
Sleep Disorders
Sudden infant death syndrome
Jul. 05, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
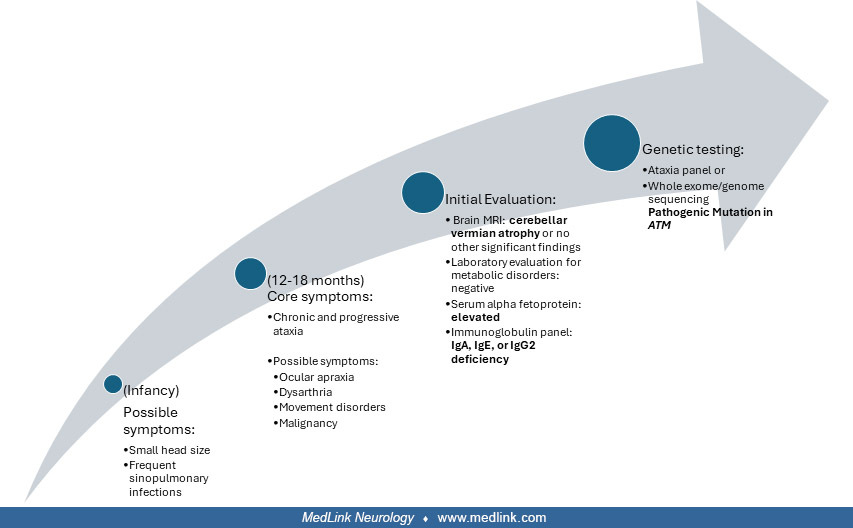
Ataxia-telangiectasia is an autosomal recessive condition of genomic instability that clinically presents in childhood as progressive ataxia and incoordination, dilated blood vessels of skin and conjunctiva, frequent sinus and pulmonary infections due to immune deficiencies, and an increased risk of certain malignancies. Elevated serum alpha-fetoprotein is seen in over 90% of patients; molecular testing of the ataxia telangiectasia mutated (ATM) gene is clinically available. Although a cure is not yet available, ongoing research is focused on developing different treatment approaches.
|
• Ataxia-telangiectasia is an autosomal recessive condition characterized by progressive childhood ataxia, oculomotor apraxia, some degree of immunodeficiency, an increased risk for certain malignancies, and telangiectasias of the skin and conjunctiva. | |
|
• Elevated serum alpha-fetoprotein is an important biochemical marker because the hallmark telangiectasias do not appear until later in the clinical course. | |
|
• The principal defect is in the ataxia-telangiectasia mutated (ATM) gene, whose end product is a protein kinase involved in cell cycle regulation. | |
|
• Patients are at an increased risk for certain malignancies, primarily leukemias and lymphomas, and are hypersensitive to ionizing radiation. | |
|
• Patients with variant ataxia-telangiectasia may present initially with extrapyramidal movement disorders but are also at an increased risk for malignancy and hypersensitivity to ionizing radiation. |
In 1926, Syllaba and Henner first described the association of oculocutaneous telangiectasias with choreoathetosis. Louis-Bar reported the combination of oculocutaneous telangiectasias with progressive cerebellar ataxia in 1941. In 1958, Boder and Sedgwick studied eight cases from five unrelated families, including six familial cases. They emphasized the heredofamilial nature of the disease and the susceptibility of these patients to sinopulmonary infections. They named this syndrome ataxia-telangiectasia (06). Ataxia-telangiectasia is now recognized as one of a group of recessive hereditary genomic instability disorders and is characterized by progressive neurodegeneration, immunodeficiency, and cancer susceptibility.
|
• Ataxia is the core neurologic feature; dysarthria, oculomotor apraxia, and extrapyramidal signs are other frequently occurring neurologic manifestations. | |
|
• The hallmark cutaneous manifestations are telangiectasias, often present by mid-childhood. | |
|
• Recurrent sinopulmonary infections are often present also starting in childhood. | |
|
• The tendency to develop lymphoreticular malignancies is a prominent cause of morbidity and mortality, with patients exhibiting a sensitivity to ionizing radiation. | |
|
• In adult-onset movement disorders, the absence of telangiectasia does not exclude ataxia-telangiectasia; serum alpha-fetoprotein remains the most cost-effective initial screen. |
Ataxia-telangiectasia is characterized by progressive cerebellar ataxia, telangiectasias of the skin and bulbar conjunctiva, progressive apraxia of eye movements, choreoathetosis, and increased susceptibility to sinopulmonary infections, lymphoreticular malignancies, and other malignant tumors. Median survival is estimated to be 25 years (16).
Neurologic features. Ataxia is the earliest clinical manifestation of ataxia-telangiectasia. The onset is insidious and is not apparent at birth or early infancy, as early motor development is usually normal. Ataxia is initially truncal and may manifest earlier than 2 years of age as postural instability with motor impersistence (32). The gait is unusual: clumsy but with a narrow base initially, and a tendency to improve with speed. Truncal ataxia is present in the majority (43 of 68 patients) by 3 years of age (79). With the rapid development of choreoathetosis, ambulation is lost by adolescence. The advancement of neurologic disease, however, may progress in a nonlinear fashion in the individual patient (27).
A small head circumference (61%) and microcephaly (17%), both acquired after the second year of life, have been recognized as a major clinical feature (44). Facial features and speech are often characteristic: a mask-like facies with excessive drooling and dysarthria. Dysarthria is common and can manifest with uncontrolled, ataxic, or involuntary movements, resulting in monotonous, unstable, slow, or hypernasal speech (74). Abnormalities of articulation and bulbar function develop early. Progressive disorders of chewing and swallowing as well as silent aspiration become increasingly important and, often, are unrecognized.
Ocular abnormalities are an early and progressive feature of ataxia-telangiectasia and include oculomotor apraxia, deficient accommodation, impaired smooth pursuit, hypometric saccades, nystagmus, absence of optokinetic nystagmus, frequent blinking, and photophobia (53). These features, especially eye movements, are important diagnostically (02). The combination of oculomotor apraxia and failure to suppress the vestibulo-ocular reflex (which is often the earliest ocular finding) is characteristic. Oculomotor apraxia involves difficulty in fixating smoothly on an object. In an attempt to visually fix on an object, the head is turned rapidly toward and past the object, often with a head thrust, causing the eyes to reach the target by means of the vestibulo-ocular reflex. Next, while maintaining fixation, the head is moved back to the target. Blinking before gaze changing can be identified before the age of 2 years (32). The head-turning technique, by engaging the vestibulo-ocular reflex, allows the patient to compensate for the difficulty in initiating voluntary refixation eye movements (saccades). This unique head-turning technique for visual fixation may not be seen in all subsets of patients, however (53). Patients will develop impairment of pursuits by school age. Increased saccadic latency, saccadic hypometria, and saccadic intrusions are also seen. Visual acuity, pupillary response, and funduscopic examination are usually normal. Poor accommodation and abnormal eye movements may lead to the reading difficulties often reported in patients with ataxia-telangiectasia (18).
The motor unit is often affected in ataxia-telangiectasia. There is wasting of distal muscles with hypotonia. Many young children have normal deep tendon reflexes that diminish by 7 to 8 years of age, with loss of the ankle jerks in later childhood. Many patients in their teens, 20s, and 30s develop progressive spinal muscular atrophy of the hands and feet, but some individuals develop severe anterior horn cell degeneration (31). Within families, the age of onset and progression of symptoms is often similar among affected relatives (79; 65). An adult-onset form has been described that features prominent distal spinal muscular atrophy in the setting of mild cerebellar signs (24). Usually, no sensory abnormalities are present in the younger patient. However, beyond adolescence, there is variable distal sensory loss of vibratory and position senses. Due in part to motor unit involvement, orthopedic manifestations can result, most commonly occurring as foot deformities and scoliosis (14).
Extrapyramidal movement abnormalities are the most variable feature; they may be seen in young patients, with increasing frequency with age. Although rare, patients are sometimes diagnosed with ataxia-telangiectasia as adults due to referrals for movement disorders. Movement disorders can present as choreiform movements, tremor, dystonia, subcortical myoclonus, and resting tremor (39).
Neuropsychological difficulties can also be present starting in childhood (26). Earlier in the course, children with ataxia-telangiectasia can show impairments in verbal intelligence, vocabulary and comprehension, processing speed, visuospatial processing, and working memory. Additionally, impairments in attention and abstract reasoning can present later. These neuropsychological impairments can often be overlooked, but they constitute an important area of necessary support.
Cutaneous manifestations. Telangiectasias are characteristic and usually noted in mid-childhood, often by 5 years or even as late as 14 years of age (36). Because of the later appearance of telangiectasias as compared to movement disorders, the correct diagnosis can be mistakenly excluded or may not be considered when a young child has ataxia (12). This is especially important because ataxia-telangiectasia is the most common cause of progressive ataxia of childhood. In the absence of telangiectasias, ataxia-telangiectasia can be suspected when either choreoathetosis or oculomotor apraxia accompanies the ataxia (especially when lymphoid tissue is hypoplastic). The telangiectasias are found on the bulbar conjunctiva and later spread to involve the rest of the conjunctiva.
Other affected areas are the bridge of the nose, ears, neck, antecubital fossae, and popliteal exposed areas of the skin along with areas of friction. The cutaneous telangiectasias originally were thought to be arterial in origin. Capillary microscopy has now shown that both ocular and cutaneous telangiectasias arise from the venous system (52).
Other cutaneous manifestations of ataxia-telangiectasia include hyperpigmentation, hypopigmentation, cutaneous atrophy, partial albinism, and premature graying of hair. Seborrheic dermatitis, follicular keratosis, dry skin, and sclerodermatous changes may also be seen (52). Cutaneous granulomatosis, seen rarely in children with primary immunodeficiencies, has also been described in children with ataxia-telangiectasia (19).
Immune system deficiencies. No single, consistent abnormality of the immune system has been identified in ataxia-telangiectasia patients. Even affected siblings can differ in the degree and profile of their immunodeficiencies. However, most ataxia-telangiectasia homozygotes manifest some degree of IgA, IgE, or IgG2 deficiency. Serum levels of IgM, IgG1, and IgG3 are usually normal (48). It should be noted, however, that 10% of patients can have elevated IgM concentrations in addition to decreased IgG and IgA levels (47). Thus, ataxia-telangiectasia should be considered in the differential diagnosis of patients presenting with hyper-IgM syndrome (46; 49). Cellular immunity may also be affected. Recurrent sinopulmonary infection is common, often occurring within the first 2 years of life and preceding neurologic manifestations (07), and often results in chronic bronchitis and bronchiectasis. Pneumococcal antibodies are especially low in these patients.
Pulmonary manifestations. Pulmonary complications observed in ataxia-telangiectasia are a significant source of morbidity and mortality. Three general categories of lung disease include recurrent sinopulmonary disease and bronchiectasis, interstitial lung disease and pulmonary fibrosis, and lung disease associated with neuromuscular and bulbar abnormalities (37). Although there are no specific guidelines for the medical management of pulmonary complications, McGrath and colleagues have published a synopsis of the 2009 multidisciplinary workshop on “Pulmonary Disease in Ataxia-Telangiectasia.” Based on available evidence and expert opinion, they highlight the need for early referral to a pulmonologist to obtain baseline pulmonary function tests and monitor for progressive lung disease, screen for sleep-associated comorbidities, and medically maximize lung function. Management is complicated due to a lack of prospective data regarding common infectious agents and colonization over time. However, reversible obstructive lung disease is common in patients with ataxia-telangiectasia, and treatment with bronchodilators and steroids may be beneficial (04). Other pulmonary complications include pneumothorax and hemoptysis.
Cancer susceptibility. The tendency to develop malignancies is well-known, with homozygotes for ataxia-telangiectasia having a risk of cancer more than 100 times that of controls. Among patients with ataxia telangiectasia, lymphoreticular malignancies, namely Hodgkin disease, leukemia, and lymphomas, are most commonly seen, especially with onset in childhood, but there is an absence of myeloid tumors (68). Solid tumors include breast carcinoma, adenocarcinoma of the stomach, basal cell carcinoma, and tumors of the pancreas, ovary, and bladder (23).
There is also an increased susceptibility for early death in heterozygote carriers of the ATM gene (about 1% of the population). In a careful analysis, heterozygote carriers had an increased mortality rate in the ages of 20 through 79 years, with mortality rates increasing with age. The increased mortality was due to cancer, particularly breast cancer, and ischemic heart disease. On average, carriers died 7 to 8 years earlier than noncarriers (64). Exposure to ionizing radiation causes an increased risk for cancer among heterozygotes, particularly female breast cancer (28). Heterozygotes who have missense mutations of the ATM gene may be at greater risk for cancer (40). In the rare T-cell prolymphocytic leukemia, the ATM gene functions as a tumor suppressor (70).
Endocrine dysfunction. Endocrine manifestations include growth failure, hypogonadism, and insulin-resistant diabetes mellitus (05). Growth failure is more likely a primary manifestation of ataxia-telangiectasia rather than a nutritional impairment (45).
Variant ataxia-telangiectasia. It has long been recognized that varying phenotypes of ataxia-telangiectasia exist. Patients with variant ataxia-telangiectasia tend to have a milder phenotype and tend to possess missense mutations in which some residual ATM protein activity is present. Patients with variant ataxia-telangiectasia tend to present initially with extrapyramidal movement disorders, often mimicking forms of primary torsion dystonias, such as dopa-responsive cervical dystonia (13), with the development of ataxia occurring later in life. Six different trajectories have been described, with initial symptoms in childhood starting with extrapyramidal features or cerebellar features; childhood to adolescent-onset with dystonia or cerebellar features; childhood to adult-onset with initial muscle weakness; or adult-onset with initial extrapyramidal symptoms (72).
The mean age of onset of dystonia in patients with variant ataxia-telangiectasia is 12 years old (57); the median age of ataxia onset is 27 years old (75). Diagnostic delay is not uncommon in patients with variant ataxia-telangiectasia: over 10 years in 68% and over 20 years in 33% (59). Many of these patients have either minimal or no ocular telangiectasias, the cutaneous hallmark of classic ataxia-telangiectasia. Although dystonia is the usual predominating movement disorder in patients with variant ataxia-telangiectasia, rarely some patients will have myoclonus as their predominant movement disorder (69). Thus, ataxia-telangiectasia should be considered in patients with an otherwise unexplained movement disorder (even in the absence of ataxia and telangiectasias), starting with screening with a serum alpha-fetoprotein level. The importance of early diagnosis is accentuated by the fact that these patients are also at higher risk for malignancy, as well as the side effects from radiation hypersensitivity, just as patients with classic ataxia-telangiectasia.
This insidiously progressive disorder, with its onset in early childhood, results in the patient being wheelchair-bound by adolescence. The mean age at loss of walking is 10 years (79). Intellectual function is normal early in life and gradually deteriorates. Long-term survival in ataxia-telangiectasia is often accompanied by an increase in physical symptoms spanning different organ systems, but especially neurologic pain due to contractures and psychosocial problems, such as mood disorders (73). Death occurs typically in early or mid-adolescence due to malignancy or bronchopulmonary infection, but rare patients reach adulthood (01). Diagnosis of extranodal lymphomas, particularly pulmonary, is sometimes delayed because of confusion with bronchopulmonary infections (42).
An 8-year-old boy with a history of frequent sinopulmonary infections was evaluated for chronic and progressive gait difficulties and dysarthria. The patient had been previously evaluated, with reportedly normal brain imaging and normal genetic and metabolic testing. The patient had prominent gait ataxia, more so than appendicular ataxia, oculomotor apraxia, palatal dysarthria, and a new finding of ocular telangiectasias. Serum alpha-fetoprotein was elevated, and a diagnosis of ataxia-telangiectasia was confirmed with genetic testing. The patient’s younger brother was subsequently diagnosed with ataxia-telangiectasia at 2 years of age, which was suspected due to delays in walking, and he subsequently developed ocular telangiectasias and progressive ataxia. Both brothers were wheelchair-bound by 10 years of age due to their progressive ataxia.
Ataxia-telangiectasia is inherited in an autosomal recessive fashion. The gene for ataxia-telangiectasia is on the long arm of chromosome 11 (11 q22.23) (20). The defective gene, designated ATM (AT mutated), has a transcript of 12 kilobases (58). The ATM gene product is a protein kinase with homology to phosphoinositol-3-kinase.
The ATM gene is expressed in a ubiquitous fashion and posited to be an important component of cell cycle surveillance and, thus, maintenance of genomic stability. ATM protein kinase is activated in the presence of DNA double-strand breaks. The subsequent cascade of phosphorylation modulates numerous signaling pathways that delay cell cycle progression allowing repair of the genome and prevention of deleterious genetic information to be passed to cell progeny (58; 70; 33; 61; 62). The ATM-deficient transgenic mouse model has typical cellular abnormalities but lacks typical neurodegeneration (15; 70). Genotypes such as truncating mutations resulting in a total absence of ATM kinase activity are associated with a classical earlier-onset phenotypic presentation with its associated higher morbidity and mortality, whereas a milder phenotype with a later onset of ataxia is associated with missense or splice site mutations resulting in the expression of ATM with some residual kinase activity (70; 41; 76). Although genotype-phenotype correlation can often be predicted, rarely mutations among family members have varying clinical manifestations (35).
Ataxia-telangiectasia homozygotes have spontaneous chromosomal instability resulting in a high in vivo occurrence of translocation involving both the small and long arm of chromosome 7 and the long arm of chromosome 14 (78). In vitro, the cells derived from these patients have defective DNA repair capability when exposed to irradiation. Serum immunoglobulins, especially IgA and IgE, are decreased. There is a reduction in T-cell numbers with reduced proliferative responses to mitogens. This is thought to be secondary to defective calcium-dependent signal transduction in the T lymphocyte (29).
The pathologic findings of ataxia-telangiectasia, as distinguished from the atrophic thymus and lymphoid hypoplasia and from the infections and various tumors to which patients with ataxia-telangiectasia are susceptible, are concentrated in the cerebellum. Macroscopically, the cerebellum is usually grossly atrophic (21). This atrophy is most prominent throughout the vermis. In some patients who survive until the fourth decade, small vascular malformations are occasionally accompanied by glial proliferation to produce gliovascular nodules. Microscopically, atrophy affects all layers of the cerebellar cortex, as evidenced by marked thinning of the molecular layer, diminution in the number of Purkinje cells, and thinning of the underlying granule cell layer (17). Neuropathologic abnormalities also include thalamic and cortical hamartomas, nerve cell loss and spheroids in brainstem nuclei (mesencephalic and trigeminal nuclei), nerve cell loss and astrocytic hypertrophy of the substantia nigra, spinal cord atrophy (especially posterior funiculi) with demyelination, gliosis and hypertrophic glial cells, dystrophic dorsal root ganglia, anterior horn cell degeneration, and neurogenic amyotrophy of striated muscles (17; 31).
A pattern of cerebellar atrophy on MRI is also described in patients with ataxia-telangiectasia. Patient disability is well correlated with the degree of cerebellar atrophy. The lateral cerebellum and superior vermis showed the earliest atrophic changes (67).
Swift and colleagues noted the incidence of ataxia-telangiectasia to be 1 in 80,000 to 100,000 live births (66). They also estimated that 1.4% of the United States white population were heterozygous carriers of an ataxia-telangiectasia gene. The gene frequency has been estimated to be between 0.003 and 0.006 (66).
Consistent with autosomal recessive inheritance, there is a 1 in 4 chance of developing the disorder in each pregnancy. Also, if the sibling of a patient with ataxia-telangiectasia decides to have a child, presuming the risk of the partner being a heterozygote is 1%, the estimated risk of their having an ataxia-telangiectasia child is 1 in 600 with each progeny (65).
In addition, ataxia-telangiectasia has been occasionally diagnosed during newborn screening for severe combined immunodeficiency disorder (SCID), which utilizes T cell receptor excision circle (TREC)-based testing (03). These patients were diagnosed with ataxia-telangiectasia with further genetic testing after a positive TREC newborn screen and ruling out severe combined immunodeficiency disorder. Further study is needed to understand how newborn screening may be able to be utilized to facilitate early diagnosis and early discussions regarding prognosis, treatment, and management of ataxia-telangiectasia.
Several conditions with ataxia can be considered in the differential diagnosis of ataxia-telangiectasia, but the most common misdiagnosis is cerebral palsy (12). By contrast, seizures or corticospinal tract dysfunction, which are common accompaniments of cerebral palsy, rarely occur in ataxia-telangiectasia and should lead to a reevaluation of the diagnosis.
Other inherited syndromes manifesting with ataxia should be included in the differential diagnosis of ataxia-telangiectasia. Friedreich ataxia, a trinucleotide repeat disorder, manifests with ataxia in addition to nystagmus, areflexia but with plantar extensor responses, sensory loss, kyphoscoliosis, and high-arched feet. Abetalipoproteinemia (Bassen-Kornzweig disease), an autosomal recessive syndrome, manifests with ataxia, areflexia, and proprioceptive loss in the second decade of life, along with a pigmentary retinopathy in most patients. Abetalipoproteinemia can lead to ataxia due to vitamin E deficiency, but vitamin E deficiency can also be inherited independently in an autosomal recessive manner, resulting in a vitamin E–responsive ataxia.
Ataxia with oculomotor apraxia constitutes a group of disorders caused by differing genetic mutations but shares some common symptoms of ataxia, oculomotor apraxia, and peripheral neuropathy, sometimes associated with an elevated level of alpha-fetal protein. Ataxia with oculomotor apraxia type 1 and type 4 usually begin in childhood, but ataxia with oculomotor apraxia type 2 tends to present in adolescence. Though the neurologic signs are similar to ataxia-telangiectasia, these patients have no telangiectasias, no evidence of multisystem involvement, and no tendency to develop infections.
Ataxia-telangiectasia-like disorder 1 (ATLD1) is an autosomal recessive disorder due to mutations in MRE11 and is characterized by ataxia, oculomotor apraxia, and nystagmus. Although telangiectasias are not a symptom and patients do not present with immunodeficiencies, patients with ATLD1 do exhibit radiosensitivity and an increased rate of malignancies. AFP levels are normal. ATLD2 is also autosomal recessive and is caused by mutations in the PCNA gene, presenting with ataxia and sometimes telangiectasias, along with a radiosensitivity and an increased risk for malignancy; however, oculomotor apraxia is less pronounced. AFP levels are also normal.
RIDDLE syndrome (radiosensitivity, immunodeficiency, dysmorphic facial features, learning difficulties) is autosomal recessive and is caused by mutations in the RNF168 gene. Ataxia can be mild in this syndrome, but patients also present with telangiectasias, recurrent sinopulmonary infections due to immunodeficiency, and an increased malignancy risk due to radiosensitivity.
Progressive ataxia is a frequent feature of mitochondrial encephalomyopathies, which usually have additional manifestations such as myoclonic seizures, retinitis pigmentosa, basal ganglia abnormalities on MRI, lactic acidosis, neurosensory deafness, or ragged-red fiber myopathies. In addition, ataxia and other extrapyramidal symptoms occur with many of the other progressive genetic encephalopathies of childhood, particularly, metachromatic leukodystrophy, Niemann-Pick disease, GM2 gangliosidoses, and Krabbe disease.
|
• Clinical suspicion should result in serum alpha-fetoprotein screening with further genetic testing to confirm the diagnosis. |
In the progressively ataxic young child of 2 to 6 years who has not developed telangiectasias, an elevated serum alpha-fetoprotein is especially useful to validate the suspicion of ataxia-telangiectasia.
Once telangiectasias have developed, the clinical diagnosis of ataxia-telangiectasia is usually confirmed, and molecular genetic testing can detect over 90% of ATM mutations.
Serum alpha-fetoprotein is elevated in at least 90% of patients with ataxia-telangiectasia (79), and levels tend to increase with age (63). Overlap with other inherited ataxia disorders is rare and is usually characterized by lower elevations of alpha-fetoprotein. Examples include ataxia with oculomotor apraxia type 2 (AOA2) and RIDDLE (radiosensitivity, immunodeficiency, dysmorphic facial features, and learning difficulties), caused by mutations in RNF168.
Several cellular and humoral immunologic defects may occur. Two-thirds or more of patients with ataxia-telangiectasia have an immunoglobulin deficiency (79). Serum levels of IgA, IgE, or IgG2 are usually absent or low. Serum IgM is normal or elevated. Total IgG isotypes are normal or decreased. There is peripheral lymphopenia and decreased responsiveness to skin antigens. The chest x-ray may show a small or absent thymic shadow. The sinus x-ray often shows sinusitis. Typically, there is decreased or hypoplastic lymphoid tissue in the nasopharynx, as seen in a lateral skull x-ray. MRI of the brain has revealed vermian atrophy and enlargement of the fourth ventricle and cisterna magna (56), and asymptomatic supratentorial vascular abnormalities (34).
|
• Treatment is supportive, though research into potential disease-modifying therapies is ongoing, and should include a multidisciplinary team. | |
|
• Live-virus vaccines and ionizing radiation should be avoided. |
The treatment of patients with ataxia-telangiectasia requires coordinated multidisciplinary care but is essentially supportive at this time. Practice guidelines in the areas of neurology, immunology, pulmonology, perioperative care, oncology, endocrinology, and nutrition have been published (71). Systemic bacterial infections, severe viral infections, and opportunistic infections are uncommon in ataxia-telangiectasia (47). However, the presence of laboratory immunologic abnormalities warrant close observation. Seven-valent pneumococcal conjugated vaccine may be protective in ataxia-telangiectasia patients; however, multiple doses of conjugated vaccine may be required (54; 60). Infections are treated with antibiotics. Intercurrent infections can be prevented with the use of intravenous gammaglobulin therapy and postural drainage (15). For malignancies, conventional doses of radiation therapy are contraindicated because these patients are hypersensitive to ionizing radiation (21). However, lymphomas should be treated with standard-course chemotherapy while remaining alert to hemorrhagic cystitis or pulmonary complications (55). Live-virus vaccines should not be used after the diagnosis of ataxia-telangiectasia (50).
No effective therapy exists for halting the progression of the ataxia. Studies suggesting that ataxia-telangiectasia cells may be in a constant state of increased oxidative stress make it likely that most antioxidants or free-radical scavengers might counteract some of the progressive neurologic deterioration of patients with ataxia-telangiectasia (77). At least one ataxia-telangiectasia mouse model study suggests that treatment with catalytic antioxidants appear to positively impact neurobehavioral deficits (09). Vitamin E has been recommended for a number of years; alpha-lipoic acid and coenzyme Q10 may also be helpful. Folic acid theoretically minimizes chromosomal fragility and the formation of double-strand DNA breaks.
One adult patient had improvement in his gait ataxia with treatment with the GABA-ergic medications pregabalin and tiagabine (22).
Some improvement (76% responder rate, defined as at least 20% improvement in the summation of the three scales used) in movement disorders has been reported with the use of amantadine sulfate (43). The successful treatment of dystonia in a patient with ataxia-telangiectasia has also been reported with trihexyphenidyl (81).
Acetyl-DL-leucine (ADLL) has been shown to improve ataxia symptoms, including improving ataxia and ocular stability in patients with ataxia-telangiectasia (10). A multinational, randomized, double-blind, placebo-controlled crossover phase 3 study is currently ongoing to evaluate the safety and clinical efficacy of N-acetyl-L-leucine, with an estimated completion date of June 2028 (https://clinicaltrials.gov/study/NCT06673056). There is also open-label evidence that restoring cellular NAD+ levels using nicotinamide precursors, such as nicotinamide riboside, is well tolerated and may improve coordination and eye movements, but further study is also needed (51).
One case report cites short-term improvement of neurologic deficits using steroids (11). Unfortunately, improvement was temporary and typical side effects associated with exogenous steroid use were seen. A follow-up study found similar positive results with clear improvement of neurologic symptoms in five of six patients treated for 10 days with oral betamethasone (08). They found improvement not only in younger patients but also in older patients many years after onset of symptoms, with the clinical benefit inversely correlated with the degree of cerebellar atrophy. Furthermore, in a multicenter, double-blind, randomized, placebo-controlled trial, oral betamethasone reduced ataxia as measured by standardized ataxia scales (80).
The long-term safety and side effect profile can limit the use of long-term steroid therapy. Thus, researchers have been investigating the use of alternative methods of delivering steroid therapy to patients with ataxia-telangiectasia, such as using engineered red blood cells to carry and deliver dexamethasone via a slow-release mechanism (38). However, the most recent multicenter, double-blind, placebo-controlled trial utilizing this method did not conclude statistical benefit for all age groups, though follow-up studies with analysis in the 6- to 9-year-old age group is ongoing (30).
Ongoing research continues in the treatment approach of antisense oligonucleotides. Although currently focused on a specific gene mutation (c.7865C> T), an ongoing phase 1/2 study is examining the safety and efficacy of intrathecally administered precision genetic therapy with atipeksen, a mutation-specific antisense oligonucleotide (https://clinicaltrials.gov/study/NCT07215416). If successful results are obtained, this study could serve as a foundation for advancing the development of additional precision genetic therapies for other patients with ataxia-telangiectasia.
There is hope that gene vector delivery therapy could someday be a curative option for patients with ataxia-telangiectasia, as the lack of a functioning ATM kinase protein due to mutations in ATM is a targetable treatment option with gene therapy. Although ATM is a large protein, exceeding the traditional capacity of commonly used adenoviral vectors, alternative vector packaging methods are a possible way to solve this problem (25). Thus, there is continued progress and research towards the goal of clinically significant symptomatic treatment or a cure for ataxia-telangiectasia in the future.
The opinions and assertions expressed herein are those of the author(s) and do not reflect the official policy or position of the Uniformed Services University of the Health Sciences or the Department of War.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

David T Hsieh MD
Dr. Hsieh of the Uniformed Services University of the Health Sciences has no relevant financial relationships to disclose.
See Profile
Alcy R Torres MD FAAP
Dr. Torres of Boston Medical Center and Boston University Chobanian and Avedisian School of Medicine has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Sleep Disorders
Jul. 05, 2026
General Child Neurology
Jun. 24, 2026
General Child Neurology
Jun. 10, 2026
Epilepsy & Seizures
Jun. 02, 2026
Neuro-Oncology
May. 27, 2026
General Neurology
May. 13, 2026
Epilepsy & Seizures
May. 08, 2026
General Child Neurology
Apr. 29, 2026