Sleep Disorders
Sleep-related leg cramps
Jul. 03, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Movement disorder emergencies refer to neurologic conditions where movement disorders are the main feature. These emergencies develop acutely or subacutely and require immediate medical attention. Failure to promptly diagnose and treat these conditions can lead to significant morbidity and even mortality (34). These include neuroleptic malignant syndrome, parkinsonism-hyperpyrexia syndrome, acute decompensation of parkinsonism, serotonin syndrome, lethal catatonia, dystonic storm, and malignant Tourette syndrome.
|
• Prompt diagnosis and management of movement disorder can reduce morbidity and mortality. | |
|
• The mainstay of movement disorder emergencies involves the cessation of the offending agent combined with aggressive supportive therapy. |
A movement disorder emergency is characterized by an acute onset of abnormal movements or the rapid evolution of any movement disorder; the failure to appropriately diagnose and treat the patient can result in morbidity or even mortality. Most movement disorder emergencies evolve over hours to days, prompting the patient to seek immediate medical attention. Neurologists will encounter these patients as consultations in the emergency department, inpatient wards, or intensive care unit (ICU). Early recognition of the syndromes and implementation of appropriate therapy is critical. Therefore, a neurologist should always consider these conditions when approaching patients with an acute onset of or rapidly evolving abnormal movements. Patients should be cared for in a closely monitored setting, with special care if the syndrome occurs during pregnancy or while undergoing anesthesia or surgery. Some patients with movement disorders, such as dystonia, experience respiratory problems associated with dystonic respiratory dysregulation (77) and other respiratory problems (76).
There are minimal epidemiologic data on movement disorder emergencies. Few large-scale randomized studies have been performed to assess the efficacy of the recommended treatment strategies. The mainstay of therapies includes aggressive supportive care and specific treatment, such as discontinuation of the offending agent in case of neuroleptic malignant syndrome or serotonin syndrome or treatment of underlying psychiatric disorders in case of catatonia. Common presenting symptoms include hyperpyrexia, confusion, rigidity, and immobility, with additional disease-specific presentations. It is important to rule out other emergency conditions that can mimic these syndromes as they may require different, specific, life-saving interventions and precautions. The differential diagnosis includes malignant hyperthermia, heatstroke, encephalitis, drug intoxication, thyrotoxicosis, acute porphyria, tetany, akinetic mutism, locked-in syndrome (basilar artery stroke, central pontine myelinolysis), and a variety of movement disorders that can result from acute stroke (78) and other systemic disorders (23; 24). In addition to neuroleptic malignant syndrome, parkinsonism-hyperpyrexia syndrome, acute decompensation of parkinsonism, serotonin syndrome, lethal catatonia, dystonic storm, and malignant Tourette syndrome are discussed in this article.
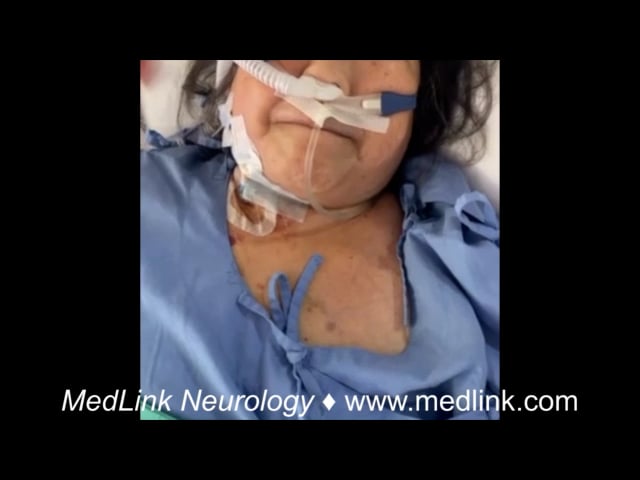
Neuroleptic malignant syndrome is an iatrogenic disorder resulting from exposure to dopamine (D2) receptor-blocking medications, first described by Delay in the 1960s (27). Neuroleptic malignant syndrome is associated with both typical and atypical neuroleptics, but other medications such as tetrabenazine, levomepromazine, prochlorperazine, metoclopramide, droperidol, valproic acid, and promethazine have also been implicated (124; 140). Cases of neuroleptic malignant syndrome have been reported in association with the use of newer-generation antipsychotics, such as lurasidone (94). Neuroleptic malignant syndrome most frequently develops within the first 24 to 48 hours after the introduction of a dopamine receptor blocking agent, although rare cases of neuroleptic malignant syndrome have been reported to occur after long-term stable therapy, adjustment of a dosage, or addition of medications such as antiemetics, donepezil, lithium, and serotonin reuptake inhibitors (87). Young males are at the highest risk for developing neuroleptic malignant syndrome (40). The pathogenesis of neuroleptic malignant syndrome is not well understood; however, abrupt alterations in central dopamine transmission and proinflammatory cytokine activation have been suggested as playing a pathogenic role (92). A high index of suspicion is necessary to diagnose neuroleptic malignant syndrome due to the low incidence rates (0.02% to 3%) (73). Early detection and treatment are crucial, as delayed management significantly increases the risks of complications and mortality. Data suggest the mortality rates for neuroleptic malignant syndrome have decreased from historical estimates of 20% to 30% to 4.7% at 30 days, 9.9% at 90 days, and up to 15.1% at 1 year (135). Neuroleptic malignant syndrome should be considered in any patient who presents with acute-onset rigidity, altered mental status, fever, and a history of neuroleptic exposure.
Neuroleptic malignant syndrome is a clinical syndrome, commonly presenting with fever, rigidity, and elevated creatine kinase (CK) with concurrent autonomic dysfunction, altered consciousness, and leukocytosis.
In rare cases, rigidity may not be present (142; 93). A reversible isolated lesion in the splenium of the corpus callosum has been reported (139; 55). Several diagnostic criteria have been proposed, which can indicate a high likelihood of neuroleptic malignant syndrome in the appropriate clinical context (see Table 1) (101). New diagnostic criteria utilizing numerical scoring have been proposed and validated through international consensus, demonstrating an 83.2% sensitivity and 94.6% specificity (41; 43) (see Table 1). A review of the Mayo Clinic experience from 1995 to 2023 found that only 6% of cases coded as neuroleptic malignant syndrome met DSM-5 criteria, highlighting the challenge of accurate diagnosis (67).
|
(65) |
International Expert Consensus Neuroleptic Malignant Syndrome Diagnostic Criteria (41) |
Priority Score | |
|
Major Criteria |
Fever |
Exposure to dopamine antagonist or dopamine agonist withdrawal within the past 72 hours |
20 |
|
Minor Criteria |
Tachycardia |
Hyperthermia (> 100.4°F) on at least two occasions, measured orally |
18 |
|
Rigidity |
17 | ||
|
Altered mental status |
13 | ||
|
Creatine kinase elevation (four or more times the upper limit of normal) |
10 | ||
|
Sympathetic nervous system lability (at least two of the following): (a) Blood pressure elevation (systolic or diastolic 25% or more above baseline) (b) Blood pressure fluctuation (20 mmHg or more diastolic change or 25 mmHg or more systolic change in 24 hours) (c) Diaphoresis (d) Urinary incontinence |
10 | ||
|
Hypermetabolism (increased heartrate of more than 25% above baseline) AND increased respiratory-rate (50% or more above baseline) |
5 | ||
|
Negative work-up for infectious, toxic, metabolic, or neurologic causes |
7 | ||
|
Diagnostic requirements |
All major criteria AND at least two minor criteria must be present |
A score of 64 or more out of 100* | |
|
| |||
Risk factors for neuroleptic malignant syndrome include psychomotor stress, dehydration, the use of intramuscular deposition of neuroleptics, adjustments of neuroleptic dose and loading rate, or concomitant use of antiparkinsonian drugs and lithium carbonate (84); rarely, this syndrome can occur while on chronic stable neuroleptic therapy (59). In a retrospective case-control study of older adults, neuroleptic malignant syndrome was associated with higher antipsychotic doses, and first-generation antipsychotics carried a higher risk than second-generation agents (98). However, atypical antipsychotics are not risk-free and can also precipitate neuroleptic malignant syndrome. Neuroleptic malignant syndrome typically progresses over 48 to 72 hours and lasts 7 to 14 days. Chronic, irreversible medical conditions may complicate recovery, such as renal failure secondary to rhabdomyolysis and respiratory failure secondary to decreased chest wall compliance or aspiration pneumonia (76). Other complications due to immobility, such as deep vein thrombosis and pressure ulcers, have been reported. The presence of multiorgan failure increases the risk of mortality (84). Long-term neurologic complications include cognitive impairment, specifically executive dysfunction; gait instability and dystonia due to cerebellar and basal ganglia injury joint contracture; sensory disturbance; and residual catatonia (84; 63; 70). Hyponatremia secondary to cerebral salt wasting and the syndrome of inappropriate antidiuretic hormone have been reported with neuroleptic malignant syndrome and may precipitate seizures (64). Risk factors for mortality associated with neuroleptic malignant syndrome are older age, acute respiratory failure at presentation, elevated creatinine kinase level, and mean platelet volume (79; 104).
Early aggressive supportive therapy is the mainstay of care in neuroleptic malignant syndrome, which includes admission to the ICU, discontinuation of causative medication, and treatment with antipyretics and dopaminergic agents, such as levodopa, amantadine, bromocriptine, apomorphine, or other dopamine agonists such as pramipexole, ropinirole, or rotigotine patch (99; 129). Dantrolene, a nonspecific muscle relaxant, reduces muscle rigidity and minimizes rhabdomyolysis (125). Carbamazepine was shown to be effective in treating symptoms in a case report of two patients (123). The introduction of dopamine agonists or levodopa has also been suggested (97; 17). Electroconvulsive therapy can be considered in patients where pharmacotherapy is not successful (130). Treatment should last 7 to 10 days, depending on the half-life of the causative agent, and combination therapy can effectively reduce symptoms. In patients who require the continuation of neuroleptic medication, the same medication can be reintroduced, but no sooner than 2 weeks after complete recovery. Low doses of low-potency neuroleptics are preferred, and gradual titration with close monitoring is recommended. Neuroleptic malignant syndrome recurrence rates are as high as 33% if the neuroleptic is introduced too quickly (102). For those patients who require immediate treatment of psychosis during this recovery period, electroconvulsive therapy may be an effective solution for symptom control (137).
Parkinsonism-hyperpyrexia syndrome is a rare and potentially fatal complication of Parkinson disease. First described in the 1980s, physicians observed this syndrome in patients admitted to the hospital for an abrupt withdrawal or dramatic reduction in dopaminergic medications (known as “levodopa holidays”) (33). Several patients were observed to have an acute onset of fever and rigidity identical in presentation to neuroleptic malignant syndrome without a history of neuroleptic exposure. Symptoms develop between 18 hours and 7 days, consisting of pyrexia and altered consciousness, followed by autonomic dysfunction. Though typically associated with Parkinson disease, parkinsonism-hyperpyrexia syndrome has been reported in other parkinsonian disorders, such as multiple system atrophy, progressive supranuclear palsy, vascular parkinsonism, and Lewy body dementia (115). The pathophysiology is similar to neuroleptic malignant syndrome. An abrupt reduction of central dopamine in combination with disruption of central and peripheral sympathetic outflow and central serotonin levels has been implicated (80). It has been suggested that severe loss of presynaptic dopaminergic neurons may be associated with increased risk of developing this syndrome (53). Mortality rates as high as 4% have been reported; therefore, it is important to recognize potential risk factors to avoid precipitating this syndrome.
Though levodopa holidays are no longer recommended, patient noncompliance and abrupt changes in medication can still precipitate this condition in the absence of planned withdrawal (31). A common scenario in which aggressive reduction in medications frequently occurs is after deep brain stimulation surgery. Neurologists should be aware of this potentially fatal complication and carefully reduce dopaminergic medications after implantation. It is important to note that deep brain stimulation does not protect against parkinsonism-hyperpyrexia syndrome (29; 126) and may even provoke this syndrome. A case report describes parkinsonism-hyperpyrexia syndrome in a Parkinson disease patient treated with only bilateral subthalamic nucleus deep brain stimulation (STN-DBS). Deep brain stimulation was discontinued due to stimulation-related side effects, and subsequently, a sudden onset of hyperpyrexia, rigidity and decreased consciousness occurred (54; 06). Additionally, parkinsonism-hyperpyrexia syndrome can occur due to deep-brain stimulation malfunctions, such as neurostimulator battery depletion (28). In general, provoking factors for parkinsonism-hyperpyrexia syndrome include concomitant use of neuroleptics, dehydration, hot temperatures, wearing-off phenomenon, infections, and changes in intestinal absorptions (ie, ileus). Treatment of parkinsonism-hyperpyrexia syndrome is mainly supportive, with admission to the ICU and reintroduction of dopaminergic therapy either orally, via nasogastric tube, via a transdermal route (rotigotine patch), or parenterally (levodopa 50 to 100 mg infused over 3 hours); this can be repeated until the patient tolerates oral medications (46). Bromocriptine (5 to 10 mg three times per day) and dantrolene sodium (10 mg/kg per day in three to four divided doses) may be added as adjuvant supportive therapy (101). Adding methylprednisolone 1 g per day for 2 days was an effective adjuvant therapy in a small randomized control trial (105). Additionally, a case report suggests that electroconvulsive therapy may be efficacious in the treatment of refractory parkinsonism-hyperpyrexia syndrome (75). Complications include the development of rhabdomyolysis and subsequent renal failure. Aggressive intravenous fluid hydration is recommended to prevent these complications. Patients are at risk for disseminated intravascular coagulation and deep vein thromboses, which can increase morbidity and mortality (86). In general, maintenance of some dopaminergic therapy (ideally levodopa-based) during medication adjustments is an important consideration to prevent parkinsonism-hyperpyrexia syndrome. Furthermore, treating the autonomic symptoms is critical in achieving a favorable outcome (49).
Disorders associated with parkinsonism are typically chronic neurodegenerative disorders with a gradual subacute progression; however, a sudden change in symptom control may lead to emergency room visits or hospitalization. In an analysis of the Nationwide Inpatient Sample (NIS) database, pneumonia, urinary tract infection, and septicemia were the most common causes of infection (71). Parkinsonism may worsen in the setting of general medical illness, most notably urinary tract infections, which may not always be symptomatic.
A physician should always rule out infection and other medical comorbidities, such as metabolic disorders and occult malignancy, when there is an acute deterioration in parkinsonian symptoms. It is routine for patients with parkinsonism, especially in advanced stages, to have fluctuations of motor symptoms and periods of suboptimal control. Motor complications, falls, and hallucinations were also implicated in hospitalization for idiopathic Parkinson disease (58). In general, adjustments to the medication regimen will resolve the issue; however, it is important to first consider other causes for decompensation.
Acute and subacute subdural hematomas have been associated with marked deterioration of parkinsonism (ie, worsening of bradykinesia, gait and balance impairments, and confusion) (134). Patients with parkinsonism have gait and balance impairments, which significantly increase their risk for falls, leading to traumatic subdural hematoma. A clinician should have a low threshold to obtain neuroimaging, specifically a CT scan, even if a history of falls is not elicited. Early identification of this condition and surgical evacuation of hemorrhage can be lifesaving. Another cause of deterioration in gait and balance is a compressive spinal cord lesion (100).
Additionally, life-threatening onset of severe dyskinesias as a complication of dopamine therapy can occur. Two cases of hyperthermia, rhabdomyolysis, hallucinations, and confusion as associated with severe dyskinesias in a Parkinson patient (dyskinesia-hyperpyrexia syndrome) were reported in advanced Parkinson disease. Both improved with aggressive supportive therapy and the gradual discontinuation of the dopamine agonist. Levodopa was continued for the duration of the syndrome (38; 07).
Respiratory compromise is rarely associated with parkinsonism, with the exception of multiple system atrophy. Multiple system atrophy is associated with an increased risk for sudden death from upper airway obstruction (14). The presence of snoring and intermittent nocturnal stridor is an early sign of vocal cord dysfunction. This is followed by an increased risk of developing sleep apnea due to central hypoventilation (76), and patients may present with snoring and stridor due to posterior cricoarytenoid muscle dysfunction (vocal cord abductor paresis) (11). These symptoms can be present without evidence of clinical laryngeal dysfunction, such as hoarseness or dysarthria. It is prudent to directly question a bed partner or caregiver. The presence of stridor has been associated with less than a 2-year survival, and the onset of daytime stridor is particularly ominous (109). Prompt referral to an otolaryngologist for a fiber optic laryngoscopy is required to determine if laryngeal dysfunction is present. Patients with evidence of partial abductor paresis are at increased risk for sudden death and require definitive treatment with tracheostomy (108). Those who decline tracheostomy can be treated with continuous positive air pressure (CPAP) at night (47). Despite these interventions, sudden death has been reported in patients even after tracheostomy placement (52).
Serotonin syndrome is an iatrogenic disorder arising from increased biological activity of serotonin mediated by brainstem 5HT2A receptors (37). Serotonin syndrome was first described by Oates in the 1960s in a patient treated with monoamine oxidase inhibitor (MAOI) monotherapy (89). Serotonin syndrome typically arises from an excessive accumulation of serotonin and occurs in a dose-dependent manner. In clinical practice, serotonin syndrome usually occurs in patients taking two or more drugs with serotonergic actions. Medication combinations such as serotonin reuptake inhibitors with nonselective MAOIs and tricyclic antidepressants are typical offending agents. A single case of abrupt withdrawal of clozapine, a serotonin receptor antagonist, in the setting of continued administration of a selective serotonin reuptake inhibitor has been reported (112). However, other medications and illicit substances such as dextromethorphan, lithium, meperidine, fentanyl, tramadol, atypical neuroleptics, linezolid, triptans, dietary supplements, and recreational drugs such as ecstasy can significantly increase the risk of serotonin syndrome (see Table 2) (101; 48; 95; 60; 13). Though nonselective MAOIs are rarely used today, there is still some risk of serotonin syndrome, even with selective MAOIs (30). The risk of serotonin syndrome from combinations between triptans and selective serotonin reuptake inhibitors or selective norepinephrine reuptake inhibitors has been debated (37). In a registry study of nearly 48,000 patients prescribed triptans, the incidence of serotonin syndrome among those also taking selective serotonin reuptake inhibitors or selective norepinephrine reuptake inhibitors was found to be very low, ranging from zero to four cases per 10,000 person-years (91). Despite a 2006 United States Food and Drug Administration warning about the risk of serotonin syndrome with these drug combinations, it has been proposed that serotonin syndrome is rare and may not warrant avoiding effective treatment for patients with coexisting affective disorders and migraines.
|
• 3,4 Methylenedioxymethamphetamine (MDMA or ecstasy) |
Serotonin syndrome encompasses a spectrum of clinical features, many of which overlap with those of neuroleptic malignant syndrome. High levels of central serotonin lower central dopamine levels, which may lead to a hypodopaminergic state similar to neuroleptic malignant syndrome (101). Core symptoms include fever, myoclonus, and altered mental status (elevated mood, restlessness, disorientation).
Additional signs include hyperreflexia and clonus, rigidity, clonus, tachycardia, diaphoresis, and seizures. Hyperreflexia, clonus, and myoclonus are important features that can help differentiate serotonin syndrome from neuroleptic malignant syndrome. At least three distinct sets of diagnostic criteria exist for identifying serotonin syndrome, namely the Sternbach criteria, Radomski criteria, and Hunter criteria (133). The Sternbach criteria allow for the identification of at-risk patients with milder symptoms (see Table 3). Serotonin syndrome may represent a clinical spectrum of excessive serotonergic activity, ranging from subclinical and mild forms to chronic “smoldering” serotonin syndrome, rather than a purely acute toxidrome (83). Polypharmacy involving serotonergic agents, drug interactions that increase serotonergic activity, and genetic variability in drug metabolism (eg, CYP2D6 polymorphisms) may all contribute to its development. Unlike neuroleptic malignant syndrome, serotonin syndrome frequently resolves in 24 hours, but mortality is as high as 11% (123). Complications include disseminated intravascular coagulation, rhabdomyolysis, metabolic acidosis, renal failure, or respiratory failure (36).
|
• Agitation | |
|
A. At least three of the following clinical features are present: in the setting of, coincident addition of, or increase in the dosage of a known serotonergic agent to an established medication regimen. | |
|
B. Other etiologies (ie, infection, metabolic, substance abuse, or withdrawal) have been ruled out. | |
|
C. A neuroleptic has not been started or increased in dosage prior to the onset of signs and symptoms listed above. | |
|
| |
Early recognition of serotonin syndrome is important. Patients with mild symptoms of serotonin syndrome and effective control of mood disorder should not have a second serotonergic medication added to their regimen. A clinical pharmacist may be able to identify at-risk patients and prevent the addition of another serotonergic medication that may exacerbate this syndrome (57). In more severe presentations, treatment is primarily supportive, with admission to the ICU and discontinuation of the offending medication. It is important not to add new medications with serotonergic action (see Table 2). Medications such as propranolol, diphenhydramine, chlorpromazine, diazepam, and methysergide have been used to treat serotonin syndrome. Cyproheptadine (12 mg starting dose, followed by 2 mg every two hours until appropriate clinical response), a 5HT2A antagonist and antihistamine, can reverse clinical symptoms in hours and may be initiated prophylactically when the addition of a third serotoninergic agent is required in specific circumstances (26). Benzodiazepines can also be considered, particularly to alleviate anxiety and agitation (22). Supportive and intensive care measures remain paramount, especially in severe cases. Of note, with the increased use of atypical antipsychotics in combination with multiple serotonergic medications, a delayed presentation of serotonin syndrome and/or a mixed clinical presentation of serotonin syndrome and neuroleptic malignant syndrome may occur (66; 113). Transitioning from one selective serotonin reuptake inhibitor to another or to an MAOI should be approached carefully to prevent serotonin syndrome, particularly when switching from an SSRI with a prolonged elimination half-life such as fluoxetine, which may require 5 weeks or more for sufficient washout (56).
Catatonia is a syndrome characterized by extreme negativism, motor immobility alternating with agitation, and behavioral abnormalities closely associated with developmental, mood, psychotic, toxic, and somatic disorders in children and adults (136). Malignant catatonia is a potentially lethal complication with a presentation similar to neuroleptic malignant syndrome (rigidity, fever, and obtundation), without a history of exposure to dopamine receptor-blocking medication. Malignant catatonia typically begins with intense agitation, catatonia, stereotypies, psychosis, and autonomic instability.
Other clinical features include motor symptoms (waxy flexibility, freezing, and stereotypies), abnormal speech (perseveration, echolalia, or mutism), reduced function (loss of independent activities of daily living), negativism (senseless refusal to comply), and reduced food and fluid intake. Frequently underdiagnosed, catatonia is a relatively common condition among psychiatric patients, with a reported frequency of 10% to 13%, and even higher rates in at-risk populations such as pervasive developmental and dysthymic disorders (19), and is often misdiagnosed as neuroleptic malignant syndrome in the ICU (103). In a study of 1719 schizophrenia patients admitted to an inpatient psychiatric facility with comorbid medical complications, catatonic patients had a 4-fold higher mortality risk than those without catatonic symptoms (35). Catatonia is frequently observed in the context of psychiatric conditions, particularly mood and psychotic disorders, but it can also manifest in less common clinical scenarios. These include autoimmune encephalitis, such as anti-NMDA receptor encephalitis (16), or as a result of withdrawal from certain medications, such as clozapine (121). Though the etiology of catatonia is unknown, low gamma-aminobutyric acid (GABA) activity in the frontal cortex in combination with low dopamine activity in the basal ganglia and/or elevated glutamatergic activity in the parietal cortex has been hypothesized. The abnormal neurotransmission has a widespread effect, which contributes to the broad range of clinical features in catatonia and may be associated with sudden discontinuation of dopamine receptor blocking agents (15). Additionally, the anterior thalamus has been implicated in malignant catatonia (90).
On average, malignant catatonia symptoms may last 8 days, with temperatures as high as 110°F, followed by severe rigidity and lack of movement, stupor, and death (100). It is important to recognize malignant catatonia as a distinct syndrome to initiate appropriate therapy, which can result in the complete resolution of symptoms within hours to days (32). Several diagnostic criteria have been proposed and are effective in screening patients for possible catatonia with high sensitivity. Unfortunately, most of these scales are not specific in establishing the diagnosis of malignant catatonia (see Table 4) (116).
|
(1) Catalepsy - the individual maintains a fixed/frozen posture | |
|
(2) Waxy flexibility - slight, even resistance to bodily manipulation | |
|
(3) Stupor - no conscious mental activity is witnessed within the person’s environment | |
|
(4) Agitation - emotionally restless; not as a result of external stimuli | |
|
(5) Mutism - little to no verbal response; cannot be explained by aphasia | |
|
(6) Negativism - opposition or unresponsiveness to external stimuli or instructions | |
|
(7) Posturing - spontaneous and active maintenance of a posture against gravity | |
|
(8) Mannerism - exaggerated or repetitive gestures or expressions | |
|
(9) Stereotypies - repetitive movements without obvious purpose | |
|
(10) Grimacing - displaying contorted facial expressions | |
|
(11) Echolalia - mimics another’s speech | |
|
(12) Echopraxia - mimics another’s movements | |
|
*3 or more symptoms are required to make the diagnosis | |
|
| |
Several published reports have demonstrated the efficacy of low-dose benzodiazepines (eg, intravenous lorazepam 0.5 to 2 mg) as both diagnostic and therapeutic agents. If symptoms of catatonia improve, the benzodiazepines should be continued until the resolution of symptoms is complete. Oral zolpidem and electroconvulsive therapy have also been reported to be effective in the management of refractory malignant catatonia symptoms (118; 143). Electroconvulsive therapy should be considered early or urgently when intravenous benzodiazepines are ineffective or when life-threatening autonomic instability is present because delayed treatment is associated with reduced efficacy of electroconvulsive therapy (45). Therefore, timely electroconvulsive therapy can be life-saving. Studies have also demonstrated that N-methyl-D-aspartate receptor (NMDA) antagonists such as memantine, amantadine, and ketamine can improve symptoms of malignant catatonia (88; 18; 90; 09). The use of atypical neuroleptics is controversial. Several studies failed to demonstrate improvement of symptoms, with an additional risk for developing neuroleptic malignant syndrome (127; 90); only one study showed mild improvement (96). In general, neuroleptics are not recommended in the treatment of malignant catatonia.
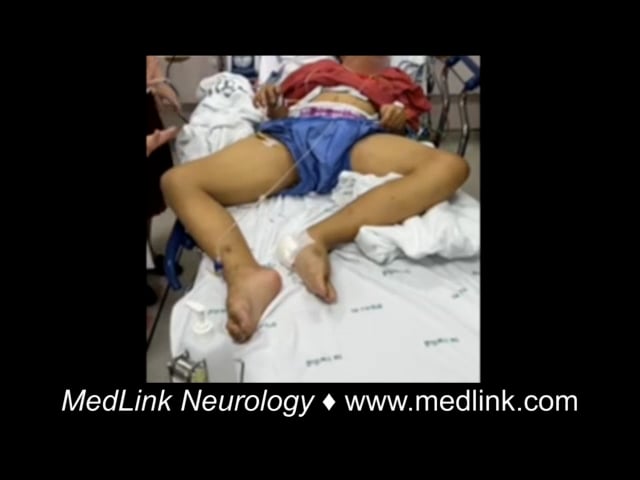
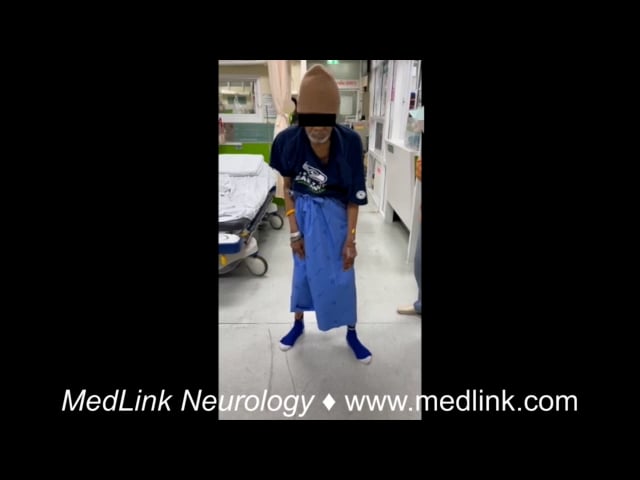
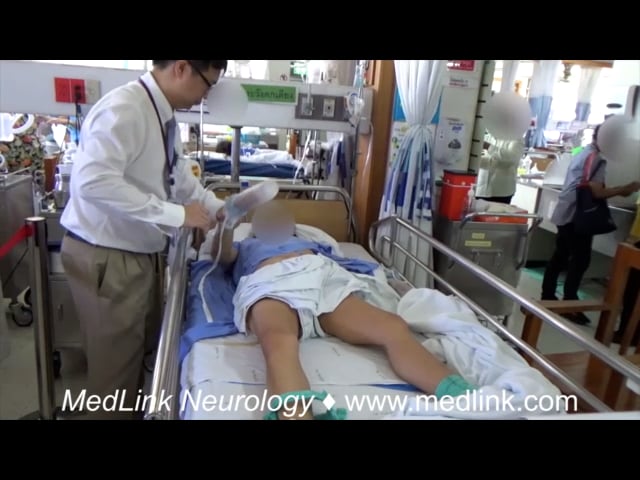
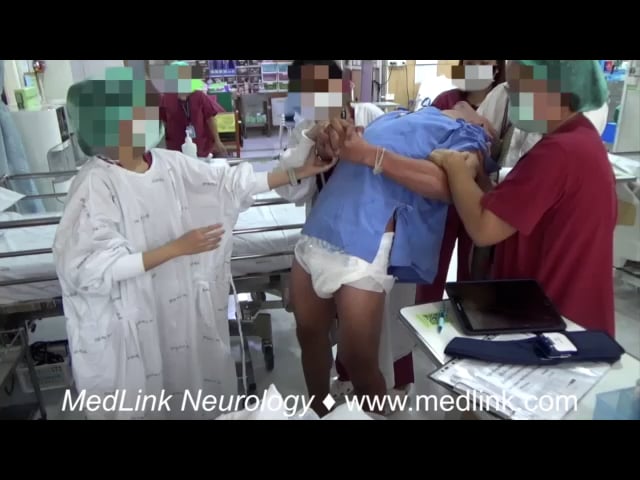
Dystonia is a movement disorder characterized by sustained muscle contractions causing irregular movements and postures. Patients with primary or secondary generalized dystonia can develop an unremitting life-threatening spasm called “status dystonicus” or “dystonic storm” (51).
The incidence and prevalence are unknown. Dystonic storm can be triggered by infections, medication adjustments, hyperthermia, trauma, dehydration, or respiratory compromise. There has been a significant increase in the number of reported episodes of dystonic storm, with cases more frequently associated with complications from deep brain stimulation, particularly battery exhaustion, rather than medication changes (68). This trend is likely influenced by advancements in the identification of genetic etiologies of dystonia, exemplified by conditions such as GNAO1, which are amenable to deep brain stimulation therapy. The pathogenesis remains poorly understood. A study using local field potential recordings in the globus pallidus interna (GPi) in 10 children with refractory dystonic storm demonstrated increased pallidal beta-band activity, suggesting that dystonic storm may represent a distinct neurophysiological state rather than simply the extreme end of severe dystonia (10).
These unrelenting spasms lead to hyperpyrexia, dehydration, respiratory failure, and rhabdomyolysis with renal failure. Thus, patients should be admitted to the ICU for close monitoring. In general, spasms are refractory to medications that are typically used to treat dystonia, such as anticholinergics (trihexyphenidyl), oral baclofen, and dopamine blockers or dopamine depleters, such as tetrabenazine (72; 49; 50; 03). Chloral hydrate and clonidine can be considered in children with dystonic storm to reduce dystonic spasms by facilitating sedation without respiratory depression (69). Benzodiazepines can be effective in suppressing spasms; however, this medication can cause respiratory depression and should be used with caution (72). Refractory cases may require general anesthesia, paralyzing agents, and intrathecal baclofen (25; 44). Case reports of pallidotomy and bilateral deep brain stimulation of the globus pallidus pars interna (GPi) or subthalamic nucleus (STN) may be useful in intractable cases (85; 05; 39; 03; 117; 12). There has been a paradigm shift toward considering neurosurgical interventions, such as deep brain stimulation, earlier in the treatment of severe dystonic storms, particularly for patients with DYT1 generalized dystonia, other primary dystonias, as well as some secondary dystonias, rather than reserving them as a last-resort option (120; 141).
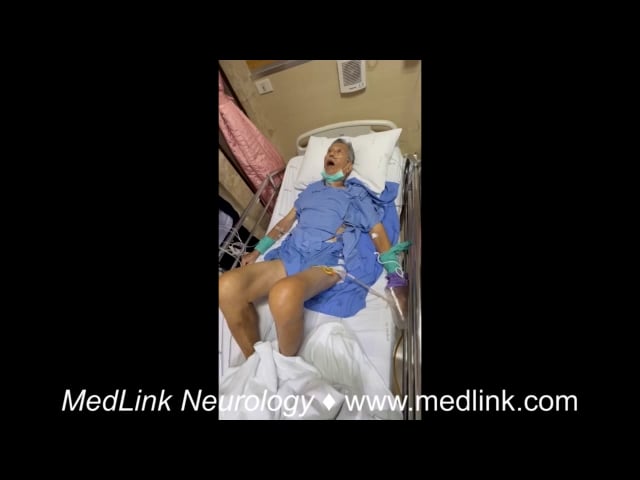
Acute dystonic reaction is a drug-induced movement disorder emergency characterized by sudden onset of dystonia, typically presenting as painful, sustained, or intermittent muscle contractions. Symptoms most commonly develop within the first few days of exposure to the offending medication, although they may occur up to the first week of treatment (128). The most frequent triggers are dopamine receptor blocking agents, including both typical and atypical antipsychotics. Other medications with dopamine blocking properties, such as metoclopramide and prochlorperazine, are also well-recognized causes (20; 107). Agents that can trigger acute dystonic reaction overlap with those implicated in neuroleptic malignant syndrome.
Phenomenologically, acute dystonic reaction most often involves the neck, trunk, as well as cranial including ocular muscles, although the extremities may also be affected. Characteristic postures include retrocollis and truncal extension dystonia.
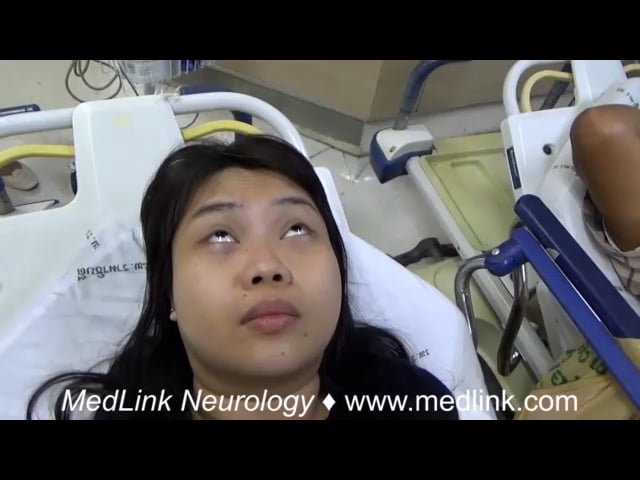
Lower cranial (aka, oromandibular) dystonia may occur and can interfere with speech and swallowing. In severe cases, involvement of the laryngeal muscles may lead to respiratory compromise, making early recognition essential. Oculogyric crisis is considered part of the spectrum of acute dystonic reaction and involves sustained dystonic contraction of the extraocular muscles. The eyes typically deviate upward or laterally, although in some patients the gaze may appear fixed straight ahead.
Episodes may also be accompanied by autonomic features, including tachycardia, diaphoresis, hypertension, or labile blood pressure.
Early recognition of acute dystonic reaction is important to prevent progression to laryngeal involvement that may compromise the airway. As with other neurologic emergencies, initial management should follow general principles, with attention to airway, breathing, and circulation (ABC) (119).
The mainstay of treatment is parenteral anticholinergic therapy. Acute dystonic reaction is believed to result from dopamine receptor blockade in the basal ganglia, leading to relative dopaminergic deficiency and an imbalance between dopaminergic and cholinergic activity (114). Anticholinergic medications restore this balance by counteracting the relative excess of cholinergic activity. Benztropine administered intravenously or intramuscularly is commonly used and typically results in rapid improvement of symptoms. Intravenous administration is preferred because it provides faster onset of action and secures vascular access should additional therapy be required. Intravenous diphenhydramine is also effective because of its anticholinergic properties. If these medications are unavailable, benzodiazepines such as lorazepam or diazepam may be used (138). Supportive care, including management of autonomic instability or fever, should also be provided when necessary.
Patients should be monitored closely after treatment because the effects of parenteral therapy may be short-lived, and recurrence of dystonia may occur. Observation for at least 24 hours is recommended, ideally in a monitored or intensive care setting when symptoms are severe. To reduce the risk of recurrence, short-term maintenance therapy with oral anticholinergic medications, such as trihexyphenidyl, is commonly continued for approximately 4 to 7 days (128).
The offending medication should be discontinued. If ongoing antipsychotic therapy is required, reintroduction should generally be deferred until symptoms have completely resolved, often waiting several days to one week after the acute event. Agents with lower risk of drug-induced movement disorders, such as quetiapine or clozapine, may be preferred.
Although oculogyric crisis most commonly occurs in the context of medication exposure, it can also be seen in other neurologic conditions. These include neurotransmitter disorders such as aromatic L-amino acid decarboxylase deficiency or tyrosine hydroxylase deficiency, postencephalitic parkinsonism, anti-N-methyl-D-aspartate (NMDA) receptor encephalitis, Kufor-Rakeb syndrome, and Perry syndrome (110). The dystonic patterns observed in acute dystonic reactions, including retrocollis, truncal extension dystonia, and oromandibular dystonia, may also resemble the phenomenology in tardive dystonia (132). However, tardive dystonia represents a delayed complication of chronic dopamine receptor blocking agent exposure and is beyond the scope of this article.
Tourette syndrome is a common neuropsychiatric disorder that presents with motor and phonic tics in childhood, in combination with comorbid attention deficit disorder and obsessive-compulsive behavior. The pathophysiology is largely unknown, though basal ganglia dopaminergic dysfunction has been implicated. Symptoms of Tourette syndrome naturally fluctuate throughout its clinical course, and most patients experience some degree of remission by the end of adolescence. In rare cases, Tourette syndrome and its comorbid behavioral symptoms can result in potentially life-threatening complications secondary to severe tics (ie, self-injurious tics, coprolalia, and copropraxia), extreme rage attacks, depression with suicidal ideation, and disabling self-injurious behaviors, which has been termed “malignant Tourette syndrome” (21; 04). Additionally, violent dystonic neck tics can result in myelopathy (62; 122).
These symptoms are difficult to treat with typical medications for tic suppression (ie, dopamine receptor blocking agents and dopamine depleters) or behavioral management (SSRIs and other mood stabilizers). Botulinum toxin injections can be effective in reducing focal disabling tics such as dystonic neck tics (02), and a case report of laryngeal injections demonstrated reduction of coprolalia (106). For refractory cases of malignant Tourette syndrome, deep brain stimulation appears to be a promising treatment for tic reduction. The ideal stimulation target, globus pallidus pars interna (GPi) versus centromedian-parafascicular (CM-Pf) complex of the thalamus, continues to be debated (01; 74; 131; 82). Other targets include centromedian nucleus-nucleus ventrooralis internus (CM-Voi) complex; ventral anterior and ventral lateral (VA/VL) nuclei of the thalamus, globus pallidus externa (GPe), anterior limb of internal capsule/nucleus accumbens (ALIC/NAc) area (08). A case of tics status was reported in a patient who developed continuous disabling vocal and motor tics on premature withdrawal of medications. These tics caused pain and disrupted sleep, later resulting in mild hyperthermia, tachycardia, and rhabdomyolysis, which required intravenous sedation. The patient required 5 days of sedation with ventilation before tics remitted to baseline (61).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Pichet Termsarasab MD
Dr. Termsarasab of Ramathibodi Hospital, Mahidol University has no relevant financial relationships to disclose.
See Profile
Robert Fekete MD
Dr. Fekete of New York Medical College received consultation fees from Acadia Pharmaceutical, Acorda, Adamas/Supernus Pharmaceuticals, Amneal/Impax, Kyowa Kirin, Lundbeck Inc., Neurocrine Inc., and Teva Pharmaceutical, Inc.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Sleep Disorders
Jul. 03, 2026
Movement Disorders
May. 24, 2026
Movement Disorders
May. 18, 2026
Movement Disorders
May. 18, 2026
Movement Disorders
May. 18, 2026
Movement Disorders
May. 18, 2026
General Child Neurology
Apr. 24, 2026
Movement Disorders
Apr. 12, 2026