















Methylmalonic acidemia
Jun. 01, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Adenylosuccinate lyase deficiency is a rare, autosomal recessive defect of purine metabolism affecting purinosome assembly and reducing metabolite fluxes through both de novo purine synthesis (DNPS) and purine nucleotide recycling pathways. The purinosome is a multienzyme complex of DNPS enzymes (including adenylosuccinate lyase) that cells transiently assemble in the cytosol to address both depletion of and increased demand for purines. Clinically, adenylosuccinate lyase deficiency is generally categorized into three phenotypes: a fatal neonatal form, a severe form (type I), and a milder form (type II). Neurologic symptoms are the most common and prominent clinical problems associated with adenylosuccinate lyase deficiency. In the severe form of the disease, neurologic symptoms and signs are typically evident soon after birth. Common neurologic presentations include acute encephalopathy, chronic encephalopathy, and behavioral abnormalities in various nonspecific combinations with seizures and developmental delay or regression. Diagnosis of adenylosuccinate lyase deficiency may be delayed or missed because patients can present with nonspecific features, such as developmental delay, autism spectrum disorder, or epilepsy. Adenylosuccinate lyase deficiency can be diagnosed by detection of elevated metabolites along the DNPS pathway (succinylpurines) in body fluids. No specific FDA-approved treatment is available for adenylosuccinate lyase deficiency.
|
• The clinical presentation of adenylosuccinate lyase deficiency varies greatly with respect to age of onset, clinical manifestations, and rate of disease progression. | |
|
• Patients with adenylosuccinate lyase deficiency can present with nonspecific symptoms, such as developmental delay, autism spectrum disorder, or epilepsy, including infantile spasms. | |
|
• Due to the lack of specific features and later onset of symptoms, diagnosis is difficult. | |
|
• Selective screening for adenylosuccinate lyase deficiency should be performed in infants who have neurologic disease without clear etiology, especially if supportive MRI findings are also present (eg, delayed or lack of myelination, abnormal white matter signal, and atrophy of the cerebrum or cerebellum). | |
|
• Detection of succinylpurines in body fluids by high-performance liquid chromatography or liquid chromatography-tandem mass spectrometry is the preferred biochemical test for adenylosuccinate lyase deficiency. | |
|
• No specific FDA-approved treatment is available for adenylosuccinate lyase deficiency. |
Origins of purine biochemistry. The word purine was coined by the German chemist Emil Fischer (1852-1919), who derived the German chemical term purin from the Latin purum uricum, for pure + uric acid; this was subsequently modified to an -ine ending in English, giving purine.
Fischer, working at the University of Würzburg, synthesized purine for the first time in 1898. Purine is a specific, water-soluble, aromatic organic compound that consists of two heterocyclic rings (ie, a 6-member pyrimidine ring and a 5-member imidazole ring) fused together. Given the name that Fischer chose, it is perhaps not surprising that he employed uric acid as the starting material for the reaction sequence that led to purine.
Uric acid had been known for a century, having been isolated from kidney stones in 1776 by Swedish-German pharmaceutical chemist Carl Wilhelm Scheele (1742-1786).
In 1891, 7 years before his synthesis of purine, Fischer, in conjunction with German chemist Oscar Piloty (1866-1915), had synthesized the pentose L-ribose.
Piloty started studying chemistry under Adolf von Baeyer (1835-1917) at the University of Munich in 1888 but transferred to the University of Würzburg in 1889 to work with Fischer on the chemistry of sugars. Piloty transferred after Baeyer awarded him failing marks on an exam, but the low exam score did not reflect Piloty's knowledge or abilities and was apparently instead Bayer's adverse reaction when he learned that Piloty had fallen in love with his daughter. Piloty earned his PhD under Fischer in 1890, helped Fischer synthesize L-ribose from L-arabonic acid in 1891, then married Baeyer's daughter in 1892, and that same year followed Fischer to the University of Berlin. A family reconciliation ultimately occurred, and Piloty and his wife returned to Munich in 1900 when his father-in-law offered Piloty a position at the University of Munich (even though Fischer had made him a better offer).
Fischer received the 1902 Nobel Prize in chemistry (only the second ever awarded to this point) "in recognition of the extraordinary services he has rendered by his work on sugar and purine syntheses" (22). Baeyer subsequently received the 1905 Nobel Prize in chemistry "in recognition of his services in the advancement of organic chemistry and the chemical industry, through his work on organic dyes and hydroaromatic compounds."
After Fischer's pioneering work, purine biochemistry advanced markedly over the first half of the 20th century, in part propelled by his students. In 1909, Lithuanian-American biochemist Phoebus Levene (1869-1940) and his assistant (later associate), American Walter Jacobs (1883-1967), recognized that D-ribose was a natural product, the enantiomer of Fischer and Piloty's L-ribose, and an essential component of nucleic acids.
Levene had spent a summer with Fischer at the University of Berlin in 1902, and Jacobs had only recently earned his PhD in chemistry under Fischer in Berlin in 1907. When Jacobs returned to New York following his doctoral work in Germany, he was appointed as a postdoctoral fellow in Levene's laboratory at the newly established Rockefeller Institute for Medical Research. Not only did Levene ultimately discover the carbohydrate components of both RNA and DNA (ie, ribose and deoxyribose, respectively), but he also correctly inferred that DNA is composed of a series of nucleotides and discovered the order of the three major components of a single nucleotide (ie, phosphate-sugar-base). Unfortunately, although widely accepted for decades, Levene had, in fact, stumbled with his overly simplistic tetranucleotide hypothesis (1909), which proposed that DNA is composed of equal parts adenine, guanine, cytosine, and thymine, resulting from a repeating linear sequence of the four nucleotides (ie, tetranucleotide repeats).
The genetic role of DNA was then unknown, and it was instead believed that Levene's proposed repeating structure excluded the possibility that DNA could store genetic information; instead, the protein component of chromosomes was then erroneously assumed to be the basis of heredity.
Several key steps beginning in the mid-1940s and extending to the early 1950s allowed a correct formulation of the structure of DNA: (1) demonstration that genes are composed of DNA, not protein; (2) discovery of "base pairing" through formulation of the Chargaff rules, which effectively disproved Levene's tetranucleotide hypothesis; (3) x-ray crystallography findings that indicated DNA had a helical structure. Work that began with Fischer ultimately culminated in the double helix model of deoxyribonucleic acid (DNA), by James Watson (b. 1928) and Francis Crick (1916-2004) in 1953.
Purine nucleosides and nucleotides. The purines as a class of molecules include the purine bases adenine (A, 6-amino-purine) and guanine (G, 2-amino-6-oxypurine), both of which can be synthesized in vivo from inosine monophosphate or can be obtained either from the diet or from breaking down and recycling existing compounds and derivatives.
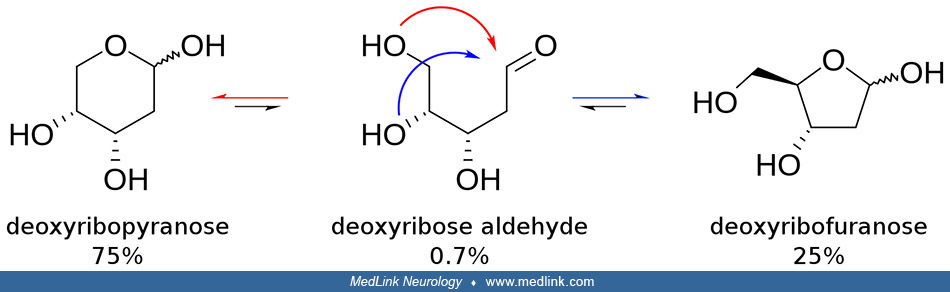
The purine bases may be attached to the pentose sugar ribose to form the ribonucleosides adenosine and guanosine, respectively. Ribose exists in equilibrium in multiple forms: an open-chain form and cyclic forms with 5- or 6-member rings.
Several of the forms of D-ribose: the open-chain form (top), and Haworth projections of two of the four cyclic forms, alpha-D-ribopyranose (middle), and beta-D-ribofuranose (bottom). Ribose exists as a mixture of cyclic forms i...
The purine bases may also be attached to the pentose sugar deoxyribose to form the deoxyribonucleosides 2'-deoxyadenosine and 2-deoxyguanosine, respectively. Ribose also exists in equilibrium in multiple forms: an open-chain form and cyclic forms with 5- or 6-member rings.
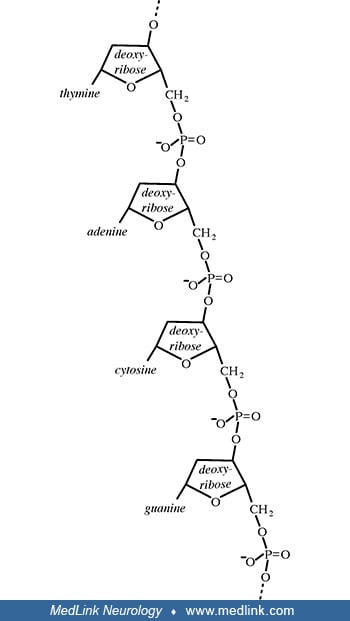
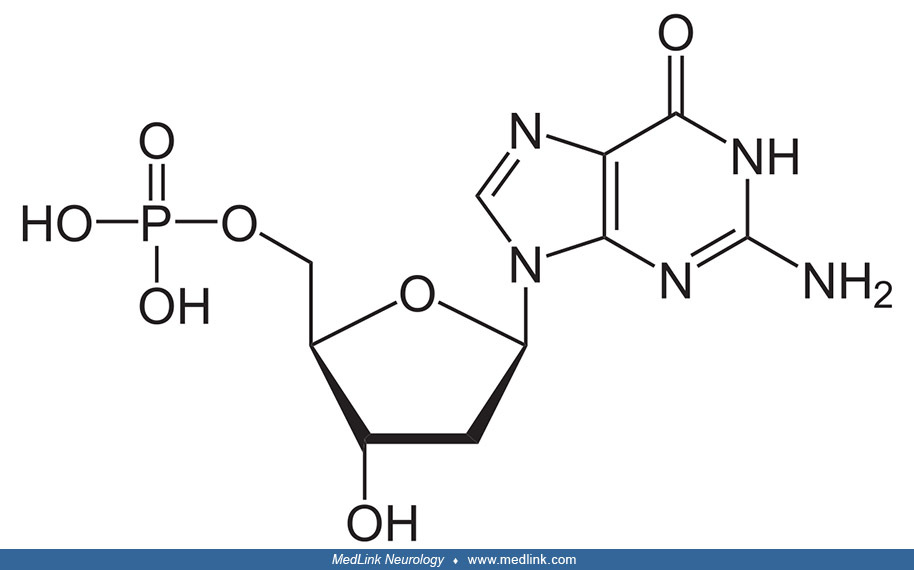
The addition of a phosphate group to the 5' carbon of ribose in the ribonucleosides adenosine and guanosine results in the formation of the ribonucleotides adenosine-5-monophosphate and guanosine-5-monophosphate, which are important metabolic and signaling molecules and building blocks of ribonucleic acid.
Addition of a phosphate group to the 5' carbon of deoxyribose in the deoxyribonucleosides 2-deoxyadenosine and 2'-deoxyadenoguanosine results in the formation of the deoxyribonucleotides deoxyadenosine monophosphate (dAMP; also known as deoxyadenylic acid or deoxyadenylate) and deoxyguanosine monophosphate (dGMP; also known as deoxyguanylic acid or deoxyguanylate), which are building blocks of deoxyribonucleic acid.
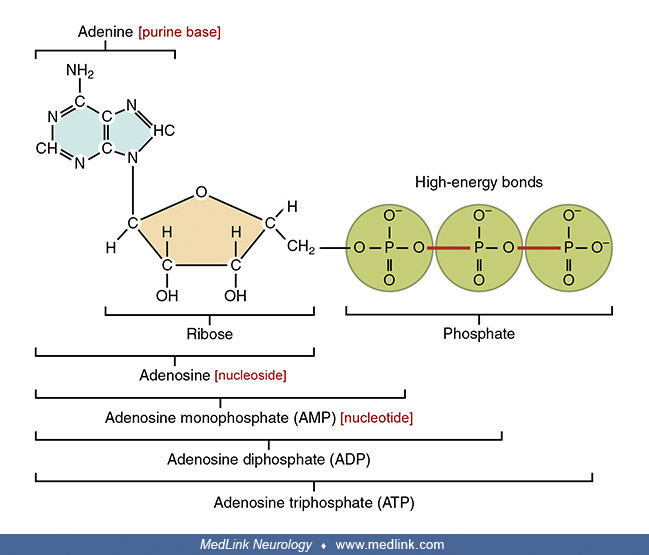
Examination of the structural components of adenosine triphosphate, the energy-carrying molecule that provides energy to drive many processes in living cells, will illustrate the components of nucleotides and nucleosides and how they are linked.
Adenylosuccinate lyase deficiency. Adenylosuccinate lyase deficiency, the first enzyme deficiency reported in the DNPS pathway in man, was discovered during a systematic study of amino acids in cerebrospinal fluid before and after acid hydrolysis (32). In three children with severe psychomotor retardation and autistic features, this procedure released abnormally large, equimolar amounts of aspartate and glycine. The additional identification by gas chromatography of an equimolar amount of ribose led to a search for purine compounds. Anion-exchange high-pressure liquid chromatography of deproteinized but not hydrolyzed cerebrospinal fluid, plasma, and urine revealed the presence of two UV-absorbing compounds that were undetectable in control samples: succinyl aminoimidazole carboxamide riboside (SAICA-riboside) and succinyl-adenosine (S-Ado). These succinylpurines are the products of the dephosphorylation by 5'-nucleotidases of succinyl aminoimidazole carboxamide ribotide (SAICAR) and succinyladenosine monophosphate (S-AMP, sometimes abbreviated SAMP), respectively.
|
• Although the clinical picture of adenylosuccinate lyase deficiency is extremely variable, descriptive classification systems have generally identified three phenotypes: a fatal neonatal form, a severe form (type I), and a milder form (type II). | |
|
• Neurologic symptoms are the most common and prominent clinical problems associated with adenylosuccinate lyase deficiency. | |
|
• In the severe form of the disease, neurologic symptoms and signs are typically evident soon after birth. | |
|
• Common neurologic presentations include acute encephalopathy, chronic encephalopathy, and behavioral abnormalities in various nonspecific combinations with seizures and developmental delay or regression. |
Although the clinical picture of adenylosuccinate lyase deficiency is extremely variable, descriptive classification systems have generally identified three phenotypes: a fatal neonatal form, a severe form (type I), and a milder form (type II) (39; 42). However, the symptoms and signs of adenylosuccinate lyase deficiency vary along a continuum, and there are no fixed parameters to define phenotypic categories (41). Most adenylosuccinate lyase-deficient children are born after uncomplicated pregnancies with normal birth and family history. Neurologic symptoms are the most common and prominent clinical problems associated with adenylosuccinate lyase deficiency. In the severe form of the disease, neurologic symptoms and signs are typically evident soon after birth (15). Common neurologic presentations include acute encephalopathy, chronic encephalopathy, and behavioral abnormalities in various nonspecific combinations with seizures and developmental delay or regression (59; 15). Epilepsy is present in around 80% of the patients, with polymorphic and often intractable seizures (15). Less common features can include prolonged episodes of multifocal sustained myoclonic tremor (04).
In a study of clinical and molecular data from 18 patients of 13 unrelated families, age at onset ranged from birth to the first 3 years (median age 0.63 years), and age at diagnosis varied from 2 months to 17 years (median age 6.4 years) (58). The presenting sign was variously psychomotor delay (44%), epilepsy (17%), psychomotor delay and epilepsy (17%), and apneas, hypotonia, or nystagmus in single cases. One patient (sibling of a previously diagnosed child) had a presymptomatic diagnosis. All patients were definitively diagnosed with adenylosuccinate lyase deficiency based on pathogenic variants or biochemical assessment. The diagnosis was made by exome sequencing in approximately one third of cases. One patient (6%) had the fatal neonatal form, seven (39%) had type I, and nine (50%) had type II. Eighteen different variants, distributed along the entire adenylosuccinate lyase coding sequence, were predicted to variably impair either proper homotetramerization or catalytic activity of the enzyme. All but two variants were missense mutations.
Fatal neonatal form. Patients with a neonatal form presented with fatal neonatal encephalopathy with a lack of spontaneous movement (“floppy infant”), respiratory failure, and intractable seizures, which resulted in early death within the first weeks of life (84; 61; 39; 42; 49; 74). Clinical and radiologic results include severe epileptic encephalopathy and leukodystrophy with a vacuolar appearance of the brain (74). Adenylosuccinate lyase deficiency may also have prenatal manifestations with impaired intrauterine growth, microcephaly, fetal hypokinesia (with all its consequences up to arthrogryposis and pulmonary hypoplasia), and loss of fetal heart rate variability (61).
Type I adenylosuccinate lyase deficiency (severe). Most patients reported so far have adenylosuccinate lyase deficiency type I (severe form) presenting within the first months of life with a purely neurologic clinical picture characterized by severe psychomotor retardation, early onset of seizures that may be intractable, and postnatal microcephaly (50; 73; 84; 44; 63; 39; 42; 04; 05; 20). Further clinical evolution is characterized by developmental arrest, epileptic seizures, hypotonia (or hypertonia), abnormal involuntary movements (including paroxysmal dyskinesias, dystonias, tremor, and limb ataxia), autism spectrum disorder, and in some patients, coma vigil (ie, apparent unconsciousness with eyes open) (34; 46; 81; 44; 39; 42; 26; 49; 38; 08; 04; 05). Severe cortical visual impairment and impaired eye fixation and tracking, corresponding to the degree of encephalopathy, may also be a feature of the severe phenotype (49). With the exception of microcephaly, there are usually no dysmorphic features, although two patients have been reported with brachycephaly associated with a long, flat philtrum and thin upper lip (29).
Type II adenylosuccinate lyase deficiency (mild or moderate severity). Patients with adenylosuccinate lyase deficiency type II (moderate or mild form) who develop symptoms within the first years of life usually suffer from slight to moderate psychomotor retardation (34; 85; 39; 42; 90). There is often mild neurologic involvement with infrequent autistic features (40). Seizures were present in about 30% of these patients and were never the first sign of the mild adenylosuccinate lyase deficiency (40). Seizures, if present, appear later than in type I disease, often between the second and fourth year of life (12; 36; 90), although they may begin as late as the ninth year (26). Speech and language impairment regarding receptive and nonverbal communication skills are common (26). Progression of ataxia may cause worsening gait. Delayed motor milestones were evident in all patients, ranging from mild to moderate. Long-term follow-up of these patients showed no obvious signs of disease progression (40).
The prognosis of adenylosuccinate lyase-deficient patients is variable. Those presenting with epilepsy in the neonatal period or early infancy may die within the first months of life. In patients with severe cognitive impairment, further evolution is characterized by stagnation or even regression of psychomotor development and persistence of autistic behavior. Patients with less severe cognitive impairment have reached adult age.
Case 1. The patient was the second daughter of unrelated Polish parents. Her older sister is healthy. Pregnancy and delivery were normal. Weight was 3550 grams, and length was 58 cm. Hypotonia was noted shortly after birth, and she had difficulty feeding. At age 7 days, she developed tonic-clonic seizures and was hospitalized. Over the following days, she had recurrent episodes of apnea and status epilepticus. She remained on artificial ventilation until her death at 2.5 months of age. Brain MRI revealed generalized atrophy of the cerebrum, an abnormally thin corpus callosum, and a generalized lack of myelination. Analysis of CSF revealed an accumulation of succinylpurines, with S-Ado/SAICA-riboside ratios of 0.92. Mutation analysis on the neonatal screening card of the patient revealed compound heterozygosity for the p.Y114H and p.T242I mutations. The fatal neonatal type of adenylosuccinate lyase deficiency was diagnosed.
Case 2. The female patient was the second child of unrelated, healthy Belgian parents. Their first child was healthy. Pregnancy and delivery were normal. Weight was 3750 grams, and length was 53 cm. At age 2 years 8 months, she was admitted for evaluation of psychomotor retardation, autistic features, and failure to thrive (weight 12.4 kg, length 93 cm, head circumference 48.1 cm). Developmental age (Gesell) was 9 months for gross motor skills but 5 to 6 months for other developmental domains. Autistic behavior was striking, with impaired eye contact, repetitive manipulation of toys, grimacing, and incessant crying without apparent reason. Funduscopic examination was normal. She ground her teeth (bruxism) and bit herself. There was moderate axial hypotonia with normal tendon reflexes. Routine biochemical analyses of blood and urine and the workup for metabolic disorders were unrevealing. Auditory, somatosensory, and visual-evoked responses were normal. Nerve conduction velocities and electromyography were normal. CT and MRI of the brain showed hypoplasia of the cerebellar vermis. Analysis of urine and CSF revealed an accumulation of succinylpurines, with S-Ado/SAICA-riboside ratios of 1.1 to 1.5. Adenylosuccinate lyase deficiency type I was the diagnosis.
Case 3. The female patient was the second child of unrelated Dutch parents. Neurologic impairment was first suspected during the neonatal period, and at age 2 years 11 months, she was admitted for investigation. Weight was 12 kg, but weight gain had been decelerating since the age of 2 years. Height was 93.5 cm, and head circumference was 50.5 cm. Motor development was at the level of 1.5 years, and speech development was at the level of 1 year. Eye contact was impaired, and reaction to auditory stimuli was poor, but auditory evoked potentials were normal. At the age of 3 years, EEG showed diffuse slowing without signs of epilepsy. CT and MRI of the brain showed slight cerebral hypotrophy. On re-evaluation at age 4 years, psychomotor development was at the level of 2.5 years, and EEG was "normal." Analysis of urine and CSF revealed an accumulation of succinylpurines, with S-Ado/SAICA-riboside ratios of 3.7 to 4.7. Adenylosuccinate lyase deficiency type II was the diagnosis.
|
• Adenylosuccinate lyase catalyzes two steps in the synthesis of purine nucleotides: (1) the conversion of succinyl aminoimidazole carboxamide ribotide into aminoimidazole-carboxamide ribotide, in the de novo purine synthesis pathway; and (2) the formation of adenosine monophosphate from succinyladenosine monophosphate, the second step in the conversion of inosine monophosphate to adenosine monophosphate. | |
|
• Adenylosuccinate lyase deficiency associated with mitochondrial dysfunction, including increased fragmentation, impaired respiration, and reduced ATP production. | |
|
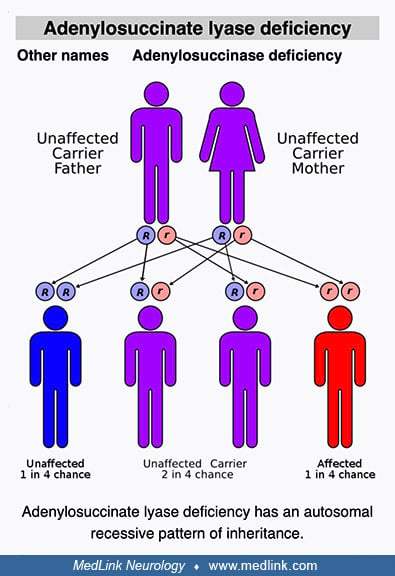
• Adenylosuccinate lyase deficiency is inherited as an autosomal recessive trait. | |
|
• Most identified mutations are missense mutations, and most patients are compound heterozygotes. | |
|
• Adenylosuccinate lyase deficiency disrupts the purinosome, a multienzyme complex of de novo purine synthesis enzymes (including adenylosuccinate lyase) that cells transiently assemble in their cytosol on depletion of, or increased demand for, purines. | |
|
• The signs and symptoms of adenylosuccinate lyase deficiency may result from neurotoxic effects of accumulating succinylpurines. |
Biochemistry. Adenylosuccinate lyase (also termed adenylosuccinase) catalyzes two steps in the synthesis of purine nucleotides: (1) the conversion of succinyl aminoimidazole carboxamide ribotide (SAICAR) into aminoimidazole-carboxamide ribotide (AICAR), in the DNPS pathway; and (2) the formation of adenosine monophosphate (AMP) from succinyladenosine monophosphate (S-AMP), the second step in the conversion of inosine monophosphate to adenosine monophosphate. Both reactions release fumarate. The term "ribotide" is a concatenation of ribonucleotide, as "ribotide" is a concatenation of ribonucleoside.
Adenylosuccinate lyase deficiency is a defect of purine metabolism that destabilizes purinosome assembly (vide infra) and reduces metabolite fluxes through both DNPS and purine nucleotide recycling pathways (07; 16).
In profoundly intellectually disabled patients with adenylosuccinate lyase deficiency, CSF concentrations of both succinylpurines are 100 to 200 µmol/l, and S-Ado/SAICA-riboside ratios are between 1 and 2. In adenylosuccinate lyase-deficient patients with milder clinical pictures, CSF concentrations of SAICA-riboside are in the same range, but those of S-Ado tend to be higher. This results in S-Ado/SAICA-riboside ratios above 2, and possibly as high as 4 to 5 (34). Urinary succinylpurine concentrations can reach 5 µmol per mg creatinine, with similar ratios to those measured from CSF. The magnitudes of S-Ado/SAICA-riboside ratios in body fluids do not predict phenotype severity, nor do they indicate inherent properties of the specific enzyme mutations, but instead reflect patient age, development, and other factors (94; 66; 67).
Individual intermediates of DNPS have potent regulatory and cytotoxic properties and, therefore, are (under physiologic conditions) either undetectable or present in very low (micromolar) concentrations in cellular extracts and body fluids (45). The efficiency of DNPS at such low concentrations of individual intermediates is ensured by the dynamic assembly and disassembly of the cytosolic multienzyme complex, the “purinosome” (03; 62). Impairment of the conversion of the two substrates of the enzyme, SAICAr and S-AMP, leads to the formation of their dephosphorylated derivatives, SAICA-ribotide and S-Ado, respectively. These metabolites accumulate in cerebrospinal fluid and urine and, to a minor extent, in plasma.
Adenylosuccinate lyase deficiency is associated with mitochondrial dysfunction, including increased fragmentation, impaired respiration, and reduced ATP production (09). The amount of mitochondrial impairment correlates with the severity of pathology, particularly in mitochondria-dependent tissues. Defects in mitochondrial dynamics and transport are linked to ERK2 and AKT suppression, whereas overexpressing constitutively active ERK2 or supplementing purine intermediates partially rescues the mitochondrial phenotype. ERK and AKT are protein kinases that act as signaling pathways regulating cell growth, proliferation, and survival/apoptosis, with their deregulation often linked to diseases like cancer. The ERK pathway (also known as the MAPK pathway) and the AKT pathway (part of the PI3K/AKT pathway) are distinct but often converge on the same downstream targets, such as mTORC1 and TSC2, to promote cellular processes like migration and invasion.
The purine-synthesis pathway is tightly regulated both enzymatically and transcriptionally (09). ERK and AKT are not only essential for controlling cell proliferation, differentiation, and apoptosis, these kinases regulate purine biosynthesis by phosphorylating phosphoribosylformylglycinamidine synthase (PFAS) and transketolase (TKT), respectively, which are two enzymes that are upstream from adenylosuccinate lyase (09). ERK2 activation, through PFAS, enhances de novo purine synthesis pathways, which augments the availability of essential intermediates such as AMP and GMP (09). ERK2 and AKT also regulate mitochondrial energy production, by influencing glucose uptake, respiratory efficiency, and ATP production (09).
Genetics. Adenylosuccinate lyase deficiency is inherited as an autosomal recessive trait.
In humans, the adenylosuccinate lyase (ADSL) gene spans approximately 23 kb on chromosome 22 (22q13.1q13.2) (25). It consists of 13 exons, and its promoter has the typical features of housekeeping genes. The ADSL gene is transcribed in most tissues into two transcript variants produced by alternative splicing of exon 12. The full-length variant encodes an active ADSL protein composed of 484 amino acids. The alternatively spliced variant encodes a catalytically inactive variant protein missing 59 amino acids (residues 397-456) (43).
Over 50 ADSL mutations have been identified in individuals with adenylosuccinate lyase deficiency. Most identified mutations are missense mutations, and most patients are compound heterozygotes (80). The most commonly identified mutation, c.1277G>A (p.Arg426His), accounts for about one third of patient alleles (15); however, there is significant phenotypic variability even among 14 reported patients homozygous for this variant (19). Many patients have private variants unique to their families. Other types of pathogenic mutations have also been identified, including splice-site mutations (95; 51) and a promoter mutation, which was found in three unrelated patients (56).
Most characterized mutations lead to structural instability of the enzyme without modifications of its kinetic properties. The severity of the clinical symptoms correlates with residual activity of the enzyme. A R303C mutation, found in two independent, mildly cognitively impaired type II patients, is thermostable and displays markedly less activity with S-AMP than with SAICAR. This explains the markedly higher S-Ado/SAICA-ribotide ratios in type II patients (83; 65).
Purinosome. The purinosome is a multienzyme complex of the DNPS enzymes (including adenylosuccinate lyase) that cells transiently assemble in their cytosol on depletion of, or increased demand for, purines (03).
Studies of purinosome formation in skin fibroblasts of patients with adenylosuccinate lyase deficiency showed considerable interindividual variation in purinosome assembly, with the ability to form purinosomes inversely associated with phenotype severity (07). Studies utilizing CRISPR-Cas9 editing of HeLa cells confirmed that purinosome formation is disrupted in adenylosuccinate lyase deficiency (06). The phenotypic severity of adenylosuccinate lyase deficiency and formation and stability of the purinosome are mainly determined by the structural stability and residual catalytic capacity of the corresponding mutant adenylosuccinate lyase protein complex (94; 88).
Pathophysiologic mechanisms. Hypotheses regarding the pathogenesis of adenylosuccinate lyase deficiency include toxicity of high levels of SAICAR, S-AMP, or their metabolites, deficiency of purine synthesis through the DNPS pathway, or disruption of the purine cycle in muscle and brain (41).
Possible toxic effects of intermediates. The signs and symptoms of adenylosuccinate lyase deficiency may result from neurotoxic effects of accumulating succinylpurines. Less severe intellectual impairment in patients with similar SAICA-riboside levels, but S-Ado/SAICA-riboside ratios above 2, might suggest that SAICA-riboside is the offending neurotoxic compound and that S-Ado could protect against the toxic effects of SAICA-riboside. However, when the activity of the enzyme against both substrates was measured separately (noncompetitively), patients exhibited a proportional decrease in enzyme activity against both substrates (43). When both substrates were present and competing against each other for the same residual enzyme activity, S-Ado/SAICA-riboside ratios varied as a function of the specific mutations (67). Alternatively, the observed abnormal ratios might be explained by differential transport of these succinylpurines across membranes (43); for example, it is known that SAICA-riboside is transported by ABCC5 (35).
Attempts to demonstrate neurotoxicity of the succinylpurines have failed, with very limited exceptions: (1) infusion of SAICAr (SAICA-riboside) to rats induced neuronal damage in specific regions of the hippocampus, consistent with the hypothesis that SAICAr is neurotoxic (78); (2) accumulation of SAICAr in a C. elegans model of adenylosuccinate lyase deficiency altered cholinergic neurotransmission (23). Although S-Ado and SAICAr are analogs of adenosine and glutamate, they neither modify the CNS effects of adenosine nor disrupt glutamatergic neurotransmission (79).
Curiously, HeLa cells with adenylosuccinate lyase deficiency (induced via CRISPR-Cas9) accumulate AICAR (06), which is quite puzzling, given that AICAR is a metabolite distal to the enzymatic block. AICAR might be provided via an alternative histidine biosynthesis pathway, which is known to crosstalk with the DNPS pathway in yeast (68); however, no pathway for histidine biosynthesis is known in humans.
Deficiency of purine nucleotides. Deficient DNPS caused by adenylosuccinate lyase deficiency has been thought to have detrimental effects during embryonic development. However, normal levels of purines were measured in various tissues of affected patients (86), presumably because of residual (partial) activity of adenylosuccinate lyase (32; 34) and additional supply of purines through the purine salvage pathway, involving the enzymes hypoxanthine-guanine phosphoribosyltransferase, adenine phosphoribosyltransferase, and adenosine kinase. Curiously, a worm model of disease utilizing a double knockout of adenylosuccinate lyase and the enzyme just proximal to it in the DNPS pathway leads to a masking of the phenotype (14); consequently, these authors have advocated blockage of this proximal enzyme as a potential therapeutic approach. Nevertheless, a deficiency of purine nucleotides could potentially occur in some cell types with profound deficiency of adenylosuccinate lyase and low activity of the salvage pathway. Similarly, the expression of a trifunctional protein participating in DNPS is known to be 20 times higher in human prenatal than in postnatal cerebellum (10), and the degree of DNPS is also known to decrease dramatically after birth in rat brains (01; 02). Little is known about the actual purine levels in the living brain or about the regulation of purine synthesis during embryogenesis, both of which might be affected by defects in purine metabolism (41).
Impairment of energy metabolism. Adenylosuccinate lyase participates in the purine nucleotide cycle along with adenosine monophosphate deaminase and adenylosuccinate synthetase, and disruption of this cycle in adenylosuccinate lyase deficiency has been suggested to cause the disorder. The purine nucleotide cycle controls the levels of fumarate, a Krebs cycle intermediate, and of adenosine monophosphate, needed particularly for maintaining the ATP/AMP ratio in muscles (85).
Animal models. Cellular and nematode models of adenylosuccinate lyase deficiency could lead to an improved understanding of the disease and subsequent development of effective treatment (88; 14; 23; 60).
Homozygous knockout of the mouse adenylosuccinate lyase ortholog is lethal before weaning (17).
A Caenorhabditis elegans model of adenylosuccinate lyase deficiency has been developed (23; 60). The lack of fertility in a roundworm model of the disease resulted from reduced DNPS, even though there was no significant decrease in purine metabolite concentrations (23). In this model, adenylosuccinate lyase function contributes to regulation of spontaneous locomotion, and aspects of adenylosuccinate lyase-related dysfunction are reversible (23). Differing molecular mechanisms are responsible for the different phenotypes: the neuromuscular defect correlates with an accumulation of a purine biosynthetic intermediate, whereas reproductive deficiencies can be ameliorated by purine supplementation (23). Learning is also affected as demonstrated with a gustatory plasticity assay (60). The worms maintain a capacity for gustatory plasticity demonstrated by a behavioral change in response to cue pairing, although their behavior is distinct from that of control animals. Substrate accumulation is associated with an unexpected perturbation in tyrosine metabolism: a lack of tyramine mediates the behavioral changes through action on the metabotropic TYRA-2 tyramine receptor.
Multiple phenotypes associated with adenylosuccinate lyase depletion in developing chicken and zebrafish embryos can largely be rescued by interventions that suppress the generation of metabolites in the DNPS pathway or restore purine levels, thereby complementing defective DNPS or purine nucleotide pathway activity; thus, both reduced purine levels and impaired DNPS contribute to neurodevelopmental pathology in adenylosuccinate lyase deficiency (20).
|
• The prevalence of adenylosuccinate lyase deficiency is unknown. | |
|
• More than 80 patients with adenylosuccinate lyase deficiency have been reported in populations across the world. |
The prevalence of adenylosuccinate lyase deficiency is unknown. Known disease-causing mutations and protein-truncating variants are present in the ExAC population database at a 1 in 557 carrier frequency, giving a conservative estimate of 1 in 1.25 million disease frequency (24). The true prevalence is likely higher because many patients have missense mutations unique to the family; in addition, there are likely many such mutations that have not yet been ascertained in undiagnosed patients.
More than 80 patients with adenylosuccinate lyase deficiency have been reported in populations across the world. Most of them have been identified in Belgium, the Netherlands, Czech Republic, and Poland (32; 44; 43; 61; 39; 42). Other patients have been identified in Australia (76; 87), China (53), Colombia (12), France (29), Germany (61), Italy (63; 65), Malaysia (13), Morocco (32; 77; 26), Norway (56), Portugal (21), Spain (12; 64), Turkey (43; 75), the United Kingdom (57; 13; 49), and the United States (43; 75).
In a study of 88 cases (seven previously unreported cases and 81 cases from the literature), 14% had neonatal, 58% Type I, and 28% Type II adenylosuccinate lyase deficiency (15).
Adenylosuccinate lyase deficiency should be distinguished from (1) other neurologic disorders with intractable seizures and encephalopathy and (2) other inborn errors of purine and pyrimidine metabolism with neurologic manifestations.
Other disorders presenting predominantly with intractable seizures include the following:
|
• Angelman syndrome (OMIM 105830) | ||
|
- Isolated sulfite oxidase deficiency (SUOX, OMIM 272300) | ||
|
• Menkes disease (ATP7A, OMIM 309400) | ||
Other inborn errors of purine and pyrimidine metabolism with neurologic manifestations include the following:
|
• AICA-ribosuria due to ATIC deficiency (ATIC, OMIM 608688) | ||
|
- Lesch-Nyhan disease (OMIM 300322) | ||
|
• Molybdenum cofactor deficiency (MOCS1, OMIM 252150; MOCS2, OMIM 603708; GPHN, OMIM 615501) | ||
The wide clinical spectrum of the disease accounts for possible difficulties in differential diagnosis with neurologic disorders, especially those with intractable seizures and encephalopathy.
In the neonatal period, the differential diagnosis should include particularly the five treatable disorders that can present with intractable seizures in neonates: (1) pyridoxine-dependent epilepsy, (2) pyridox(am)ine-5'-phosphate oxidase deficiency, (3) folinic acid-responsive seizures, (4) 3-phosphoglycerate dehydrogenase deficiency, and (5) hyperinsulinemic hypoglycemia (72). Biotin-responsive holocarboxylase synthetase deficiency can also rarely present with neonatal seizures. In the first months of life, biotin-responsive biotinidase deficiency and GLUT1 deficiency (which is treated with the ketogenic diet) can also present with intractable seizures. Other inborn errors of metabolism less amenable to treatment can present in the neonatal period with severe epilepsy, including nonketotic hyperglycinemia, D-2-hydroxyglutaric aciduria, mitochondrial glutamate transporter defect, peroxisomal biogenesis defects, respiratory chain disorders, molybdenum cofactor and isolated sulfite oxidase deficiency, and Menkes disease. In infancy, the association of seizures and autistic features should prompt consideration of Angelman syndrome and Rett syndrome.
SAICAr and S-Ado elevations also occur in two other conditions: AICA-ribosuria (AICAR transformylase/inosine monophosphate cyclohydrolase deficiency) and, to a lesser degree, fumarase deficiency. One patient has been reported with AICA-ribosuria--a female infant with dysmorphic features, severe neurologic defects, and congenital blindness. She had significant elevations of SAICAr and S-Ado detectable in urine and CSF. In contrast to adenylosuccinate lyase deficiency, these findings were associated with massive excretion of AICA-riboside (AICAr), the dephosphorylated counterpart of AICAR, one of the products of adenylosuccinate lyase (55). Further studies led to the identification of a deficiency of the bifunctional enzyme that catalyzes the final steps of purine biosynthesis AICAR transformylase/inosine monophosphate cyclohydrolase (ATIC).
Fumarase deficiency has been associated with less pronounced elevations of succinylpurines in one patient who had dysmorphic features, brain malformations, severe developmental delay with progressive hypotonia, hepatosplenomegaly, and neutropenia (92). S-Ado (8.4 umol/l) and SAICAr (7.6 umol/l) were detected in CSF but not in urine, likely due to dilution below the detection limit of the test. These succinylpurine levels were clearly abnormal, with S-Ado six times the upper limit of the reference range but lower than the 100 to 200 umol/l concentrations typical for adenylosuccinate lyase deficiency. Succinylpurine elevations in fumarase deficiency are thought to reflect inhibition of adenylosuccinate lyase by excessive fumarate because fumarate is the first product released in both of the adenylosuccinate lyase-catalyzed reactions and acts as a weak inhibitor of this enzyme in vitro. The authors suggested that an accumulation of succinylpurines in the CNS might contribute to the pathogenesis of fumarase deficiency (92).
|
• Head-imaging abnormalities include atrophy of the cerebral cortex, corpus callosum, and cerebellar vermis; delayed or lack of myelination; anomalies of the white matter; and lissencephaly. | |
|
• The combination of hypomyelination with psychomotor retardation should raise suspicion of this disorder in children with consistent clinical features. | |
|
• Biochemical diagnosis requires demonstration of succinyl aminoimidazole carboxamide riboside (SAICAr) and succinyladenosine (S-Ado) in extracellular fluids, such as plasma, cerebrospinal fluid, or urine, using HPLC with UV detection or HPLC-MS. | |
|
• Genetic diagnosis requires mutation analysis, specifically the genomic or cDNA sequencing of adenylosuccinate lyase gene and characterization of mutant proteins. | |
|
• Diagnosis is supported by (1) evidence of an appropriate clinical phenotype; (2) demonstration of SAICAr and S-Ado in extracellular fluids, such as urine, CSF, or plasma; and (3) confirmation at an accredited laboratory of large decrease of adenylosuccinate lyase enzyme activity in cultured skin fibroblasts. |
Neuroimaging. Head-imaging abnormalities include atrophy of the cerebral cortex (with frontal predominance), corpus callosum, and cerebellar vermis; delayed or lack of myelination; anomalies of the white matter; and lissencephaly (81; 44; 63; 76; 21; 57; 49; 37; 15). The combination of hypomyelination with psychomotor retardation should raise suspicion of this disorder in children with consistent clinical features. Diffusion restriction may be present in bilateral basal ganglia, thalamus, and periventricular white matter (05).
Magnetic resonance spectroscopy of white matter abnormalities can suggest adenylosuccinate lyase deficiency if specific metabolites are detected but has not been tested sufficiently to rule out the diagnosis. In patients with white matter abnormalities on MRI, proton magnetic resonance spectroscopy may help suggest the diagnosis as the abnormal-appearing areas may contain abnormal peaks corresponding to S-Ado at 8.3 ppm and SAICAR at 7.5 ppm (28; 95). However, these metabolite peaks typically fall outside of the usual chemical shift range evaluated during clinical neuroimaging procedures (0 to 4 ppm).
Electroencephalography. Common patterns of electroencephalographic abnormalities and developmental trajectories include the following: (1) poor general background organization with theta-delta activity; (2) hypsarrhythmia with infantile spasms (usually adrenocorticotropic hormone-responsive); (3) generalized epileptic discharges with frontal or frontotemporal predominance; and (4) epileptic discharge activation in sleep with an altered sleep structure (15).
Biochemical and genetic diagnosis. Biochemical and genetic diagnosis requires the following (41):
|
• Demonstration of succinyl aminoimidazole carboxamide riboside (SAICAr) and succinyladenosine (S-Ado) in extracellular fluids, such as plasma, cerebrospinal fluid, or urine, using HPLC with UV detection or HPLC-MS | |
|
• Mutation analysis--genomic or cDNA sequencing of adenylosuccinate lyase gene and characterization of mutant proteins |
Diagnosis is supported by the following (41):
|
• Evidence of an appropriate clinical phenotype | |
|
• Demonstration of SAICAr and S-Ado in extracellular fluids, such as urine, CSF, or plasma, using simple screening tests, such as Bratton-Marshall or thin-layer chromatography | |
|
• Analysis of adenylosuccinate lyase enzyme activity in cultured skin fibroblasts at an accredited laboratory to demonstrate a large decrease of adenylosuccinate lyase activity. Although adenylosuccinate lyase enzyme activity levels may differ between testing laboratories, enzyme activity in patients with adenylosuccinate lyase is generally between 2% and 20% of the lower limit of normal (69). Enzyme assay in lysates is not completely reliable due to tissue heterogeneity of the adenylosuccinate lyase defect (61). |
Biochemical diagnostic methods. The deficiency of adenylosuccinate lyase activity results in the accumulation of SAICAR and S-AMP in the cells and the presence of enormously elevated concentrations of their dephosphorylated forms, SAICA-riboside (SAICAr) and S-Ado in extracellular fluids--urine and CSF and, to a lesser extent, also in plasma (41). SAICAr and S-Ado are present in low micromolar concentrations in bodily fluids of controls (73; 45). In patients with adenylosuccinate lyase deficiency, concentrations of both metabolites range from 5 to 10 µmol/L in plasma, 100 to 200 µmol/L in CSF, and in the millimolar range in urine (32). Several methods have been described for selective screening of subjects with adenylosuccinate lyase deficiency that allow identification of either one or two of the relevant compounds in body fluids.
The preferred diagnostic methods are HPLC-DAD or LC-MS/MS because these allow detection of both SAICAr and S-Ado and their deribosylation products (47; 41). In particular, high-throughput urine screening techniques for adenylosuccinate lyase deficiency have been developed using HPLC combined with electrospray ionization tandem mass spectrometry (MS/MS) (31; 27; 87). These methods allow rapid and specific screening for disorders of purine and pyrimidine metabolism with use of liquid urine samples or urine-soaked filter-paper strips. A similar LC-MS/MS-based assay for dried blood spots has also been described (93).
Other methods used include a modified Bratton-Marshall test (48); thin-layer chromatography for identification of SAICAr (89), S-Ado (52) and both nucleosides (33); isolation of SAICAr and S-Ado with a cation exchange resin and determination of A270/A250 ratio (UV absorbance of the ammonia eluate at 270 and 250 nm) (18); capillary electrophoresis (30); and high-resolution proton magnetic resonance spectroscopy (28). However, although the Bratton-Marshall test and thin-layer chromatography with Pauly reagent detect excessive urinary SAICAr, false-negative results have been reported due to bacteria-mediated deribosylation of SAICAr and S-Ado in urine (47). In addition, because the Bratton-Marshall test detects free primary aromatic amines, false-positive results may be observed in patients who receive antibiotics, such as sulfonamides (for which the test was initially devised), anticonvulsants (including clonazepam, nitrazepam, and lamotrigine), or other medications that contain or have metabolites containing this group.
Enzymatic diagnosis of adenylosuccinate lyase deficiency is possible in liver biopsy specimens, preferably fresh tissue, because the enzyme is unstable to freezing and thawing (82). Cultured skin fibroblasts are less appropriate for diagnosis because adenylosuccinate lyase activity is variable in this tissue and only partially deficient in affected patients.
Prenatal diagnosis is limited to families having a previous child with adenylosuccinate lyase deficiency and is based on mutational analysis for at-risk fetuses. Diagnostic testing may be conducted for prenatal diagnosis on viable fetal cells from chorionic villi, cultured amniotic fluid cells, or in the newborn dried blood spots (54).
Selective screening for adenylosuccinate lyase deficiency. The wide clinical spectrum of adenylosuccinate lyase deficiency complicates the differential diagnosis with other neurologic disorders, especially those with intractable seizures and encephalopathy (11; 41). Therefore, when adenylosuccinate lyase deficiency is possible or suspected, diagnostic protocols are needed to avoid useless investigations and treatments. The marked clinical heterogeneity justifies systemic screening for the disorder in patients with the following symptoms (41):
|
• Newborns and infants with hypotonia and acquired microcephaly | |
|
• Unexplained psychomotor retardation | |
|
• Unexplained developmental delay | |
|
• Unexplained seizures, especially intractable | |
|
• MRI findings such as atrophy of the cerebral cortex, corpus callosum, cerebellar vermis, lack of myelination, delayed myelination, anomalies of the white matter |
|
• No specific FDA-approved treatment is available for adenylosuccinate lyase deficiency. | |
|
• Several supplements have been used empirically in a small number of patients, all aimed at replenishing hypothetically deficient adenine nucleotides in adenylosuccinate lyase-deficient tissues, but none of them has shown any clear benefit. | |
|
• Seizure management in adenylosuccinate lyase deficiency utilizes standard anticonvulsants. | |
|
• Use of the ketogenic diet may be helpful for patients with intractable epilepsy. |
No specific FDA-approved treatment is available for adenylosuccinate lyase deficiency.
Several supplements have been used empirically in a small number of patients, all aimed at replenishing hypothetically deficient adenine nucleotides in adenylosuccinate lyase-deficient tissues, but none of them has shown any clear benefit (34; 71; 70; 39; 42; 64; 87; 53). These include trials of 3 months to as many as 20 months with oral supplements of adenine and allopurinol (the latter to avoid conversion by xanthine oxidase of adenine into poorly soluble 2,8-dihydroxyadenine) (34), D-ribose and uridine (71; 70), D-ribose alone (39; 42; 64; 53), and S-adenosylmethionine, (which can enter the CNS) (87). Urine succinylpurines were also unchanged, suggesting that treatment did not lead to feedback inhibition of DNPS. It is unclear if the negative results of purine supplementation are due to unaddressed neurotoxicity of accumulating metabolites.
Seizure management in adenylosuccinate lyase deficiency utilizes standard anticonvulsants. Myoclonic seizures have been treated with valproic acid in combination with other drugs, such as clobazam and levetiracetam (51; 53). Infantile spasms may be resistant to treatment (49; 95). However, patients have had cessation of spasms with therapies including high-dose prednisolone (91) and vigabatrin, which normalized EEG in one patient (87; 59), although they subsequently developed other types of seizures.
Use of the ketogenic diet for intractable epilepsy has been reported in five patients with adenylosuccinate lyase deficiency, four of whom had some improvement in seizure control. One patient with neonatal-onset epilepsy was seizure-free on the ketogenic diet from ages 2 to 5 years but developed a metabolic hyperchloremic acidosis with Fanconi syndrome, which resolved a month after cessation of the diet (38). A patient with infantile spasms did not respond to the ketogenic diet (95). Another patient with generalized tonic and clonic seizures had a 95% reduction in seizure frequency when the ketogenic diet was started at 7 years of age, with 2 years of follow-up (40). Finally, a pair of monozygotic twins with neonatal onset of seizures started on the ketogenic diet at 3 months of age and had 14 months of seizure freedom, even with no other anticonvulsants, although seizures subsequently recurred (53). One patient also had improvement of nonepileptic paroxysmal dyskinesias and dystonias on the ketogenic diet (08).
Surgical biopsy of liver and kidney obtained under general anesthesia has reportedly been well tolerated.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Douglas J Lanska MD MS MSPH
Dr. Lanska of the University of Wisconsin School of Medicine and Public Health has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurogenetic Disorders
Jun. 01, 2026
Neurogenetic Disorders
May. 08, 2026
Neurogenetic Disorders
Apr. 30, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026