






Sleep Disorders
Sleep-related urologic dysfunction
Jul. 06, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Limited literature exists regarding the unique features of cerebral edema in developing children. In this article, the authors describe the known mechanisms of cerebral edema, as well as etiologies for the development of cerebral edema in the pediatric population. They describe the existing literature, which underscores the challenge of managing cerebral edema in childhood. The role of dynamic changes in cerebral blood flow and brain compliance in the pathogenesis of cerebral edema across the pediatric age range is emphasized, in addition to the need for designing management strategies to mitigate against secondary brain injury.
|
• Cerebral edema can be a manifestation of vasogenic, cytotoxic, hydrostatic, or osmotic mechanisms. | |
|
• Various advanced monitoring and data analysis techniques allow for real-time measurements of intracranial pressure in addition to model-based indices, including brain compliance and cerebrovascular pressure reactivity. |
Pediatric clinicians and researchers continue to be challenged in recognizing how the developing brain responds differently than the mature brain to a variety of insults.
Brain edema often clinically manifests as encephalopathy but is often subclassified based on specific pathologic and radiographic findings. Even so, pediatric cerebral edema can be difficult to delineate categorically, as many patients follow a changing and progressive clinical course. A cross-sectional study of the 2016 Kids’ Inpatient database documented 4903 pediatric cases among 2,210,263 children discharged (2.2 of 1,000), with a 29.4% mortality rate (34). This article describes commonly identified causes of cerebral edema in children; key pathophysiologic processes involved in the development of childhood cerebral edema; and monitoring and treatment strategies for cerebral edema in this population.
Fundamentally, cerebral edema represents an increase in brain volume that is contained within the brain interstitial space. Four different forms of cerebral edema represent different pathophysiologic mechanisms by which interstitial brain volume increases: vasogenic edema, cytotoxic edema, hydrostatic cerebral edema, and osmotic cerebral edema (Table 1) (38). Although these subtypes of cerebral edema are important to differentiate from the standpoint of pathophysiology, clinical management, and prognostic implications, it is also important to note that in the setting of acquired brain injury, multiple forms of cerebral edema can coexist and compound one another.
Table 1 outlines major forms of cerebral edema in childhood and mechanisms of injury and common pediatric conditions associated with each form of cerebral edema.
|
Form of cerebral edema |
Mechanism of injury |
Common conditions |
|
Vasogenic edema |
Blood-brain barrier breakdown; increased water permeability into brain interstitia |
Central nervous system tumors; diabetic ketoacidosis; posterior reversible encephalopathy syndrome; acute hepatic failure |
|
Cytotoxic edema |
Cellular metabolic crisis; cell death and apoptosis; influx of ions and water from extracellular to intracellular space |
Cardiac arrest; traumatic brain injury; neonatal hypoxic-ischemic encephalopathy; mitochondrial crises; acute hepatic failure |
|
Hydrostatic edema |
Net movement of cerebral spinal fluid from the ventricular space into brain interstitia |
Obstructive hydrocephalus |
|
Osmotic edema |
Isolated osmotic gradient between brain parenchyma and cerebrovascular system |
Dialysis disequilibrium syndrome; osmotic demyelination syndrome |
|
Source: (38) |
Vasogenic edema manifests as a result of blood-brain barrier breakdown (38). The blood-brain barrier represents a barrier between the brain and the systemic circulatory system that is formed by endothelial cells interconnected by tight junctions, in addition to astrocyte and pericyte cells. Astrocytes are intimately involved in controlling the flux of water across the blood-brain barrier, which is primarily composed of astrocytes and astrocytic foot processes interfacing with the tight junctions between endothelial cells (07). The astrocyte endfeet are separated by gaps of approximately 20 µm (52). Pericytes, mesenchymal cells that contribute to cerebral blood flow regulation and increase the tightness of the blood-brain barrier, are embedded within the endothelial basement membrane (52).
The blood-brain barrier is a dynamic barrier that alters its properties in response to neuronal activity (52). Astrocytes control water homeostasis through a number of cell membrane molecules, the most well-studied of which is the water channel, aquaporin 4 (AQP4). AQP4 facilitates water movement into and out of the brain, accelerates astrocyte migration, and alters neuronal activity (53). Gunnarson and colleagues demonstrated that glutamate can enhance astrocytic water permeability through a complex metabolic effect that can be inhibited by ablation of AQP4 (22). In addition, a study by Lichter-Konecki and colleagues demonstrated co-location of AQP4 with the gap junction channel connexin 43 (CX43) and the astrocytic inward-rectifying potassium channel (KIR) genes (KIR4.1 and KIR 5.1) in a hyperammonemic mouse model (37). The authors were able to show that hyperammonemia can cause a direct effect of downregulating these three channels, resulting in aberrant homeostasis of water and potassium, thus leading to astrocyte swelling and brain edema. In a related experiment, selective inhibition of AQP4 with AER-271 following pediatric asphyxial cardiac arrest was found to block early edema (57).
Cytotoxic edema is fundamentally the result of a cellular metabolic crisis when metabolic demand is insufficiently supported by energy substrate delivery (38). Cerebral cellular death causes brain cells to lose their ionic gradient. This results in an influx of ions and water, first, from the extracellular space to the intracellular space and, subsequently, from the vascular space to the extracellular space, resulting in increased brain volume. Because cytotoxic edema results from energy failure, it is commonly seen in conditions such as hypoxic-ischemic brain injury, traumatic brain injury, and refractory status epilepticus. In children with brain contusions after traumatic brain injury, intramyelinic cytotoxic edema has been described manifesting as central white matter diffusion restriction on brain magnetic resonance imaging (43).
Hydrostatic cerebral edema is the result of the movement of the CSF from the ventricular space into brain interstitia as the result of obstructive hydrocephalus (38). In obstructive hydrocephalus, an increase in hydrostatic pressure forces movement of the CSF through ependymal cells lining the ventricles and into brain parenchyma.
Osmotic cerebral edema results from an isolated osmotic gradient that can form between brain parenchyma and the cerebrovascular space that favors water entry into the brain. This can result in unique circumstances like dialysis disequilibrium syndrome or osmotic demyelination syndrome (38). Osmotic edema often occurs when there is preexisting blood-brain barrier disruption and either preexisting vasogenic or cytotoxic edema. As such, osmotic cerebral edema can be challenging to distinguish on standard neuroimaging. However, there are specific conditions, such as central pontine myelinolysis, in which this form of insult is inferred to have occurred.
The recognition of the development of cerebral edema is important in pediatric neurocritical care, given its role in secondary brain injury propagation. In children older than 12 months of age who have fontanelle closure, the intracranial space is relatively fixed. In such situations, the Kellie-Monroe doctrine can be applied, which states that the intracranial space contains three primary compartments: CSF space, brain parenchyma, and intracranial blood volume (38). According to the Kellie-Monroe doctrine, any increase in the volume of any space, or the introduction of an additional space (eg, brain tumor, epidural hematoma), will allow compression of other compartmental elements within the intracranial space and a subsequent rise in intracranial pressure.
Brain compliance, which relates changes in volume to pressure, can initially be stable with mild increases in total intracranial volume. CSF can act as an initial buffer to increases in intracranial volume, with drainage out of the intracranial vault occurring either down the third and fourth ventricles or through the cerebral sinus system, ultimately passing through to the lumbar space. To a lesser degree, intracranial vasculature can also act as a buffer against increased intracranial pressure through compression of the venous or arterial circulation. Compression of intracranial vasculature, however, carries an increased risk of secondary brain injury, as impedance of cerebral venous flow can limit CSF drainage, and impedance of cerebral arterial flow increases the risk of cerebral ischemia. At a critical level of increased intracranial pressure, intracranial compliance can be exhausted, and changes in brain volume result in an exponential increase in intracranial pressure. In such situations, brain herniation can ensue, leading to permanent regional brain damage and cerebral ischemia in all regions in which cerebral arterial blood flow is compromised.
The tentorium cerebelli distinguishes the supratentorial and infratentorial spaces within the intracranial vault. Increases in supratentorial volume with decreased brain compliance carry a risk for uncal, central, and falcine herniation (38). Uncal herniation occurs when the ipsilateral medial temporal lobe is displaced through the tentorium cerebelli to compress the ipsilateral midbrain and ipsilateral cranial nerve III. This manifests with increased ipsilateral pupil size with decreased reactivity, contralateral hemiplegia, posturing, and eventually coma.
Central herniation produces bilateral medial temporal lobe displacement against the diencephalon, manifesting with bilateral pupillary dilatation, posturing, and coma. Falcine herniation occurs when there is medial displacement of the cerebral hemisphere against the falx cerebri, with the cingulate gyrus being displaced under the falx, which manifests with contralateral leg weakness. Due to the decreased likelihood of impacting the diencephalon, falcine herniation is less likely to result in coma or pupillary dilatation. Increases in infratentorial volume can result in ascending transtentorial herniation or tonsillar herniation. Ascending transtentorial herniation represents upward displacement of the cerebellum through the tentorium cerebelli, resulting in dorsal midbrain compression. This can manifest with vertical gaze palsy, coma, and obstructive hydrocephalus. Tonsillar herniation represents downward displacement of the cerebellar tonsils through the foramen magnum, with compression of the cervicomedullary junction. This can manifest with obstructive herniation, quadriparesis, and coma.
Brain compliance may be increased in infants with open fontanelles compared to older patients, given an increased ability for total intracranial volume to change. In such patients, examination of the fullness of the anterior and posterior fontanelles may aid in identifying the presence of intracranial pressure.
Because of the need to avoid intracranial hypertension and the blunting of clinical signs in comatose patients, invasive intracranial pressure monitoring is often utilized in severe acute brain injuries, such as traumatic brain injury and hemorrhagic stroke. Multiple techniques are available to monitor intracranial pressure (03). The gold standard approach is the external ventricular drain, whereby a catheter is inserted via a frontal burr hole into the lateral ventricle. The external ventricular drain provides a global measure of intracranial pressure and confers a therapeutic advantage in also being able to drain CSF, augmenting the buffering capacity of CSF drainage as a measure to improve brain compliance. The disadvantages of the external ventricular drain are that it is invasive, it only measures intermittent intracranial pressure values when clamped, and it carries an increased risk of infection.
The intraparenchymal probe is a fiberoptic catheter that allows for continuous intracranial pressure monitoring and is smaller and less invasive than the external ventricular drain. Disadvantages are that it measures intracranial pressure regionally where it is placed and is prone to drifting, particularly when placed for very extended periods. Epidural bolts are also available for intracranial pressure monitoring but are used less commonly.
Noninvasively, several studies have demonstrated the value of transcranial Doppler ultrasound as a screening method to investigate for intracranial pressure (48). Fundamentally, transcranial Doppler ultrasound is utilized to investigate cerebral blood flow measured through the major basal arteries (middle, anterior-posterior, basilar, and vertebral arteries). Evidence shows that decreased flow velocities and elevated pulsatility indices on transcranial Doppler ultrasound can be sensitive for detecting increased intracranial pressure. Several studies incorporating transcranial Doppler ultrasound waveforms with arterial blood pressure waveforms have helped develop models that predict intracranial pressure, including in pediatric patients. A prospective, multicenter diagnostic accuracy study investigated the accuracy of a physiological index (ICPtcd), incorporating both transcranial Doppler ultrasound flow velocities and mean arterial blood pressure to determine evidence of increased intracranial pressure, and demonstrated that transcranial Doppler ultrasound has a high negative predictive value in ruling out intracranial hypertension (47). Currently, guidelines from the European Society of Intensive Care Medicine indicate that transcranial Doppler ultrasound can be used as a basic ultrasound skill for intensivists to screen against intracranial pressure (49). Some work has suggested that transcranial Doppler ultrasound is integrated with multimodality neurologic monitoring, and novel biomarkers, such as critical closing pressure and diastolic closing margin, may be related to intracranial pressure and functional outcomes (23).
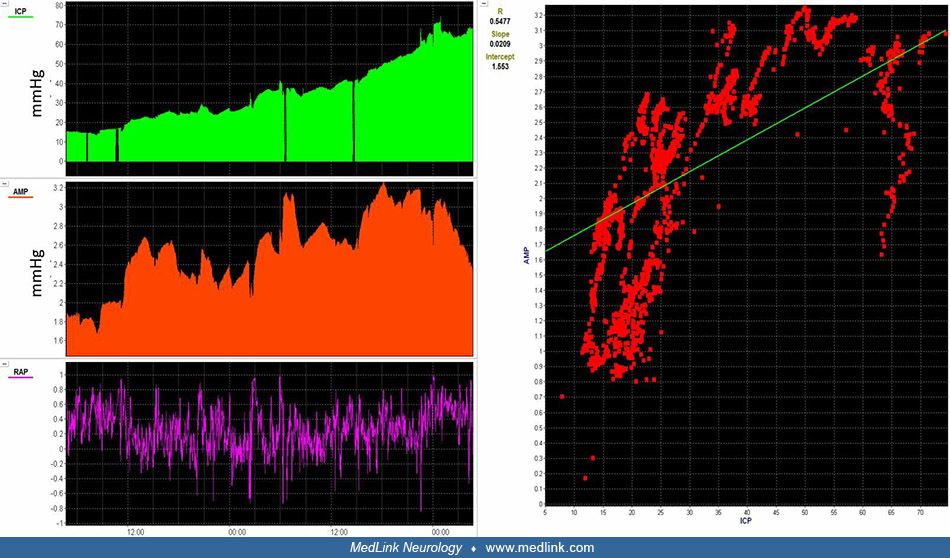
In addition to assessing intracranial pressure values, there are various other methods to investigate for evidence of decreased intracranial compliance and increasing cerebral edema. Intracranial pressure monitoring waveforms contain three pulses: P1, P2, or P3. P1 represents the percussion wave, reflecting the arterial input during systole. P2 is considered the tidal wave, reflecting the venous pulsation before closure of the aortic wave. P3 represents the dicrotic wave in which venous drainage occurs immediately after closure of the aortic valve. When brain compliance is adequate, P1 is typically of a higher amplitude than P2 or P3. When brain compliance decreases, P2 will typically increase in amplitude beyond P1 or P3. At critically high intracranial pressure and low brain compliance, there is a loss of distinction of P1, P2, or P3 as the intracranial pressure waveform morphology evolves toward one wave.
Another way to investigate the correlation of changes in intracranial pressure pulse amplitude with changes in intracranial pressure values themselves is the pressure-volume compensatory reserve index (55). In high brain compliance, intracranial pressure pulse amplitude has a minimal positive association with intracranial pressure. As brain compliance is compromised, increases in intracranial pressure are directly associated with rises in intracranial pressure pulse amplitude. At a critical threshold of intracranial pressure, there is deranged physiology in which the association of intracranial pressure and intracranial pressure pulse amplitude is lost.
A 15-year-old male with refractory intracranial hypertension after severe traumatic brain injury. Intracranial pressure steadily rises and initially correlates with intracranial pressure pulse amplitude. RAP, which correlates i...
In pediatrics, another brain compliance index is the intracranial pressure-partial pressure of carbon dioxide compliance index (PCI), which represents a moment-to-moment correlation between changes in intracranial pressure and changes in end-tidal carbon dioxide. Good compliance is reflected by a lack of correlation, and poor compliance is reflected by a positive correlation between the two physiological inputs (61). Another index, the pulse shape index (PSI), classifies the intracranial pressure pulse-wave configuration into four categories representing incremental states of brain compliance. PSI has been studied within a multicenter cohort study of 98 pediatric patients with traumatic brain injury and carried an association with intracranial pressure, worsened cerebrovascular pressure reactivity, and functional outcomes within univariable analyses (10). Other modalities may be complementary in understanding the evolution of cerebral edema. Partial pressure of brain tissue oxygenation (PbtO2) can be assessed using a micro-Clark electrode. Values under 10 mmHg have been associated with worsened outcomes after pediatric traumatic brain injury, and intrasubject decreases in PbtO2 values could reflect worsening cerebral edema, among other secondary brain injury mechanisms (17). Cerebral edema may also be assessed using thermal diffusion flowmetry techniques. Although thermal diffusion flowmetry has historically been used for the assessment of regional cerebral blood flow in adult patients, there is evidence that thermal conductivity is highly correlated with estimated water content and that estimated brain water content from thermal conductivity measurements is associated with neuroimaging evidence of cerebral edema in comatose patients (29).
Appropriate cerebral blood flow and cerebral perfusion pressure are needed to provide adequate neuroprotection in cerebral edema. Cerebral perfusion pressure represents the driving pressure that supplies cerebral arterial circulation perfusion. Although it cannot be directly measured in the critical care environment, it is derived by subtracting intracranial pressure from mean arterial blood pressure. Avoidance of low cerebral perfusion pressure is necessary to reduce the risk of cerebral ischemia from acute cerebrovascular insufficiency. Because cerebral perfusion pressure is derived from intracranial pressure values, it can be limited to estimating cerebral perfusion pressure in a regional manner in locations near where intracranial pressure monitoring is located. Furthermore, the cerebral perfusion pressure calculation assumes that changes in intracranial pressure are directly related to a loss of cerebral perfusion, which may not apply when increases in intracranial pressure are directly related to changes in intracranial arterial blood volume. Current pediatric traumatic brain injury guidelines support maintaining intracranial pressure under 20 mmHg and cerebral perfusion pressure above 40 mmHg (31). These thresholds are based on population-based studies across the entire pediatric age range, and more precision is often needed. A retrospective analysis of children undergoing intracranial pressure monitoring identified mean intracranial pressure thresholds associated with in-hospital mortality as being below 20 mmHg and age-dependent cerebral perfusion pressure thresholds as being above 40 mmHg (63).
An important intrinsic neurophysiologic mechanism that regulates cerebral blood flow is cerebrovascular pressure reactivity, often referred to as cerebral autoregulation. Cerebrovascular pressure reactivity is the mechanism by which cerebral small vessel arterioles will vasoconstrict or vasodilate in response to slow-wave fluctuations in cerebral perfusion pressure to maintain a steady state rate of cerebral blood flow (11). Decades of work in animal models and adult humans have produced the Lassen curve, whereby efficiency of cerebrovascular pressure reactivity appears over mean arterial blood pressure approximated between 50 to 100 mmHg. Beyond these lower and upper limits, cerebrovascular pressure reactivity mechanisms are less efficient, and cerebral blood flow is pressure-passive. Below the lower limit of cerebrovascular pressure reactivity, reductions of cerebral perfusion pressure lead to oligemia and an increased risk of cerebral ischemia and cytotoxicity. Exceeding the upper limit of cerebrovascular pressure reactivity leads to hyperemic cerebral blood flow, which may compound preexisting vasogenic edema.
Continuous monitoring of cerebrovascular pressure reactivity can be assessed by correlating systemic arterial blood pressure with a surrogate of cerebral blood flow. These surrogates can include transcranial Doppler ultrasound flow velocities, intracranial pressure values, brain tissue oxygenation values, or cerebral regional oximetry values (15). The most well-studied index of cerebrovascular pressure reactivity is the pressure reactivity index, which functions as a moving Pearson correlation coefficient between slow-wave fluctuations in intracranial pressure and mean arterial blood pressure (65). If intracranial pressure acts as a surrogate of cerebral blood volume, the assumption is that with efficient cerebrovascular pressure reactivity, changes in mean arterial blood pressure are negatively associated with cerebral blood volume and, thus, intracranial pressure, and they positively associate with inefficient cerebrovascular pressure reactivity. Thus, positive pressure reactivity index values approximating +1 are theorized to represent inefficient cerebrovascular pressure reactivity, whereas negative values approaching -1 are associated with efficient cerebrovascular pressure reactivity. When plotting pressure reactivity index values or other cerebrovascular pressure reactivity indices over a range of systemic blood pressures or cerebral perfusion pressures, there are ways to extract putative “optimal” pressures where cerebrovascular pressure reactivity is most efficient, as well as the upper and lower limits of cerebrovascular pressure reactivity.
Multiple studies have associated inefficient cerebrovascular pressure reactivity with worsened outcomes in pediatric conditions, such as traumatic brain injury, hemorrhagic stroke, and cardiac arrest (36; 04; 05; 28; 51; 59), in addition to increased time below either the optimal calculated value of cerebrovascular pressure reactivity or time below its lower limit. These studies used either the pressure reactivity index or other indices of cerebrovascular pressure reactivity derived from intracranial pressure waveforms or other surrogates of cerebral blood flow. A multicenter prospective observational study across 10 pediatric intensive care units in the United Kingdom affirmed that elevated cerebrovascular pressure reactivity index values (PRx) were associated with unfavorable outcomes after pediatric traumatic brain injury (02). Furthermore, hourly dose and percentage time spent below the lower limit was independently associated with worsened outcome in that multicenter cohort study (50). Although prospective studies are needed to validate whether cerebrovascular pressure reactivity monitoring can help guide cerebral perfusion pressure targets for individualized patients, this process may be promising given that such targets are likely to shift dramatically with respect to age.
Existing pediatric traumatic brain guidelines guide neurocritical care management to alleviate the impact of cerebral edema (31). Active normothermia is utilized to avoid the impact of hyperemia and increased metabolic demand to worsen cerebral edema. Three class II clinical trials have been performed to assess the utility of therapeutic hypothermia in pediatric traumatic brain injury, with no differences in functional outcomes and suggestions of a trend of increased mortality (25; 01; 08). Clinical trials in in-hospital and out-of-hospital pediatric cardiac arrest have not demonstrated a therapeutic benefit of therapeutic hypothermia compared to active normothermia, although the presence of hyperthermia has been associated with unfavorable outcomes (41; 40).
Sedative pharmacotherapy and neuromuscular blockade decrease metabolic demand and promote brain homeostasis. Critical care ventilatory management strategies maintain the arterial content of carbon dioxide (PaCO2) within a range of 35 to 45 mmHg to avoid oligemic or hyperemic cerebral blood flow. Existing pediatric traumatic brain injury guidelines support using 3% hypertonic saline for treating intracranial hypertension. Notably, however, no class I evidence demonstrates that pediatric traumatic brain injury care interventions improve functional outcomes (31). Mannitol is also available as a therapeutic option against cerebral edema. A multicenter comparative study evaluated the effect of bolus hypertonic saline and mannitol in treating intracranial hypertension. Bolus administration of hypertonic saline was associated with superior intracranial pressure and cerebral perfusion pressure outcomes compared to mannitol, particularly during intracranial pressure crises (30). Evidence suggests that hyperosmolar therapy is more likely to be effective when there is evidence of efficient cerebrovascular pressure reactivity (18; 58; 66).
Various candidate biomarkers that target the blood-brain barrier or receptors of aquaporin-4, vasopressin V1a, and other genes responsible for cellular swelling are actively being studied (27). However, these are not utilized as standard treatments for managing cerebral edema. Currently, the strongest level of evidence for the management of cerebral edema comes from pediatric traumatic brain injury. Further work is needed for other pediatric conditions, such as arterial ischemic stroke, intracranial hemorrhage, and cardiac arrest, among other pediatric disease states (13).
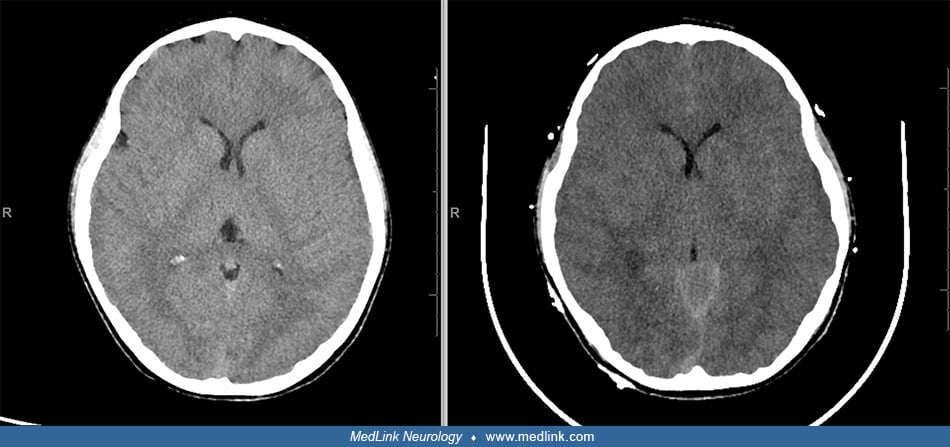
Hepatic encephalopathy. Fulminant hepatic failure and its association with brain edema as a cause of significant mortality has been recognized in 50% to 81% of patients undergoing necropsy (35). The severity of cerebral edema measured by noncontrast CT scan correlates with the degree of hepatic encephalopathy (60). Although the cause of this association was enigmatic for many years, severity of encephalopathy in fulminant hepatic failure has been correlated with arterial ammonia levels (45). Ammonia in the brain is detoxified by astrocytes in a process leading to glutamate accumulation, causing the astrocyte to swell and release inflammatory cytokines (12). Additionally, the citric acid cycle fails, leading to an accumulation of lactate within the brain. Cerebral hyperemia has also been implicated in the evolution of cerebral edema in fulminant hepatic failure (09).
Diabetic ketoacidosis. Although relatively uncommon, cerebral edema as a complication of diabetic ketoacidosis remains the most frequent diabetes-related cause of death in children (62); 95% of cases occur in children less than 20 years of age (46). Clinical cerebral edema occurs in 0.3% to 1% of episodes of diabetic ketoacidosis in children (39). Most patients with cerebral edema have subclinical signs and remain asymptomatic throughout their treatment course. However, once cerebral edema manifests into clinical sequelae, approximately 20% to 50% of cases are fatal (19). Cerebral edema can present on a continuum from simple headaches with progressive changes in level of consciousness to signs of central herniation. The current diagnostic markers of cerebral edema rely on detailed systemic and neurologic evaluation. Neuroimaging studies such as MRI and CT are recommended by the International Society for Pediatric and Adolescent Diabetes, but only after starting treatment to confirm diagnosis. A study by Jeziorny and colleagues found that transorbital ultrasonography and pachymetry in children with ketoacidosis can help to assess the increased risk of cerebral edema (26). Diabetic ketoacidosis–related cerebral edema remains poorly understood, and fluid treatment has been implicated as a potential exacerbating factor in its pathogenesis. One theory that investigators hypothesize is that rapid decline in serum osmolality with vigorous fluid resuscitation has promoted osmotically mediated fluid shifts into the brain. Additional theories postulate that the intracellular accumulation of sodium and water via the activation of the sodium/hydrogen ion exchange from cerebral acidosis is a contributing factor. In addition, using a bicarbonate buffer has been implicated as causing a paradoxical central acidosis, contributing to central hypoxia. Hypocapnia caused by metabolic acidosis may lead to central vasoconstriction, which may lead to cerebral ischemia and subsequent cytotoxic edema (46). Most investigators now believe that the development of cerebral edema is a cellular response to injury rather than a result of treatment. Studies to date suggest that administering intravenous fluids in volumes up to 20 mL/kg does not contribute to cerebral edema in diabetic ketoacidosis (39). New evidence suggests that early isotonic fluid therapy does not confer increased risk and may be beneficial for select patients (21).
Clinical data demonstrate that patients can manifest cerebral edema on head CT performed before any treatment has been initiated. Some patients have died at home due to complications of cerebral edema secondary to diabetic ketoacidosis. Hom and Sinert undertook a systematic review to evaluate the strength of the data implicating fluid therapy as a cause of cerebral edema in diabetic ketoacidosis (24). They concluded there was insufficient evidence to implicate either the rate or volume of fluid administration as a cause of cerebral edema in this patient population. As noted above, many patients with diabetic ketoacidosis have cerebral edema, with only a few progressing to neurologic injury with symptoms. Although the pathophysiologic mechanism responsible for cerebral edema is unknown, there are many theories beyond osmotic changes due to treatment. To identify those at-risk patients with ketoacidosis, several studies have described possible clinical predisposing factors that enhance the propensity to develop cerebral edema (Table 2). Durward and colleagues showed that one of the earliest warning signs for the development of cerebral edema in children with diabetic ketoacidosis is failure of the glucose-corrected serum sodium to rise during treatment (16). Fluid refractory shock has been shown to be an independent risk factor for cerebral edema in these patients in a resource-poor environment (56).
|
Yes |
No | |
|
Initial increase in blood urea nitrogen level |
x |
|
|
Source: (19; 16; 56) | ||
Acute necrotizing encephalopathy. Acute necrotizing encephalopathy is a rare condition that manifests after acute febrile, often viral, illnesses (64). Viral symptoms such as fever, cough, and gastroenteritis are often followed by dramatic deterioration toward seizures, altered consciousness, and hepatic dysfunction. Magnetic resonance brain imaging characteristically demonstrates cytotoxic edema that focuses over the bilateral thalami in addition to brainstem tegmentum, putaminal, and cerebellar regions.
Acute necrotizing encephalopathy has been observed to occur sporadically within families or reoccur, with familial and recurrent cases being linked to pathogenic variants of the Ran binding protein 2 (RANBP2) gene and classified as “infection-induced encephalopathy 3” (IIAE; OMIM 608033) (42). There are no consensus guidelines for management against acute necrotizing encephalopathy, although some evidence suggests that immunomodulatory therapies, such as glucocorticoids and tocilizumab, may provide benefit (44; 32; 06).
Mitochondrial disorders. Primary mitochondrial disorders can manifest with cytotoxic edema due to energy failure (20). Mitochondria are intracellular double-membrane organelles that reside within nucleated cells and are responsible for oxidative phosphorylation (20). When primary mitochondrial disorders manifest with dysfunction in mitochondria, situations in which metabolic demand rises, such as seizures, hyperthermia, or viral illnesses, may trigger an increase in metabolic demand such that energy substrates may not be well prepared to compensate. Such edema may manifest in gray matter, where energy metabolism is greatest. Leigh syndrome can manifest with lesions in the basal ganglia, particularly in the striatum, diencephalon, and brainstem (substantia nigra, periaqueductal gray matter, inferior olivary nuclei) (54; 20).
Deoxyribonucleic acid polymerase gamma (POLG)-related disease relates to pathogenic variants of POLG. POLG is a nuclear gene that encodes DNA polymerase y, which is responsible for the mitochondrial genome (14; 20). Patients with this condition can manifest with stroke-like changes and T2-weighted and FLAIR sequence abnormalities in cortical regions as well as cytotoxic edema, in addition to lesions in the bilateral thalami. Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) manifests similarly often, with episodes of headache, seizures, or focal neurologic weakness accompanying focal acute brain lesions with corresponding cytotoxic edema (33; 20). The stroke-like episodes are speculated to result from dysfunctional mitochondria in endothelial and smooth muscle cells of blood vessels, which results in inefficient cerebrovascular pressure reactivity.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Rebecca Silverstein DO MS MBA
Dr. Silverstein of Phoenix Children's Hospital has no relevant financial relationships to disclose.
See ProfileBrian L Appavu MD
Dr. Appavu of University of Arizona College of Medicine has no relevant financial relationships to disclose.
See Profile
Nina F Schor MD PhD
Dr. Schor of the University of Rochester School of Medicine and Dentistry has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Sleep Disorders
Jul. 06, 2026
Sleep Disorders
Jul. 05, 2026
General Child Neurology
Jun. 24, 2026
General Child Neurology
Jun. 10, 2026
Epilepsy & Seizures
Jun. 02, 2026
General Neurology
May. 13, 2026
General Child Neurology
May. 12, 2026
Epilepsy & Seizures
May. 08, 2026