
Neuro-Oncology
Turcot syndrome
May. 27, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Ganglioglioma is an uncommon, usually low-grade, central nervous system tumor composed of cells resembling neuronal tissue and glial cells. In this article, the authors discuss the diagnosis and treatment of ganglioglioma.
|
• Gangliogliomas are well-differentiated, slow-growing, mixed neuronal-glial tumors composed of dysplastic ganglion cells (neurons) and neoplastic glial cells. | |
|
• Gangliogliomas should be suspected in any child or young adult with temporal-lobe epilepsy and a temporal lobe cortical-based lesion on imaging, although these tumors can be found throughout the central nervous system. | |
|
• No formal criteria for WHO grade 2 gangliogliomas have been established. | |
|
• Gross total resection of the tumor remains the only effective treatment in most cases, and prognosis is excellent when a WHO grade 1 ganglioglioma is grossly, totally resected. | |
|
• BRAF V600E mutation status represents a potential biomarker for stratifying seizure recurrence risk and may influence surgical strategy. |
Gangliogliomas and gangliocytomas comprise a spectrum of low-grade tumors characterized by dysplastic neuronal (or ganglion) cell types. In gangliogliomas, the dysplastic neuronal cells are accompanied by neoplastic glial cells, whereas in gangliocytomas, large, multipolar, binucleated, well-differentiated neurons are the unique neoplastic component.
The term ganglioglioma was originally coined in 1926 (40). The diagnosis was initially based on gross histopathology and light microscopy with additional use of electron microscopy. Histochemistry and immunocytoreactivity are used to demonstrate the ganglion cell component. The oncofetal marker CD34 and detection of BRAF V600E mutation or other MAPK pathway alterations characterize gangliogliomas.
|
• The majority of gangliogliomas present by 30 years of age, most often with focal seizure; BRAF V600E-mutant gangliogliomas present at a younger age. | |
|
• Over 90% of gangliogliomas are found in superficial hemispheres, especially the temporal lobes. |
Ganglioglioma is the top differential diagnosis in any patient presenting with chronic, medically refractory epilepsy during the first 3 decades of life, in which a temporal mass isointense to gray matter on T1-weighted images is found on brain MRI. Eighty percent of all gangliogliomas present by 30 years of age, with the majority (63%) after age 10 years and less than 5% before the first year of life (09). Median age at diagnosis is 19 years old for low-grade gangliogliomas, with a slight male predominance for either grade.
Patients with BRAF V600E-positive gangliogliomas present at a younger age, with seizure onset occurring at a median age of approximately 9 years compared to approximately 15 years in BRAF V600E-negative cases (52).
Gangliogliomas can occur anywhere in the brain or spinal cord but are most commonly found in superficial hemispheres (greater than 90%), especially the temporal lobes (greater than 45%). They have been reported in the pineal and sellar regions, basal ganglia, ventricles, and, less frequently, infratentorially and within the cranial nerves. Spinal gangliogliomas are mostly found in the cervical or cervico-thoracic segment (39). Multifocal intracranial gangliogliomas at diagnosis have also been described (30). Rarely, leptomeningeal dissemination of ganglioglioma with anaplastic features can occur (33).
Focal seizure is the most frequent presenting symptom of cerebral ganglioglioma (occurring in greater than 75% of patients), and long-standing epilepsy is not uncommon; in fact, ganglioglioma represents one of the most common epilepsy-associated tumors and may even be an incidental finding on temporal lobectomies performed for epilepsy management (34). Molecular studies have identified BRAF V600E mutation as a significant risk factor for seizure recurrence, particularly in patients with radiologically complete tumor resection (52).
Otherwise, the locus of the ganglioglioma in the brain often determines the presenting symptoms or signs. In posterior fossa and intraventricular gangliogliomas, obstructive hydrocephalus, with symptoms of increased intracranial pressure such as headache or depressed consciousness, is the most frequent presentation. For spinal gangliogliomas, the most common presenting symptom is extremity weakness, followed by sensory impairment, gait problems, urinary disturbances, and back pain (13; 47; 55; 39).
Prognostic factors associated with aggressive behavior include older age, higher grade histology, and loss of p16 or CDKN2A mutation (53).
LT, a 30-year-old, right-handed mechanical engineer, had a history of focal seizures since 12 years of age; these seizures were characterized by odd smell, spreading discomfort, and “déjà-vu” lasting less than a minute, with postictal anxiety and no altered consciousness. Seizure frequency increased over the years from a few annually to a few daily and went unmanaged.
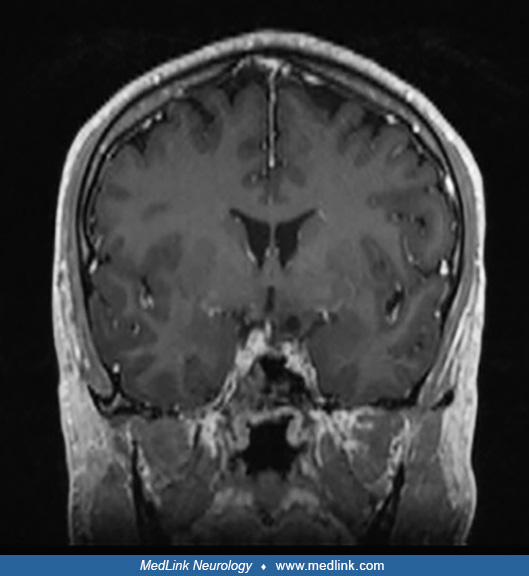
Over the next several years, he experienced intermittent short-term memory loss and increasing word-finding difficulties. At 30 years of age, he had a generalized tonic-clonic seizure with postictal speech production difficulty. On ensuing brain MRI, he was found to have a non-enhancing left mesial temporal mass. The lesion was hyperintense on T2/FLAIR weighted imaging and contained a small cyst.
Imaging differential diagnosis included dysembryoplastic neuroepithelial tumor and ganglioglioma. He was started on antiepileptic drugs and referred to neurosurgery. A preoperative comprehensive neuropsychological testing reported baseline impaired naming and reduction in auditory attention, suggesting some compromise of the left temporal lobe. An interictal EEG showed continuous left temporal slowing.
Given his course, the patient proceeded with surgical resection. He was considered at risk of postoperative language deficits because of the location of the lesion; thus, awake craniotomy with speech mapping and intraoperative electrocorticography was done to localize extralesional epileptiform activity, and the lesion was completely resected. The pathology was consistent with a WHO grade 1 ganglioglioma. The patient experienced postoperative transient word-finding difficulties that resolved with time. Post-surgery, he remained seizure-free for 3 years, though remained on antiseizure medication; he continued semiannual MRI follow-up without recurrence to date.
|
• Gangliogliomas are well-differentiated, slow-growing, mixed neuronal-glial tumors composed of dysplastic ganglion cells (neurons) and neoplastic glial cells. | |
|
• Gangliogliomas are characterized by mitogen-activated protein kinase (MAPK) pathway activation through different genetic alterations, most commonly BRAF V600E mutation (20%-80% of gangliogliomas). BRAF V600E status should be determined for possible targeted therapy in case of residual or recurrent tumor. | |
|
• Identification of IDH1/2, TP53, or PTEN mutations or of CDK4 or EGFR amplification strongly argues against the diagnosis of ganglioglioma. | |
|
• Somatic BRAF V600E mutation in developing neurons has been implicated in the intrinsic epileptogenicity in ganglioglioma. |
The etiology and the molecular pathogenesis of ganglioglioma remain unclear. The cell of origin is suggested to be a common glioneural precursor with clonal neoplastic proliferation of the glial cell population.
Although no specific mutation has been identified as a solely causative genetic aberration, gangliogliomas have been characterized by alterations that activate the MAPK signaling pathway, similar to pilocytic astrocytomas, pleomorphic xanthoastrocytomas, dysembryoplastic neuroepithelial tumors, rosette-forming glioneural tumors, polymorphous low-grade neuroepithelial tumors, and multinodular and vacuolating neuronal tumors (38). In a cohort of 40 gangliogliomas on which targeted next-generation sequencing was performed, 36 had mutations activating MAPK signaling pathway (18 BRAF V600E mutation, five had other BRAF mutation, four BRAF fusion, two KRAS mutation, one RAF1 fusion, one NF1 biallelic mutation, and five FGFR1/2 alterations). In most cases, genetic alteration with the MAPK pathway was the solitary genetic alteration identified. None had IDH, TP52, ATRX, TERTp, CIC, or FUBP1 mutation; MYB-MYBL1 rearrangement; or TSC1-TSC2 mutation (38).
Somatic BRAF V600E mutation in developing neurons has been implicated in the intrinsic epileptogenicity in ganglioglioma (18; 06). The BRAF V600E-induced epileptogenesis seems to be mediated by RE1-silencing transcription factor, which is a regulator of ion channels and neurotransmitter receptors associated with epilepsy (18). In that study, seizure control in mice was significantly improved with a BRAF inhibitor, vemurafenib.
Molecular analyses have identified additional genetic alterations that may have prognostic implications in gangliogliomas. A study found that alterations in FGFR4, KIT, and PEG3 were correlated with shorter overall survival in astrocytoma, including gangliogliomas. Additionally, as with other primary central nervous system tumors, TERT alterations were associated with poor prognosis in glioneuronal and neuronal tumors (12).
Genetic susceptibility. Rare cases of gangliogliomas have been described in patients with cancer predisposition syndromes such as neurofibromatosis types 1 and 2 and Peutz-Jeghers syndrome (STK11 mutation) (44; 07; 45; 11; 05; 39).
Pathology. Macroscopically, gangliogliomas are well-circumscribed solid or cystic lesions with minimal mass effect and occasional calcifications. Microscopically, gangliogliomas, as suggested by their name, consist of a combination of two cell populations in varying proportions:
(1) Ganglion cells (ganglio-) are large, mature dysplastic neurons. Dysplastic neurons are characterized by lack of cytoarchitectural organization, cytomegaly, perimembranous aggregation of Nissl substance, and binucleated or clustering appearance.
(2) Neoplastic glial cells (-glioma) can resemble pilocytic astrocytoma, fibrillary astrocytoma, or oligodendroglioma.
Other prominent features include perivascular lymphocytic infiltrates, important capillary networks, dystrophic calcifications, and mild to moderate cellularity (occasional mitoses and small foci of necrosis are compatible with the diagnosis). In less than 5% of cases, these tumors show aggressive histopathological features such as increased cellularity, nuclear pleomorphism, increased mitosis, vascular proliferation, and necrosis. In such cases, exclusion of diffuse glioma with entrapped neurons is mandatory. Also, note that the 2021 WHO classification does not define criteria for WHO 2 gangliogliomas and has abolished WHO 3 gangliogliomas (28). Indeed, as most cases of grade 3 gangliogliomas from previous series lacked molecular analyses to exclude other high-grade glioma subtypes, the 2021 WHO Classification states that “further studies are needed to confirm the existence of anaplastic ganglioglioma.”
The neuronal elements are demonstrated by positive immunostaining for neuronal markers such as microtubule-associated protein 2, neurofilament, chromogranin-A, synaptophysin, and CD34. CD34 is positive in about 80% of gangliogliomas and is not present in adult brain neurons (27). Glial elements are demonstrated by cytoplasmic positivity for GFAP. The mean MiB1 labeling index, calculated from the glial component, is less than 3%.
Genetic profile. About 30% of gangliogliomas have chromosomal abnormalities, with gain of chromosome 7 being the most frequently described. Firm associations between cytogenetic data and clinical outcome are lacking (27). BRAF V600E mutation is the most common genetic alteration found in 20% to 60% of gangliogliomas (37). BRAF V600E is not specific for ganglioglioma because this mutation is also found in pleomorphic xanthoastrocytoma, pilocytic astrocytoma, dysembryoplastic neuroepithelial tumors, and epithelioid glioblastoma. Identification of IDH1/2, TP53, or PTEN mutations or of CDK4 or EGFR amplification strongly argues against the diagnosis of ganglioglioma. CDKN2A/B inactivation has been shown to play a significant role in ganglioglioma malignant transformation. Importantly, DNA methylation profiling showed that primary and recurrent tumors clustered in different methylation classes, indicating fundamental epigenetic reprogramming during malignant progression (54).
|
• Gangliogliomas account for less than 1.5% of all central nervous system tumors. | |
|
• A majority of gangliogliomas are diagnosed in children and young adults. |
Gangliogliomas account for 0.4% to 1.3% of all central nervous system tumors (27) and for up to 10% of all pediatric central nervous system tumors (16; 35). In population-based epidemiological data on newly diagnosed and histologically confirmed gangliogliomas from 2006 to 2011 in France, the worldwide adjusted incidence rate was estimated to be less than 0.2 of 100,000 person-years for WHO grade 1 ganglioglioma (08). About 80% of WHO grade 1 gangliogliomas are diagnosed in patients younger than 30 years of age (48). Median age at diagnosis is 19 years for low-grade ganglioglioma, but they can occur at any age. Spinal gangliogliomas appear to affect children and young adults and most frequently involve the cervicothoracic spine (55). There is a slight male predominance (1.14:1) (08).
No preventive measure is known.
Clinical presentation and imaging. The clinical differential diagnosis of ganglioglioma is broad and varies with patients and imaging characteristics. Most importantly, ganglioglioma should be considered high on the list of causes in any child/young adult presenting with temporal lobe epilepsy with an intra-axial temporal lesion seen on MRI, especially if it is a cortically-based, partially cystic, enhancing mass. However, ganglioglioma should also be included in the differential of all lesions with unusual imaging characteristics seen in the optic pathway, sellar, pineal, intraventricular, and cerebellar-pontine angle regions. Ganglioglioma may also present with atypical intracerebral hemorrhage without vascular abnormality on cerebral angiogram in a young adult (23).
Supratentorial tumors. In supratentorial tumors, the main differential diagnosis includes dysembryoplastic neuroepithelial tumor, pleomorphic xanthoastrocytoma, pilocytic astrocytoma, desmoplastic infantile astrocytoma and ganglioglioma, diffuse astrocytoma, and oligodendroglioma. If the lesion is hemispheric and cortically based, the differential diagnosis is further restricted to dysembryoplastic neuroepithelial tumor, pleomorphic xanthoastrocytoma, and oligodendroglioma. Dysembryoplastic neuroepithelial tumor can be differentiated from ganglioglioma due to the dysembryoplastic neuroepithelial tumor’s multi-cystic “bubbly” appearance and milder, infrequent contrast enhancement; oligodendroglioma may be more heterogeneous due to frequent calcifications and hemorrhagic components; pleomorphic xanthoastrocytoma usually shows prominent contrast-enhancement of both the cystic wall and its mural nodule abutting the pia. In the case of a superficial mass associated with skull remodeling, meningioma also needs to be considered. Desmoplastic infantile astrocytoma and ganglioglioma, as its name implies, most commonly occurs in children younger than 2 years of age and manifests as an exceptionally large cerebral hemispheric cystic and solid mass, most commonly in the frontal and parietal lobes.
Infratentorial tumors. In infratentorial tumors, the differential diagnosis includes pilocytic astrocytoma, diffuse astrocytoma, medulloblastoma, and hemangioblastoma.
Histology. Histologically, the neoplastic glial component of the tumor can resemble pilocytic astrocytoma, fibrillary astrocytoma, or oligodendroglioma and may, therefore, be mistaken for such tumors, among others. The presence of anaplastic features such as mitoses, increased proliferative activity, necrosis, or vascular proliferations expands the differential diagnosis to include high-grade astrocytic tumors. In a pathological review of 22 previously-termed “anaplastic gangliogliomas,” nine (41%) cases were reclassified (51). In the case of midline tumors, the H3K27M mutation must be searched, as cases of H3K27M-mutant diffuse gliomas have been reported to phenotypically mimic “anaplastic” ganglioglioma (20).
|
• Supratentorial gangliogliomas are sharply demarcated lesions, either as a solid-only or a cystic or solid-cystic mass. | |
|
• The majority of gangliogliomas demonstrate contrast-enhancement of various patterns. | |
|
• Approximately 30% of gangliogliomas show calcifications. | |
|
• Peritumoral edema is distinctly uncommon in ganglioglioma, arguing against the diagnosis. |
As with all neoplasms, the workup for ganglioglioma includes cerebral imaging (CT or MRI) followed by surgical resection. Imaging characteristics are highly variable due to the various patterns of growth and possible locations of these tumors. Supratentorial gangliogliomas present as a sharply demarcated lesion, either as a solid-only (in approximately 50% of cases) or as a cystic or solid-cystic mass. Over 60% demonstrate contrast enhancement, with either a nodular, rim, or solid enhancement pattern. Intraventricular gangliogliomas often have cystic degeneration and calcifications (47). Compared to supratentorial gangliogliomas, infratentorial gangliogliomas have less cystic changes (about 20% compared to 50%) and more contrast-enhancement (over 70% compared to less than 50%), especially when located in the cervicomedullary junction (100% are enhancing) (26; 15).
On CT, approximately 30% of gangliogliomas show calcifications, and bony remodeling can be seen (27). Gadolinium-enhanced MRI is the imaging modality of choice, and both the brain and spine should be performed if leptomeningeal dissemination or ventricular seeding of ganglioglioma is suspected. On MRI, the solid component is iso- to hypointense to grey matter on T1-weighted imaging (T1WI), hyperintense on T2WI, and the solid component shows variable enhancement intensity and pattern on T1WI post-contrast imaging. The cystic component has variable T2WI signal depending on the presence of blood products and the amount of proteinaceous material. Peritumoral T2/FLAIR edema is distinctly uncommon in ganglioglioma, arguing against the diagnosis. On susceptibility-weighted imaging, about 30% of gangliogliomas will show blooming signal loss corresponding to calcified areas and rarely to blood products. Higher-grade (formerly anaplastic) gangliogliomas seem to lose the typical radiological characteristics of grade 1 gangliogliomas, especially their cystic and well-circumscribed features, and they gain characteristics of grade 3 gliomas and glioblastomas, such as necrosis, surrounding cerebral edema, and mass effect (51).
It must be emphasized that the diagnosis of a ganglioglioma cannot be excluded by a negative CT or MRI examination only. There are reports of histologically confirmed ganglioglioma without a recognizable tumor on either CT or MRI images.
|
• Gross total surgical resection is the treatment of choice for ganglioglioma. | |
|
• In cases of subtotal resection, adjuvant radiotherapy should be considered to improve local tumor control. | |
|
• Targeted therapy against the BRAF/MEK pathway represents a novel and promising therapeutic option in gangliogliomas harboring BRAF V600E mutation. |
Surgical resection. Gross total surgical resection followed by close observation with serial MRI is the treatment of choice for ganglioglioma, as the extent of resection is considered the single most important prognostic factor (15). Fluorescence-guided tumor resection has been used to improve intraoperative visualization and resection extent during the resection of gangliogliomas (14). In cases of subtotal resection, the 2022 EANO-EURACAN-SNO guidelines on rare glial and neuroglial tumors recommend considering the use of adjuvant radiotherapy to improve local tumor control, as well as the use of BRAF or MEK inhibitors in ganglioglioma with BRAF alteration (46). On recurrence or tumor regrowth, surgical resection should be repeated whenever feasible.
Radiation therapy. After gross total resection, adjuvant radiation therapy has no clear benefit (41). For those with subtotal resection or biopsy, the role of radiation is controversial, as it may improve progression-free survival but not overall survival in both low- and high-grade gangliogliomas; this point was illustrated in a large study comparing four therapies for local control and overall survival in 402 patients with ganglioglioma (42). The 10-year local control and overall survival rates were 89% and 95% after gross total resection, 90% and 95% after gross total resection plus radiotherapy, 52% and 62% after subtotal resection, and 65% and 74% after subtotal resection plus radiotherapy, respectively. After subtotal resection, irradiation significantly improved local control but not overall survival. After gross total resection, irradiation did not significantly improve local control or overall survival. Given that most gangliogliomas are found in young adults and are in the temporal lobe, the benefit of radiation must be carefully considered against long-term toxicity, especially its neurocognitive impact. In addition, some in the field have expressed concern that radiation therapy may contribute to a transformation to a more anaplastic tumor (50). As such, adjuvant radiation after a subtotal resection should be reserved for those with a high risk of recurrence, such as gangliogliomas with anaplastic features, in whom a retrospective review showed a three times increased rate of recurrence among gangliogliomas with highly anaplastic features, compared to those with moderate anaplastic features (21). However, there are insufficient data to make a firm recommendation about the role of radiation therapy after subtotal resection of low-grade gangliogliomas.
Stereotactic radiosurgery appears to be a safe and effective treatment option for patients unfit for surgery and for surgically inaccessible, recurrent, and residual gangliogliomas (31).
Chemotherapy. Chemotherapy may also be considered to prevent ganglioglioma recurrence, although the agent of choice and the optimal duration are unknown. Various chemotherapeutic agents have been used as adjuvant therapy, including temozolomide, vincristine, procarbazine, and nitrosoureas.
Targeted therapies. Targeted therapy with oral selective BRAF V600E inhibitors represents a promising therapeutic option in the 20% to 60% gangliogliomas harboring BRAF V600E mutation. BRAF is a member of the RAS/RAF/MEK/ERK pathway, and the BRAF V600E mutation activates MERF-ERK signaling pathway, promoting cell proliferation, differentiation, and survival. BRAF V600E mutation in some tumors can be sensitive to BRAF V600E mutant-specific inhibitors such as vemurafenib and dabrafenib. The status for BRAF V600E mutation should be tested for all gangliogliomas in case of tumor regrowth or as recommended by the National Comprehensive Cancer Network Guidelines for central nervous system cancers (19).
Vemurafenib and dabrafenib have shown efficacy in this setting. A basket trial of vemurafenib in BRAF V600-mutant non-melanoma cancers reported an objective response rate of 66.7% in gangliogliomas (01).
Combination therapy with BRAF and MEK inhibitors has shown improved efficacy and tolerability. Dabrafenib (BRAFi) and trametinib (MEKi) combination is FDA-approved for patients with tumors harboring BRAF V600E mutations.
A randomized phase 2 trial demonstrated significantly higher response rates and longer progression-free survival with dabrafenib plus trametinib compared to standard chemotherapy in pediatric low-grade gliomas with BRAF V600 mutations (03). As 90% of ganglioglioma harbor mutations activating the MAPK pathway (38), acquired resistance to vemurafenib may result from persistent MAPK activation downstream to BRAF V600E, and the combination of BRAF/MEK inhibition could overcome resistance.
Tovorafenib, an oral, selective, central nervous system-penetrant, type II RAF inhibitor, has shown promising results in the treatment of BRAF-altered pediatric low-grade gliomas. In the phase 2 FIREFLY-1 trial, tovorafenib demonstrated efficacy in various types of pediatric low-grade gliomas with BRAF alterations. On April 23, 2024, the Food and Drug Administration granted accelerated approval to tovorafenib for patients 6 months of age and older with relapsed or refractory pediatric low-grade glioma (LGG) harboring a BRAF fusion or rearrangement, or BRAF V600 mutation. This represents the first FDA approval of a systemic therapy for treating patients with pediatric low-grade glioma with BRAF rearrangements, including fusions. Although further studies are needed, tovorafenib may represent a promising therapeutic option for patients with BRAF-altered gangliogliomas, particularly in refractory or recurrent cases (17).
A Canadian consensus for the treatment of BRAF V600E-mutated pediatric and adolescent/young adult (AYA) gliomas, including gangliogliomas, recommends considering BRAF inhibitors (BRAFi) with or without MEK inhibitors (MEKi) as first-line systemic therapy in most patients. The consensus suggests that BRAFi monotherapy could be reasonable in some patients to facilitate compliance and reduce costs. The optimal duration of treatment remains uncertain, but the consensus suggests treating with BRAFi ± MEKi for a total of 36 months for pediatric low-grade gliomas and 60 months for high-grade gliomas. When discontinuing treatment, a slow taper is recommended to mitigate the risk of rapid tumor progression (10).
Bevacizumab, an anti-VEGF antibody, has been described to rapidly shrink the cystic component of a low-grade ganglioglioma (24).
Symptoms management. Lastly, adequate symptom management of increased intracranial pressure and epilepsy is important. When hydrocephalus is present, ventricular shunting may be necessary. Although seizures due to ganglioglioma can usually be controlled with appropriate antiepileptic medications, surgical resection of the tumor is curative in most patients (02).
The typical patient with ganglioglioma generally follows a “manageable” clinical course, but tumor recurrence, malignant progression, and secondary glioblastoma have been observed in some cases. Good prognosis has been associated with lower grade, temporal location, epilepsy, and gross total resection. A greater extent of resection is the most important variable associated with long-term disease-free survival (29; 42; 04; 15).
The overall prognosis of patients with low-grade ganglioglioma is favorable, with a 95% 7.5-year recurrence-free survival rate (09). Survival rates of 93% at 5 years, 98% at 7.5 years, and 94% at 15 years have been reported from large series (29; 04; 09).
Brainstem and spinal cord gangliogliomas tend to have a worse prognosis than supratentorial tumors because of increased subtotal resection and operative morbidity (22). In one retrospective review including only patients with brainstem gangliogliomas, the 3-year survival rate was 91%, with a 3-year event-free survival rate of 68% and a mean recurrence-free survival rate of 5.5 months (36). The 5-year survival rate for brainstem gangliogliomas compared to non-brainstem gangliogliomas was reported in two large studies showing 79% to 81% versus 93% to 97%, respectively (22; 09). Brainstem gangliogliomas also exhibited a 3-year event-free survival rate and a 5-fold increased risk of recurrence (22).
In a large population-based study of 703 adults with low-grade ganglioglioma, younger age, female gender, temporal lobe location, and gross total resection were positive prognosis factors (25).
Anaplastic ganglioglioma. The previous 2016 WHO Classification included an anaplastic variant for those gangliogliomas with features such as high mitotic activity, microvascular proliferations, or necrosis in the glial component, often associated with aggressive clinical behavior. However, as most cases of anaplastic gangliogliomas lacked molecular analyses to exclude other high-grade glioma subtypes, the 2021 WHO Classification has abolished WHO grade 3 gangliogliomas. Indeed, in the largest multicenter retrospective analysis of 43 adults with pathologically confirmed anaplastic ganglioglioma, survival was only slightly improved compared to glioblastomas despite gross total resection and adjuvant therapy (51). It is possible that some prior reports of anaplastic gangliogliomas included higher-grade glial tumors.
Epilepsy. The prognosis for epilepsy in the setting of ganglioglioma is excellent. The meta-review of patients after ganglioglioma resection on epilepsy outcomes, including 20 retrospective articles since 1995, with a total of 553 patients, concluded that resection leaves most patients seizure-free and many with freedom from antiepileptic therapy, regardless of patients’ demographic, tumor characteristic, and operative variables (02). Although reporting standards vary widely, the most consistent finding across studies was the correlation of increased seizure freedom with increased extent of resection. Other prognostic factors associated with seizure freedom included shorter duration of epilepsy and younger age at surgery. Reported rates of seizure freedom following surgery ranged from 63% to 100%, with mean follow-up intervals ranging from 3 to 10 years. Many of the included studies had follow-up times of longer than 5 years, suggesting a durable response. Between 17% to 77% of seizure-free patients were weaned from antiepileptic drugs in the studies reporting this measure (02; 32).
No data are available to suggest any correlation of this tumor with pregnancy. Non-enhanced MRI is the imaging modality of choice and has been demonstrated to be safe for both the mother and the fetus as gadolinium crosses the placenta, although it has not been associated with birth defects at conventional doses (49). Symptomatic relief of headaches and seizures can be accomplished with well-known medications accepted as being of low fetal risk. Hydrocephalus, if serious enough to produce papilledema or other significant effects, can be surgically corrected during pregnancy, as demonstrated in one review (43).
No special anesthetic precautions are mandated by the presence of this tumor. General symptoms of increased intracranial pressure may require an alteration in patient preparation for anesthesia.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Sarah Lapointe MD
Dr. Lapointe of the University of Montreal received consultant fees from Alexion, Bayer, Novocure, and Servier.
See ProfileNicholas Butowski MD
Dr. Butowski of the University of California, San Francisco, has no relevant financial relationships to disclose.
See Profile
Rimas V Lukas MD
Dr. Lukas of Northwestern University Feinberg School of Medicine received honorariums from Jazz Therapeutics, Novocure, and Servier for speaking engagements, honorariums from Cardinal Health, Catalyx, Merck, and Novocure for advisory board membership, research support from BMS as principal investigator, and an honorarium from GT Medical Technologies for DSMB membership.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuro-Oncology
May. 27, 2026
Neuro-Oncology
May. 27, 2026
Neuro-Oncology
May. 23, 2026
Neuropharmacology & Neurotherapeutics
May. 11, 2026
Stroke & Vascular Disorders
May. 03, 2026
Neuro-Oncology
Apr. 30, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026