
Neuro-Oncology
Turcot syndrome
May. 27, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
The authors provide an updated summary of spinal ependymomas, highlighting new molecular features of the most common primary intraspinal tumor of adults as well as imaging characteristics. The update includes published epidemiological data, surgical treatment options, comments on the occurrence of these tumors in pregnancy, and updates in the evolving imaging modalities.
In 1887, Horsley performed the first reported successful removal of an intradural, extramedullary tumor (34). With the help of Sir William Gowers, Horsley removed a "fibromyxoma" overlying the spinal cord at the T4 level. Postoperatively, the patient developed a debilitating pain syndrome but later experienced a full neurologic recovery. In the following 50 years, pioneering neurosurgeons such as Elsberg, Frazier, and Cushing took particular interest in extramedullary tumors, recognizing their frequently benign nature and often dramatic recovery from profound neurologic deficit. In 1911, Elsberg and Beer reported the first successful removal of an intramedullary spinal cord tumor. Frazier also commented on the potential for the removal of encapsulated intramedullary neoplasms. However, early attempts at removal of intramedullary spinal cord tumors were associated with serious operative morbidity and mortality, such as complete paralysis.
For the next several decades, there was little impetus to modify the approach of biopsy, dural decompression, and radiation therapy, despite the recognition that after a relatively short remission, serious disability or death ensued. This "traditional" attitude was based on the assumption that it was not feasible to carry out extensive removal of tumors from within the center of the spinal cord without inflicting additional neurologic injury (20; 40). In 1954 Greenwood, with the aid of bipolar cautery and loupe magnification, reported six patients who underwent complete removal of intramedullary ependymomas (36). By 1963 Greenwood had treated nine patients with surgery alone. There was no tumor recurrence in his surviving patients (7) with a mean follow-up of 9 years (37). With time, it has become clear that the majority of intramedullary spinal ependymomas can be radically excised with an acceptable morbidity and mortality, and a low incidence of recurrence (39; 66; 60; 92; 29; 40; 18; 17; 04; 61; 102; 28; 14; 24).
Sensory complaints, such as subjective sensory symptoms, objective dysesthesia, and back pain, are the most common presenting symptoms. Primary motor dysfunction at presentation is uncommon and reported in 13% patients (24). Symptoms are often nonspecific initially, and a high index of suspicion is necessary to allow early diagnosis of these lesions, especially without trauma history or prior complaints (84). Disease onset is usually insidious, and the duration of symptoms prior to diagnosis varies from several weeks up to 15 years (average of 14 months) (99). The sensory symptoms may be in the hands or in the trunk as the tumors spread along the cervical and thoracic cord with an average span of two to three segments. Unusual and falsely localizing signs and symptoms may result from subarachnoid hemorrhage (02; 88), hydrocephalus due to raised cerebrospinal fluid pressure (77), and intramedullary cyst formation (93). Rarely, bleeding from spinal ependymomas may lead to secondary superficial siderosis of the nervous system and sensorineural hearing loss (53). Hemorrhagic presentations of ependymomas have been associated with anticoagulation, epidural anesthesia, pregnancy, and trauma (72).
Patients with ependymomas arising in the cauda-equina and filum-terminale region usually present with longstanding symptoms associated with extradural cord compression include segmental, hemicord, and transverse dysfunction. Local or radicular pain is the most common symptom. Pain may be dull or sharp, constant or intermittent. Urinary dysfunction and motor weakness follow if the tumor is not diagnosed early. Less than 30% of patients with caudal ependymomas present with weakness. Early symptoms of extramedullary corticospinal tract compression or, rarely, intramedullary conus medullaris extension include stiffness, fatigue, and gait abnormalities.
A 45-year-old male presented with a 1-year history of sensory dysesthesias. Laboratory testing, including workup for diabetes and vitamin deficiency, was completely unremarkable. Neurologic exam was remarkable for sensory loss in the arms, trunk, and legs; however, there was no motor weakness. Patient described worsening gait. An MRI of the brain was unremarkable; however, an MRI of the C-spine showed an enhancing mass with a hemosiderin cap expanding the spinal cord at C6 and C7. He underwent a surgical procedure for resection of the intramedullary mass with laminoplasty. His sensory deficits worsened immediately postoperatively; he remained otherwise at his neurologic baseline. At his 3-months follow-up visit, the dysesthesia improved but persisted.
Classification. The WHO 2021 classification of CNS tumors classified spinal cord ependymomas according to a combination of histopathological and molecular features as well as anatomic site (58) (see Table 1)
|
Table 1. Pathologic Characteristics of Spinal Ependymoma Subtypes | ||||
|
WHO grade |
Histologic features |
Molecular features |
Location | |
|
Spinal ependymoma |
2.3 |
Monomorphic glial cells with round to oval nuclei and speckled chromatin, pseudorosettes. IHC: immunoreactivity for GFAP, S100 protein, and vimentin, focal dot-like EMA expression. |
NF2 mutation Chromosome 22q deletion |
Cervical spine (85.5%), followed by the thoracic spine (61.9%) and the lumbar spine (7.5%). |
|
Spinal ependymoma MYCN-amplified |
Pseudorosettes High-grade features including high mitotic activity, microvascular proliferation, and necrosis. IHC: immunoreactivity for GFAP and dot like EMA expression, MYC expression. |
MYCN amplification chromosome 2p |
Cervical and thoracic with higher incidence of leptomeningeal spread | |
|
Spinal myxopapillary ependymoma |
2 |
Well-differentiated cuboidal to elongated tumor cells radially oriented around hyalinized fibrovascular cores, commonly with degeneration-derived myxoid accumulation. IHC: immunoreactivity for GFAP, S100, CD99, and CD56. Increased expression of HIF1a, HK2, PDK1, PDHA1. |
Mutations in chromosome 7 |
Filum terminale or conus medullaris |
|
Spinal subependymoma |
1 |
Small and round-to-oval nuclei clustering in a voluminous matrix of fibrillary cytoplasmic processes, perivascular pseudorosettes, IHC: immunoreactivity for GFAP, less frequent dot-like EMA expression. |
TERT mutations chromosome 6 deletion. |
Thoracic |
|
Abbreviations: GFAP: Glial fibrillary acidic protein; EMA: Epithelial membrane antigen; HIF: Hypoxia-Inducible Factor; HK2: hexokinase 2; PDK1: pyruvate dehydrogenase kinase 1; PDHA: Phosphorylation of the pyruvate dehydrogenase PDHA; NF2: Neurofibromatosis type 2; MYCN: myelocytomatosis-neuroblastoma | ||||
Adapted from (97)
|
(1) Spinal ependymomas occur along the spinal canal, are intramedullary tumors, and are commonly located in the cervical or cervico-thoracic segments. Ependymomas are unencapsulated but well-circumscribed glial neoplasms. The median age of diagnosis is 25 to 45 years. Spinal ependymoma show chromosomal alterations, with chromosome 22 loss being the most frequent. Sporadic spinal ependymomas most frequently have somatic NF2 mutation. By definition, these tumors have absence of MYCN amplification. They have a low level of mitotic activity and are assigned to CNS WHO grade 2 or 3 based on mitotic activity and invasion of the surrounding spinal cord structures. They have a favorable prognosis with a PFS of 70% to 90% and OS of 90% to 100% over 5 to 10 years. Spinal ependymomas associated with NF-2 seem to have an indolent course and may be observed or treated with surgery resection (03). | |
|
(2) Spinal ependymomas, MYCN-amplified, are intramedullary tumors occurring along the spinal canal, with an occasional extramedullary component. They are aggressive tumors with a poor prognosis. They are commonly located in the cervical or cervico-thoracic segments and rarely in the lumbar segment. They are large, involve multiple spinal segments, and frequently have leptomeningeal dissemination at presentation or during the disease course. They have high-grade histological features (microvascular proliferation, necrosis, high mitotic count) but are yet to be assigned a CNS WHO grade. A high level of MYCN amplification is present. SP-MYCN is characterized by early metastases, fast progression upon relapse, leptomeningeal spread, and poor response to multimodal therapy methods (32). | |
|
(3) Myxopapillary ependymomas are extramedullary, largely located in the conus medullaris and filum terminale, may exhibit leptomeningeal dissemination, and rarely have extra-neural metastases. They account for 27% of all spinal ependymomas and have a favorable prognosis (54). They occur at an incidence of 0.6 to 1 per million with a male:female ratio of 1.4 to 2:1. They are characterized by papillary arrangement of tumor cells around blood vessels with perivascular myxoid change and microcyst formation, as well as a Ki-67 labelling index of 2% to 3%. They are assigned to CNS WHO grade 2 (had been classified as WHO grade 1 in the preceding WHO classification system). Anaplastic myxopapillary ependymoma have regional hypercellularity and reduced mucin, in association with two of the following features: greater than 2 mitosis/mm2, Ki-67 labelling index of greater than 10%, microvascular proliferation, and spontaneous necrosis. They have a favorable clinical prognosis, with long-term OS rates surpassing 90% after whole or partial surgical resection (07). | |
|
(4) Spinal cord sub-ependymoma are well circumscribed with a lack of conspicuous nuclear atypia and are assigned to CNS WHO grade 1. They have a predilection for the cervical or cervicothoracic junction and typically present with insidious myelopathy or radicular pain (89). They are commonly seen in adults, and the mean age of presentation is 44 years. They have an excellent prognosis with rare postsurgical recurrence. A higher Ki-67 labelling index (> 1%) is associated with increased risk of recurrence. They can often have a characteristic deletion of chromosome 6q (74). |
Etiology and pathogenesis. Ependymomas arise from ependymal cells forming the lining of the ventricles and the central canal. The etiology of ependymomas is unknown. Familial autosomal dominant intramedullary ependymoma associated with the neurofibromatosis type 2 gene and without other features of neurofibromatosis type 2 has been described (103). Although rare, ependymomas have been reported in patients with neurofibromatosis type 1 as well (12). Loss of heterozygosity on 22q is inversely corrected with loss of heterozygosity of 11q. Loss of heterozygosity of 11q may be accompanied by mutations of the MEN1 gene, which is associated with malignant presentation (79). Preliminary gene expression profiling studies of spinal ependymomas suggest that they have increased expression of HOXb5, PLA2G, and CDKN2A and can be differentiated from intracranial tumors (48). Genes found to be hypermethylated include RASSF1A and TRAIL. HIC-1 was only hypermethylated in intracranial ependymoma. MGMT was rarely found to be hypermethylated. The stem cells for ependymoma have been proposed to be radial-glial (79).
The relative abundance of ependymomas of the spinal cord may be in proportion to the distribution of ependymal tissue throughout the CNS (65; 96; 61). About 50% of spinal ependymomas arise from the central canal of the cervical and thoracic spinal cord. They are the most common intramedullary spinal cord tumor in adults (24). The rest (approximately 50%) arise from the intradural portion of the filum terminale (85). Because of its neuroectodermal derivation, Slooff and Kernohan classified these tumors as intramedullary (90). From a surgical and clinical perspective, however, most of the ependymomas in the filum/cauda-equina region are extramedullary. A minority of these tumors may have their epicenter located within the tissue of the conus medullaris and, therefore, are "true" intramedullary neoplasms.
Ependymomas are relatively rare tumors occurring at an incidence rate of 0.43 patients per 100,000 population (71). They account for 1.6% of all CNS tumors, 3.1% of all malignant brain tumors, and 6.5% of all gliomas. Intracranial ependymomas generally occur in children and young adults and represent 1% to 8% of primary brain tumors. About one-third to one-half of all ependymomas arise within the spinal canal. Nearly 90% of pediatric ependymal tumors occur intracranially, and approximately 65% of adult tumors occur in the spine (62). Spinal ependymomas account for approximately 17% of all spinal cord tumors in adults (71). They represent the most common intramedullary spinal neoplasm in the adult population and account for 40% to 60% of the 2700 primary spinal cord tumors diagnosed in the United States each year (99; 15; 83; 94; 38). Intramedullary ependymomas are seen most frequently in the fourth decade of life and are rare below the age of 10 years (38; 63). In children, older age and spinal location are predictive of improved 5-year survival (62).
Filum ependymomas may arise at any age but are more common in early and middle adult years (30; 15), with a peak occurrence in the third to fifth decade (69; 90; 27). Men are slightly more commonly affected than women (63). Familial cases have been reported (82).
Based on SEER data, the incidence of ependymomas appears to have increased over the past 30 years in adults but not in children (63). However, spinal ependymomas associated with NF-2 seem to have an indolent course and may be observed or treated with surgery resection (03).
There is currently no prevention for spinal cord ependymomas.
Intramedullary ependymomas are a group of spinal cord tumors with a well-defined clinical presentation and MRI appearance. The differential diagnosis includes astrocytomas, gangliogliomas, oligodendrogliomas, subependymomas, hemangioblastomas, metastases, and benign lesions, such a lipoma (84). Clinically, the sensory phenomena, manifested by dysesthesias, are present in the great majority of patients with intramedullary ependymomas. It may be related to the common location of the tumor around the central canal. It is different from patients with spinal cord astrocytomas and gangliogliomas in whom the presenting problem is usually back pain followed by progressive motor dysfunction. When sensory phenomena are part of the initial symptomatology in patients with astrocytomas, they are usually manifested as loss of sensation rather than dysesthesias. Unlike astrocytomas, gadolinium-enhanced MRI in patients with ependymomas often shows clear demarcation of the rostral and caudal poles of the tumor. When viewed on cross-sectional images, the cord is expanded uniformly, unlike the astrocytomas in which the cord is usually "lumpy." In a comparative MRI analysis of 43 patients, the presence of syringohydromyelia appeared to be a significant factor in distinguishing ependymoma from astrocytomas (46). Furthermore, inflammatory lesions (multiple sclerosis, neuromyelitis optica, lupus, etc.) may also mimic spinal cord ependymomas.
Subependymoma is a distinct tumor entity characterized by slow growth and usually noninvasive behavior. MRI, even with enhancement, is often not distinguished between a subependymoma and the more common ependymoma (67; 73). Spinal cord subependymomas follow a benign course. The surgical treatment is identical to that for ependymomas.
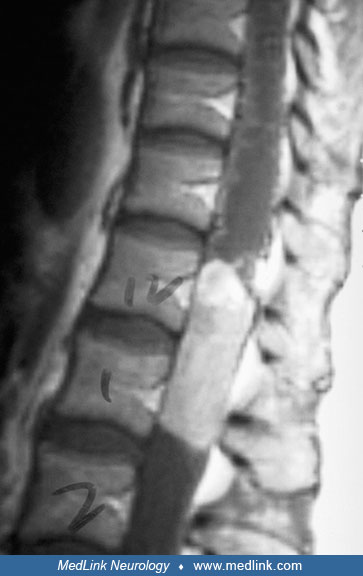
MRI is the diagnostic procedure of choice for primary and recurrent intramedullary spinal ependymomas (98; 06; 68; 47; 59). Following gadolinium injection, more than 90% of lesions enhance homogeneously and brightly, and have sharply defined rostral and caudal margins. Ependymomas originate from the region of the central canal, expand symmetrically within the spinal cord, and typically squeeze the surrounding neural tissue to only a few millimeters of thickness. About 30% of intramedullary ependymomas are associated with a rostral or caudal cyst, similar to that noted for spinal cord astrocytomas. Myxopapillary ependymomas of the conus medullaris also exhibit contrast enhancement, although not always as intensely as those within the cord parenchyma. The presence of syringohydromyelia on MRI favors the diagnosis of ependymoma and can help to differentiate it from astrocytoma (46).
These tumors may appear hyperintense on T1-weighted images as a result of the accumulation of mucin (45).
Presurgical MRI with diffuse tensor imaging to evaluate the relationship between neoplasm and white matter fiber tracts has helped in differentiating noninfiltrating neoplasms, such as spinal ependymomas, from other infiltrative and more aggressive neoplasms, which are considered not resectable. This allows an understanding of the neoplasm that is not appreciated with conventional MRI and may facilitate in preserving neurologic function after surgery (35).
There are no prospective randomized studies available to guide the management of these patients. Gross total resection should be attempted where feasible with acceptable neurologic risks (80). Post-operative MR should be performed to assess extent of resection. Leptomeningeal dissemination should be assessed for all patients due to risk of CSF dissemination after surgery. MRI complete spine with contrast and CSF cytology should be obtained no earlier than 2 to 3 weeks after surgery.
Surgery. With advances in neurosurgical techniques, the "traditional" approach of biopsy, dural decompression, and radiation therapy for intramedullary gliomas is no longer recommended. This course of treatment was based on the assumption that it is not feasible to carry out extensive removal of tumors from within the center of the spinal cord without a great likelihood of inflicting additional neurologic injury (20; 40; 76). This guideline, however, has been revised because the majority of these neoplasms are low-grade neoplasms that are surgically curable (56). Modern neurosurgical aids such as the operating microscope, the intraoperative ultrasound (25), the CO2 laser (21), and especially ultrasonic aspiration (23; 16) have dramatically improved the results and the operative morbidity of spinal ependymomas (24; 56).
Ependymomas of the cauda equina can usually be seen immediately following dural opening. The nerve roots are displaced laterally but may lie posteriorly and obscure the tumor in patients with a ventrally located tumor. In these cases, it is relatively simple to remove the tumor en bloc by dividing the distal filum terminale caudal to the tumor and then displacing the entire mass out of the cauda equina and incising the remainder of the tumor just below the conus medullaris. The relationship of the tumor to the conus medullaris is variable. In many cases, the entire mass is within the filum, and it is not necessary to pursue tumor fragments rostrally into the conus medullaris. Occasionally, however, this pursuit may be mandatory in an effort to obtain a surgical cure. It is essential that neural tissue in the conus medullaris not be manipulated in any way and that tumor fragments be "extracted" from below.
A few ependymomas of the cauda equina seem to have grown from the region of the conus medullaris and have erupted out of the filum, with tumor tissue filling the entire thecal sac below the conus medullaris. In these cases, the normal neural elements of the cauda equina are not displaced circumferentially around the mass but rather run through the tumor tissue. In these cases, it is necessary to remove the tumor bit by bit by working between and around the neural elements until all of the neoplastic tissue is removed. In such an occurrence, it is frequently necessary to extract remaining tumor fragments from within the conus medullaris. However, it is again important to leave the neural tissue undisturbed and remove tumor fragments by working through that area from which the tumor has grown into the thecal sac. Rarely, ependymomas may grow caudally into the sacrum with the tumor being mostly or entirely extradural and may reach gigantic size (13; 22; 19; 31).
True intramedullary ependymomas almost invariably have a "true" cleavage plane between the tumor and adjacent neural tissue (24). Although this contributes to "total" excision of these neoplasms, it is also a potential hazard, because it encourages the surgeon to attempt an en bloc resection and to remove the entire mass in one or two large pieces. If this is attempted, there will be excessive and unnecessary manipulation of normal neural tissue. To avoid these complications, the ependymoma should be centrally "debulked" as with an astrocytoma, and only when the core of the tumor has been removed should the surgeon develop the plane of cleavage between the tumor and adjacent tissues. This may be accomplished by retracting the remaining tumor tissue into the residual cavity and not by retracting the spinal cord from the tumor. Some authors, however, favor en bloc excision of these tumors (43; 51).
The decision to perform an instrumented fusion in addition to the laminectomy is a difficult decision. A study by Sciubba and colleagues involving 32 patients who underwent cervical laminectomy without fusion for resection of an intradural tumor (18 intramedullary and 14 extramedullary) were followed to assess for kyphotic deformity that subsequently resulted in the need for a subsequent fusion surgery (86). Each increasing number of laminectomies performed was associated with a 3.1-fold increase in the likelihood of subsequent vertebral instability. At a mean follow-up interval of 25.2 months, 33% (four of 12) of the patients who had undergone a three or more level laminectomy required subsequent fusion compared with 5% (one of 20) who had undergone a two level or less laminectomy (p = 0.03). Four (36%) of 11 patients initially presenting with myelopathic motor disturbance required subsequent fusion compared with one (5%) of 21 presenting initially with myelopathic sensory or radicular symptoms (p = 0.02). Age, the presence of a syrinx, intramedullary tumor, C-2 laminectomy, C-7 laminectomy, and laminoplasty were not associated with subsequent symptomatic instability requiring fusion (86).
Radiation therapy. Surveillance is recommended for all grade 2 ependymomas after gross total resection and local postoperative radiation therapy (45–54 Gy) for patients with subtotal resection. All WHO grade 3 ependymomas are treated with postoperative radiation therapy (45–54 Gy) irrespective of extent of resection. Craniospinal irradiation (CSI) is recommended for patients with CSF dissemination (80). There is increasing evidence that adjuvant radiation therapy increases the time-to-tumor progression after subtotal resection. Others recommend careful monitoring (with MRI) of patients with minimal residual disease and reoperation in those with substantial residual tumor.
In general, ependymomas receive approximately 5040 cGy in 180 cGy fractions, whereas malignant ependymomas and multifocal ependymomas receive between 5040 and 5400 cGy, respectively (44). There are studies suggesting the feasibility and safety of stereotactic radiosurgery in patients with spinal cord tumors (81; 08). Favorable acute toxicity profile of proton beam radiation was demonstrated in a study of eight pediatric spinal ependymoma patients (05).
The European Association of Neuro-Oncology (EANO) has similar recommendations for pediatric patients, except it is recommended that postoperative radiotherapy should be used in children older than 18 months or in children between 12 and 18 months with significant neurologic deficits (80).
Chemotherapy. Chemotherapy has no role for the majority of patients with low-grade or myxopapillary ependymomas. For patients with tumors that recur after surgery and radiation therapy, a variety of chemotherapeutic agents, including platinum agents, procarbazine, temozolomide, and etoposide have been tried with only limited success (87; 101; Balmaceda 2000; 11). There are also anecdotal data suggesting that irinotecan (95) and imatinib (26) may have activity in ependymomas.
Systemic therapy with a combination of dose dense temozolomide and lapatinib in a phase 2 study of 50 adult recurrent ependymomas (including 50% spinal ependymomas) demonstrated clinical activity with objective responses, prolonged disease control with a median progression-free survival of 7.8 months, and improvement in disease-related symptoms (33).
Myxopapillary ependymoma. These tumors are irregularly shaped, and anatomical location can make gross total resection challenging. A strong correlation between capsular violation at surgery and recurrence has been found. Observation is recommended after gross total resection with intact capsule. In cases of gross total resection with capsule violation or incomplete resection, local postoperative radiotherapy is recommended to doses of 50 or more Gy, respecting spinal cord tolerance. Craniospinal irradiation is recommended with leptomeningeal spread.
Recurrence is largely local (27%). Both distant spinal (9%) and brain (6%) recurrence has been reported (100). The recurrence rate of malignant pleural effusion after surgery is 35%, and the median time to recurrence is 36 months (50).
Five-year progression-free survival and overall survival after surgery are 74% and 99%, respectively. Addition of adjuvant radiation therapy after subtotal resection decreased failure rates (70; 50). However, the addition of radiation therapy was not beneficial after gross total resection (50).
A multi-institutional retrospective cohort study of 60 pediatric and young adult patients diagnosed with malignant pleural effusion characterized patterns of relapse and long-term outcomes after radiation (55). Five-year overall survival, progression-free survival, and the cumulative incidence of local in-field progression after radiation therapy were 100%, 60.8%, and 4.1%, respectively. The risk of recurrence after radiation therapy was low within the radiation field and high in the spine above the radiation field and intracranially.
In a retrospective study of 59 spinal myxopapillary ependymomas, recurrence-free survival was 17.2 years with gross total resection versus 5.5 years after subtotal resection (49). This study demonstrated no benefit to immediate adjuvant radiation therapy. However, radiation therapy at the time of recurrence was associated with a significantly longer time to second disease recurrence (9.5 vs. 1.6 years).
A study of 183 patients with spinal malignant pleural effusion reported better prognosis in patients aged over 36 years, receipt of gross total resection, and receipt of adjuvant radiation therapy (100).
Twelve patients with a median age of 13.5 years with disseminated spinal malignant pleural effusion were spared craniospinal irradiation and were effectively treated with limited-volume brain-sparing proton therapy (54 Gy RBE) in both the primary and salvage setting (57). Five-year actuarial rates of local control, progression-free survival, and overall survival were 100%, 92%, and 100%, respectively.
The results of surgery for caudal ependymomas are excellent. Complications are generally related to wound healing and perioperative fistulas. A joint neurologic and plastic surgery approach was developed to reduce the morbidity of wound closure following extensive and complicated laminectomy (105; 104). The fascia requires particular attention because this is usually the only watertight layer. The fascia and muscle are released both superficially from the subcutaneous tissues and deeply from the bony elements. If this is not enough to achieve closure with no tension, relaxing incisions are performed. The musculofascial layer is closed with "figure-of-8" Novafil or Prolene sutures that are tightly knotted. If there is any doubt regarding the watertightness of the closure, it is tested with injection of fluid under pressure. Improvement of the perioperative deficit is often dramatic and depends mainly on the severity and duration of the existing deficit. Filum ependymomas generally do not recur following en bloc resection. There is about a 20% recurrence rate if tumor is left behind (91). It appears that gross-total resection seems to be particularly beneficial for WHO grade II spinal ependymomas (70).
In a series by Epstein and colleagues of adult intramedullary spinal cord ependymomas treated surgically, 37 out of 38 patients had gross total removal of their tumor, as confirmed by a postoperative MRI (24). There has been no clinical or radiographic evidence of recurrence after a mean follow-up period of 24 months in any patient in whom total removal was accomplished. The morbidity of the surgical procedure was directly related to the preoperative neurologic status. Patients with little or no disability preoperatively are at little risk to sustain an injury, whereas patients with more significant neurologic dysfunction have a greater likelihood of being impaired by surgery. In one study, spontaneous regression of residual tumor after surgical resection was noted in 37% of patients (41).
For patients treated with surgery and radiation therapy, the overall 5-year survival ranges from 83% to 97%, and 10-year survival from 68% to 95% (99; 83). Patients with myxopapillary ependymomas generally have a better prognosis than other types of low-grade ependymomas (100% 5-year survival) (83). Local recurrence is the predominant pattern of failure (102; 99). Rarely, cerebrospinal fluid dissemination, spread to extravertebral soft tissues, and distant metastases may occur (64). The majority of patients who develop recurrent disease generally do so within 3 years of diagnosis, although there are numerous reports of recurrent disease developing over 10 years after treatment (99).
Nonmodifiable risk factors including age, tumor location (eg, spinal vs. intracranial), and associated mutations substantially influence the natural history of ependymomas. Poor prognostic factors for ependymomas include incomplete resection (101; 10; 01; 09), impaired preoperative neurologic function (41), age less than 20 years, the presence of three to four anaplastic features (necrosis, mitosis, vascular proliferation, cellular pleomorphism, or overlapping nuclei), and a MIB-1 proliferation index of greater than 20% (78). In children with ependymoma, older age and spinal location are correlated with improved survival (62).
There are reports in the literature of pregnant women who are discovered to have spinal ependymoma. The management of these patients presents unique surgical and anesthetic challenges (52; 42; 75). Intratumoral or subarachnoid hemorrhagic presentations of ependymoma are associated with pregnancy (72).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Jigisha Thakkar MD
Dr. Thakkar of Loyola University Medical School has no relevant financial relationships to disclose.
See ProfileJoseph Frazzetta MD
Dr. Frazzetta of Loyola University Medical Center has no relevant financial relationships to disclose.
See ProfileDaryn Cass MD
Dr. Cass of Loyola University Chicago has no relevant financial relationships to disclose.
See ProfilePaul M Arnold MD
Dr. Arnold of Loyola Medicine has no relevant financial relationships to disclose.
See Profile
Rimas V Lukas MD
Dr. Lukas of Northwestern University Feinberg School of Medicine received honorariums from Jazz Therapeutics, Novocure, and Servier for speaking engagements, honorariums from Cardinal Health, Catalyx, Merck, and Novocure for advisory board membership, research support from BMS as principal investigator, and an honorarium from GT Medical Technologies for DSMB membership.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuro-Oncology
May. 27, 2026
Neuro-Oncology
May. 27, 2026
Neuro-Oncology
May. 23, 2026
Neuropharmacology & Neurotherapeutics
May. 11, 2026
Stroke & Vascular Disorders
May. 03, 2026
Neuro-Oncology
Apr. 30, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026