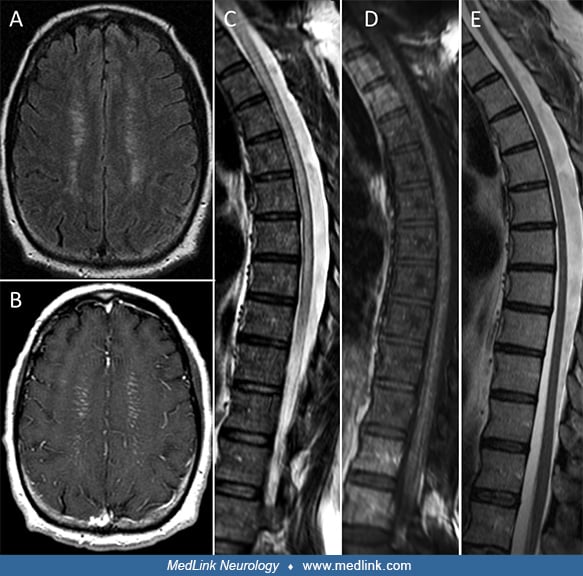
His brain MRI demonstrated T2 signal abnormalities and enhancing linear lesions in a periventricular distribution (see below image A-B) The spinal cord MRI revealed T2-hyperintense lesions involving the cervical and thoracic segments (see below image C-D). His CSF was remarkable for lymphocytic pleocytosis (212 cells/mcL [normal, ≤ 5], 97% lymphocytes), elevated protein (145 mg/dL [normal, ≤ 35]), and six CSF-exclusive oligoclonal bands (normal, < 4). A CT-scan of his entire trunk did not reveal systemic inflammation or malignancy. Histological examination of right frontal brain tissue after biopsy revealed microglial nodules and lymphocytic inflammation, without evidence of vasculitis or granulomatous disease. The patient received a course of IV methylprednisolone 1 g daily for 5 days, followed by oral prednisone taper (60 mg/kg for 1 month, taper over a total of 4 months). Five months later, during the taper (while on prednisone 10 mg per day) he had reaggravation of his symptoms. His steroids were increased again at 60 mg/day and long-term immunosuppression was initiated with mycophenolate mofetil (500 mg twice per day for 1 month followed by 1000 mg twice per day). The patient reported a complete recovery of cognitive and behavioral symptoms after 3 months whereas subtle gait sensory ataxia and diffuse hyperreflexia were still evident after two years of clinical and radiological surveillance. The steroids were eventually tapered off over 1 year starting 3 months after the initiation of mycophenolate mofetil. Extensive evaluations for malignancy were unrevealing. Follow-up brain imaging demonstrated complete resolution of enhancement and mild reduction of the T2-hyperintense lesion burden. The spinal cord follow-up MRI revealed moderate atrophy of the cervical and thoracic portions but without signal abnormalities (see image E).
This patient was retrospectively diagnosed, after autoantibody discovery, with autoimmune GFAP astrocytopathy on the basis of GFAP-IgG detection in both CSF and serum samples.
This description is of a real GFAP-IgG positive patient; some of the disease course and treatment details were altered for better disease illustration.
|
• The etiology and pathobiology of autoimmune GFAP astrocytopathy are poorly understood. The GFAP-IgG antibody itself should not be pathogenic but may be a surrogate marker of anti-GFAP T cell effectors. |
The glial fibrillary acidic protein is the main constituent of intermediate filament type 3 in astrocytes and a key component of the astrocyte’s cytoskeleton participating both in cell structure (eg, cellular integrity, resilience) and function (signal transduction) (22). GFAP is also important in astrocytic regeneration, motility, and migration (29; 19).
So far, 10 different GFAP isoforms have been identified (14). For diagnostic purposes of GFAP-IgG, the alpha and epsilon isoforms are currently used (see below in Diagnostic workup). At the Mayo Clinic, the alpha isoform alone is currently used for antigen-specific autoantibody-diagnosis (08), whereas Long and colleagues use both alpha and epsilon, as they have found some patients with GFAP-IgG specific solely for the epsilon isoform (21). The alpha isoform seems to be the most widely expressed and can be found in the astrocytes in the brain, spinal cord, and in the peripheral nervous system. The epsilon isoform is most abundant in neural progenitor niches in periventricular regions, the hippocampus, and the central spinal cord (14).
Etiology. Similar to other autoimmune diseases, in most cases of GFAP autoimmunity the disease trigger cannot be identified. Paraneoplastic cases are described and account for 25% to 30% of cases. Accompanying neoplasms, such as teratomas, are known to express GFAP, suggesting a tumor antigen-driven immune response that targets GFAP, initiated in the tumor bed and then targeting the central nervous system (05; 37). In the same context, a few cases have also been reported after immune checkpoint inhibitor therapies for cancer (08; 05).
Postinfectious cases have also been described after herpes encephalitides (herpes simplex virus type 1 or varicella zoster virus) (05; 20), suggesting that antigen release secondary to a CNS viral infection might trigger antigen-specific autoimmunity as seen in cases of NMDA-R encephalitis (18). Elevated CSF adenosine deaminase has been reported in one case of autoimmune GFAP astrocytopathy, which is consistent with (though not specific for) infectious etiology (24).
Pathogenesis. Insights into the disease pathogenesis in humans are given by the available histopathology. In the largest series to date, eight patients were reported with clinical and pathological data (36). Pathology in all revealed marked perivascular lymphocytic infiltration of cerebral white matter. In seven of eight, CD8+ cytotoxic T cells were predominant, particularly adjacent to dystrophic neurons and astrocytes. Macrophage infiltrates were also prominent (36). A prior study from the same group reported four patients reported by Long and colleagues with evidence of extensive perivascular inflammation along with microglial activation (21). The infiltrates were characterized by perivascular B cells and intraparenchymal T cells as well as antibody-secreting cells.
In an Italian cohort, Iorio and colleagues reported histopathological findings in one patient with meningeal biopsy that revealed necrotizing inflammatory lesions with infiltrate composed of CD8+ lymphocytes, macrophages, and some multinucleated giant cells (17).
The intracellular location of GFAP suggests that GFAP-IgG is a biomarker of the disease without pathogenic potential. It is postulated, even though not yet sufficiently investigated, that GFAP-specific CD8+ T cells are the immune effectors of this disease. In an animal model of GFAP autoimmunity, Sasaki and colleagues demonstrated the role of GFAP-specific CD8 T cells in causing CNS inflammation targeting both the gray and white matter of the brain and spinal cord (26). The composition and location of the lesions and the disease course were dependent on the disease trigger (spontaneous vs. viral triggers). Further studies need to be done to identify the pathogenic triggers and effectors of this autoimmune astrocytopathy. On the contrary to classic paraneoplastic cytotoxic T-cell mediated diseases, like cerebellar degeneration associated with Purkinje-cell cytoplasmic antibody type 1 (PCA-1 or anti-Yo) antibodies, patients with GFAP autoimmunity seem to respond to immunotherapy and return to the baseline, suggesting differences in the disease pathogenesis.
The cytokine profile of patients with GFAP autoimmunity is enriched for factors associated with the tumor necrosis factor-signaling pathway, as is also the case in aquaporin-4-positive NMOSD. Some cytokines such as MIP-3α have demonstrated an association with clinical severity in GFAP IgG autoimmunity (09).
GFAP astrocytopathy is a rare disease with an estimated prevalence of 0.6 per 100 000 and an incidence of 0.03 per 100 000 per year in Olmsted county, Minnesota (06). As mentioned above, the median age at symptoms onset is 44 to 50, but patients of all ages may be affected, with children accounting for 10% of cases (05). There is no clear gender predilection. Even though there were descriptions of severe disease in Asian populations, the data available at the moment do not allow us to identify any racial disparities (21; 38).
To date, effective prevention strategies are not known.
In patients with prominent meningeal involvement and encephalitis, an infectious meningitis or meningoencephalitis is high in the differential diagnosis, especially in patients with prodromal symptoms (35). Given the subacute presentation, viral or mycobacterial meningitis are more probable in the differential diagnosis than bacterial. Other considerations include neurosarcoidosis and other inflammatory CNS conditions, which can also present with similar MRI findings. Screening for inflammatory changes elsewhere in the body with a CT or PET-CT might be useful in cases of sarcoidosis. CNS lymphoma and meningeal carcinomatosis are also part of the differential diagnosis as is CNS vasculitis and lymphomatoid granulomatosis. In the past it is likely that some GFAP astrocytopathy cases have been diagnosed as “microvascular vasculitis”, with a negative angiogram and steroid-responsiveness (08). When the clinical presentation is mainly characterized by subacute cognitive decline together with movement disorders such as tremor or myoclonus, also rapidly evolving neurodegenerative diseases and Creutzfeldt-Jakob disease could be considered even though the CSF changes point toward an infectious/inflammatory disease.
In patients presenting with myelitis and blurred vision, neuromyelitis optica or MOG-IgG related autoimmunity are often considered in the differential diagnosis, as is neurosarcoidosis. Patients with GFAP-IgG-associated myelitis have a milder myelopathic presentation than AQP4-IgG seropositive patients and a more subacute course (27). In autoimmune GFAP astrocytopathy, patients present with papillitis with preserved vision. This presentation contrasts with the vision loss (at least temporary) and eye pain accompanying AQP4 and MOG autoimmune CNS disorders (03).
Association between disorders of T-cell function and GFAP autoimmunity have been reported. These include HIV infection, chronic lymphopenia, and exposure to immune checkpoint inhibitors, and they were observed in 23% of the total cohort (08; 10).
|
• CSF testing for white cell count and GFAP-IgG is critical. Serum antibody testing does not suffice. |
GFAP-IgG in the CSF of patients detected by both tissue-based immunofluorescence assays showing the characteristic pattern of immune-fluorescence on CNS tissue with filamentous staining of the ependymal/sub-ependymal and pial/sub-pial regions and the myenteric plexus as well as a confirmation by a GFAP-specific transfected-cell-based assay are necessary to make the serological diagnosis. CNS-tissue staining by indirect immunofluorescence using patients’ samples may sometimes reveal a GFAP-like pattern with linear ependymal and subependymal staining with limited parenchymal involvement and/or without myeneteric plexus staining.
These patients do not have GFAP-IgG and are negative by GFAP-specific cell-binding assays, showcasing the necessity of both testing platforms for accurate diagnosis. In a patient with the right clinical phenotype of meningoencephalomyelitis and no CSF available, serum positivity for GFAP-IgG might be sufficient. In the Mayo Clinic cohorts, all patients had antibody specificities for the GFAP-alpha isoform with or without GFAP-epsilon isoform positivity, whereas in a clinical cohort by Long and colleagues there were patients that had antibodies reacting only with the GFAP-epsilon isoform. Serum positivity alone may yield false positive results as well as only antigen-specific assays without strict detection criteria, as mentioned above. The titer does not predict disease severity (08).
CSF analysis shows abnormalities in 54% to 90% of cases (05; 17). Lymphocytic pleocytosis is reported in 85% to 100% (05; 21) and elevated protein in about 80% (05); CSF-exclusive oligo-clonal bands are found in up to 77% of patients (08; 10). When performed, EEG generally shows nonspecific abnormalities and diffuse slow waves, whereas epileptic discharges are rarely found. In a single pediatric case, extreme delta brush has been reported (30).
Brain MRI is abnormal in 42% to 51% of patients (05; 17; 10) and shows T2-hyperintensities involving the subcortical white matter, periventricular areas, the basal ganglia, hypothalamus, brainstem, and the cerebellum. In about 50% of cases, the brain MRI shows a characteristic pattern with linear perivascular gadolinium enhancement, perpendicular to the ventricles (08; 05; 17). This radial enhancement is considered to be a radiological hallmark of this disease, although not a pathognomonic sign because it can also be found in other neurologic conditions such as neurosarcoidosis, CNS vasculitis, and others. Although classically periventricular, linear radial enhancement on MRI can also be observed infratentorially within in the brainstem (23). Reversible lesions in the splenium of the corpus callosum have emerged as an additional radiological feature associated with GFAP-IgG autoimmunity (12). Ten percent of patients from a French cohort demonstrated reversible splenial lesions with restricted diffusion on MRI imaging similar to that observed in mild encephalopathy/encephalopathy with reversible splenial lesion (MERS) syndrome (10).
Leptomeningeal, punctate, serpentine, and ependymal enhancement have also been reported. In the spinal cord MRIs, the most common abnormalities seen are T2-hyperintense longitudinally extensive lesions, sometimes ill-defined with linear or hazy enhancement along the course of the central canal (27).
Every patient that is diagnosed with GFAP-IgG autoimmunity needs to have a cancer screening according to age, sex, and risk factors. A whole-body CT or PET scan should be considered, as multiple neoplasms have been found in the adult population. In women, a pelvic ultrasound should be part of the work-up, as ovarian teratomas are the neoplasms most often encountered. A mammogram in women and a testicular ultrasound in men should complete the cancer screening, when appropriate.
|
• Corticosteroids are the mainstay of treatment for autoimmune GFAP astrocytopathy. |
To date, there are no prospective studies of treatments for autoimmune GFAP astrocytopathy. However, there are some limited retrospective studies and expert opinion recommendations (class 4 evidence). This disease is characterized by remarkable steroid responsiveness. The authors recommend initial intravenous high-dose steroids (IV methylprednisolone) with subsequent oral or directly high-dose oral steroids (prednisone 1 mg/kg or equivalent). Initial high-dose steroids should be continued for 3 to 6 months before tapering, as early relapses happen if steroids are tapered prematurely, with clinical and radiological improvement documented before. For prednisone, we suggest decreasing by 10 mg/month until 10 mg, followed by 1 mg/month taper until cessation. Long-term steroid use should be accompanied by PCP prophylaxis and osteoporosis prophylaxis. Intravenous immunoglobulin and plasma exchange have been used and could be considered options in patients with an absolute contraindication to steroids, though IVIg may not be as effective in some patients (11). More than 70% of patients respond well to first-line immunotherapy (05; 16; 10).
In the acute phase for severe cases (decreased level of consciousness, dysautonomia, refractory seizures) intensive care unit admission may be required for airway protection and hemodynamic stabilization.
In those patients with relapsing-remitting courses after steroid withdrawal (20% to 50%) a steroid-sparing agent can be introduced (08; 05; 21; 16). Long-term treatments include classic immunosuppressants such as azathioprine or mycophenolate mofetil. The authors have primarily treated patients with mycophenolate mofetil. Depending on severity, availability and tolerance methotrexate and cyclophosphamide might be other considerations. A small cohort of patients had good responses to tacrolimus (32). The correct duration of the treatment is unknown, but the authors suggest 2 to 5 years before considering cessation. In cases that relapse after discontinuation of immunosuppression, long-term immunosuppression should be considered. If a concomitant neoplasm is found, cancer treatment is necessary as soon as possible to also optimize neurologic therapeutic results and outcomes.
Patients with GFAP-IgG autoimmunity when appropriately treated have a good prognosis, with a median modified Rankin score of 2 at 20 months follow-up in a Mayo Clinic cohort (08). This was corroborated by a French series in which 89% of patients had good outcome (Rankin score < 2) with a median follow-up of 14 months (10). Progressive neurologic decline and death has been reported in patients, especially if not undergoing treatment (05). The outcomes vary among different studies, and in the study by Long and colleagues there were patients with worse outcome (21); it is not clear if this was dependent on diagnostic delay, treatment modalities, or a more severe disease in an Asian population. In general, however, GFAP astrocytopathy is characterized by a marked response to steroid therapy (05; 16). In cases of coexisting autoimmunity targeting the NMDAR, the prognosis is similar to patients with NMDAR encephalitis. In paraneoplastic cases, the underlying malignancy may dictate prognosis.
The pediatric population’s manifestations and response to treatment is similar to the adults, as mentioned above.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Andrew McKeon MD
Dr. Andrew McKeon of Mayo Clinic has received research funding from Roche/Genentech and Janssen.
See ProfileMichael Gilligan MD
Dr. Gilligan of Mayo Clinic has no relevant financial relationships to disclose.
See Profile
Francesc Graus MD PhD
Dr. Graus, Emeritus Professor, Laboratory Clinical and Experimental Neuroimmunology, Institut D’Investigacions Biomédiques August Pi I Sunyer, Hospital Clinic, Spain, has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuroimmunology
Apr. 07, 2024
General Child Neurology
Mar. 29, 2024
Neuroimmunology
Mar. 24, 2024
Neuroimmunology
Mar. 24, 2024
Movement Disorders
Mar. 06, 2024
General Neurology
Mar. 03, 2024
Neuroimmunology
Mar. 02, 2024
Neuroimmunology
Feb. 29, 2024