





Neuro-Oncology
Turcot syndrome
May. 27, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Craniopharyngioma is a rare histologically low-grade (WHO grade 1) suprasellar tumor believed to originate from Rathke’s pouch. It occurs in adamantinomatous and papillary subtypes. Consistent with its location, patients experience visual abnormalities, symptoms of increased intracranial pressure, and endocrine dysfunction. If the tumor is favorably located, the most effective treatment is adequate resection. In this article, the authors present the protean clinical manifestations, suspected pathogenesis, and currently available treatment options for craniopharyngioma, with emphasis on the importance of a multidisciplinary approach. Genetic analysis has shown that adamantinomatous and papillary craniopharyngiomas have distinct tumor biology. Adamantinomatous craniopharyngiomas demonstrate activating mutations in the gene encoding β-catenin (135; 18; 46; 121). On the other hand, the majority of papillary craniopharyngiomas harbor the oncogenic BRAF V600E substitution, which can be used as a confirmatory test using immunochemistry and shows promise as targeted therapy in selected patients (13; 121).
|
• Due to its suprasellar location, patients with craniopharyngioma often present with visual and endocrine symptoms. | |
|
• There are two craniopharyngioma subtypes: adamantinomatous and papillary. | |
|
• Adamantinomatous craniopharyngiomas have an activating mutation in the gene encoding β-catenin and show bimodal peaks of incidence: first two decades and fifth-sixth decades. Papillary craniopharyngiomas often carry the BRAF V600E mutation and manifest in late adulthood. | |
|
• Most patients are treated by surgery followed by irradiation. | |
|
• The role of chemotherapy remains unclear. |
Craniopharyngioma is a rare parasellar tumor of low histological grade that came into attention towards the end of the 19th century. It was first reported by a German pathologist, Friedrich Albert von Zenker in 1857, who described a cystic suprasellar mass containing cholesterol crystals and squamous epithelium in an autopsy. In 1860, Luschka extensively studied squamous epithelial cells in the adenohypophysis.
Pioneering work by Jakob Erdheim in 1904 led to accurate histopathological characterization of craniopharyngiomas. The first successful surgical resection of a craniopharyngioma was performed by A E Halstead in 1909 by an intranasal approach. The current terminology of craniopharyngioma was introduced by Harvey Cushing in 1929 (114). Before this, craniopharyngioma was referred to as interpeduncular cyst, dysontogenetic cyst, craniobuccal cyst, Rathke’s pouch tumor, hypophyseal duct tumor, Erdheim tumor, craniopharyngeal duct tumor, and adamantinomas (82; 11). Of note, it was through the study of craniopharyngiomas that Percival Bailey demonstrated the hypothalamic origin of diabetes insipidus (122).
Most craniopharyngiomas are suprasellar in localization. Therefore, clinical findings are similar to those seen in pituitary tumors. In the original registry of Harvey Cushing, the majority of patients presented to medical attention due to (1) visual disturbance with loss of visual acuity and field cuts (80%); (2) increased intracranial pressure (66%); (3) endocrine deficiency (67%); or (4) hypothalamic dysfunction (38%) (114). A systematic review that included 84 studies in children found visual impairment at diagnosis in 50%, with decreased visual acuity being the most common in 41%, followed by visual field defects in 38% (107).
Although the classic visual dysfunction is bitemporal hemianopia from the involvement of the optic chiasm, a broad range of abnormalities, such as photophobia, diplopia due to sixth nerve paresis, strabismus, dimming of vision, loss of acuity, relative afferent pupillary defect, color desaturation, homonymous defect, and oculomotor dysfunction, have been described (66; 23). Thorough examination in children is necessary because they will likely underreport visual dysfunction.
Rostral extension of the tumor with compression of the third ventricle may lead to increased intracranial pressure, hydrocephalus, and hypothalamic dysfunction. Manifestations of hypothalamic involvement include weight gain from hyperphagia (146) and weight loss from hypothalamic anorexia (65). Behavioral problems, cognitive memory deficits, and personality changes may also suggest encroachment into the hypothalamus. In school-aged children, deterioration in academic performance is an early indication. There are also disabilities that are unseen such as issues with emotional control, altered quality of life, as well as social discrimination (30).
Endocrine dysfunction of the hypothalamic-pituitary axis is a prominent presenting symptom in patients with craniopharyngioma. Panhypopituitarism is a frequent manifestation. Patients can develop a deficiency of growth hormone (75%), gonadotropins (93%), adrenocorticotropic hormone (25%), thyroid stimulating hormone (25%), and luteinizing hormone/follicle-stimulating hormone (40%) (102). This leads to short stature, failed sexual development in children, and decreased libido and impotence in adults. Precocious puberty in children and amenorrhea in premenopausal women have been observed. Symptoms of polyuria and polydipsia often herald the development of diabetes insipidus (102). Rarely, children may present with signs or symptoms of the syndrome of inappropriate secretion of antidiuretic hormone (45; 108). Melatonin deficiency and disruption of circadian rhythm have been described in survivors of this disease, presumably from hypothalamic dysfunction (83; 146). Similarly, obesity is a complication of hypothalamic involvement or due to treatment-related hypothalamic damage (102).
Additional clinical manifestations have been described for the occasional craniopharyngiomas that form ectopically in the pharynx, optic nerve, or sphenoid bone. Further, the rare rupture of a cystic tumor may result in the development of aseptic meningitis (26).
A multicenter study of 280 patients with craniopharyngioma followed from 1966 to 2000 revealed 20-year overall survival in 0.88 +/- 0.03 and 20-year progression-free survival of 0.58 +/- 0.05 (143). Interestingly, there was no observed difference in overall and progression-free survival between patients who underwent gross total and incomplete surgical resections. However, hypothalamic involvement was significantly correlated with decreased 20-year overall survival (0.95 vs. 0.84).
Favorable prognosis is associated with smaller tumor diameter (less than 2.5 cm) and the presence of calcifications, which are suggestive of a slower growing process (118; 29; 158). Higher perioperative deaths are associated with adult adamantinomatous tumors (01).
Despite being histologically benign tumors, craniopharyngiomas have a high risk of recurrence. A study in 59 children showed tumor growth in 8.5% at a median follow-up of 44 months; however, another in 64 adults had actuarial 5- and 7-year progression-free survival rates of 71% and 63% (88; 113). In another study, 76% of recurrences/progression occurred in the role of first 2 years (range 0.25-9 years) with the majority of cases being asymptomatic at the time of detected radiological progression (39). The risk of recurrence appears to be related to an inflammatory microenvironment in the primary tumor. An abundance of M2 macrophages and higher levels of inflammatory cytokines, mainly IL-6 and IL-8, have been associated with recurrence (81; 117). A systematic review found cystic lesions and whorl-like arrays to be associated with increased recurrence (28). The role of Ki-67 as a predictor of recurrence and hydrocephalus on mortality remains unclear due to conflicting study results (102; 100). Annexin A2 is a potential biomarker for the progression and recurrence of adamantinomatous craniopharyngiomas (152). Rarely, recurrences may present distally, with a few cases of spinal recurrence reported (87).
Complications arising from the natural history of the disease and those resulting from treatment regimens are discussed under appropriate sections. Hypothalamic obesity leading to metabolic syndrome and subsequent development of nonalcoholic steatohepatitis and liver cirrhosis in some cases (102). Pituitary hormone deficiencies (98%), visual disturbances (75%), and obesity (56%) are the most commonly associated long-term complications of craniopharyngiomas (155; 93). However, at a mean follow-up of 7 years, 84% of adults and 74% of children with hypothalamic dysfunction treated with surgery were living independently and working professionals (149).
Malignant transformation of craniopharyngiomas and de novo malignant craniopharyngiomas are rare and portend a poor prognosis (151). One meta-analysis of 23 cases found that transformation can occur years following initial diagnosis and is not associated with radiotherapy (139). Transformation to squamous cell carcinoma is the most common type encountered, followed by ameloblastic or odontogenic ghost cell carcinomas (96).
Case 1. A 14-year-old boy was referred to the neurosurgical service for an opinion concerning an MRI demonstrating a suprasellar lesion. He initially presented to his primary physician for headache and nausea. In consideration of his family history, a provisional diagnosis of migraine was given. The boy’s parents became more concerned with the development of poor performance at school and the progressive nature of the headache. He was subsequently referred to a psychologist and eventually to a neurologist. The examining neurologist found papilledema and requested a brain MRI. The imaging study revealed a suprasellar mass composed of solid and cystic components with patchy enhancement. An open craniotomy with a bifrontal approach was performed for gross total resection. Postoperatively, the patient developed diabetes insipidus and partial hypopituitarism but experienced relief from headache and nausea. Surgery was followed by conformal external-beam radiation therapy delivered as 1.8 Gy fractions to a total dose of 54 Gy. He remains disease-free at 5 years following initial treatment.
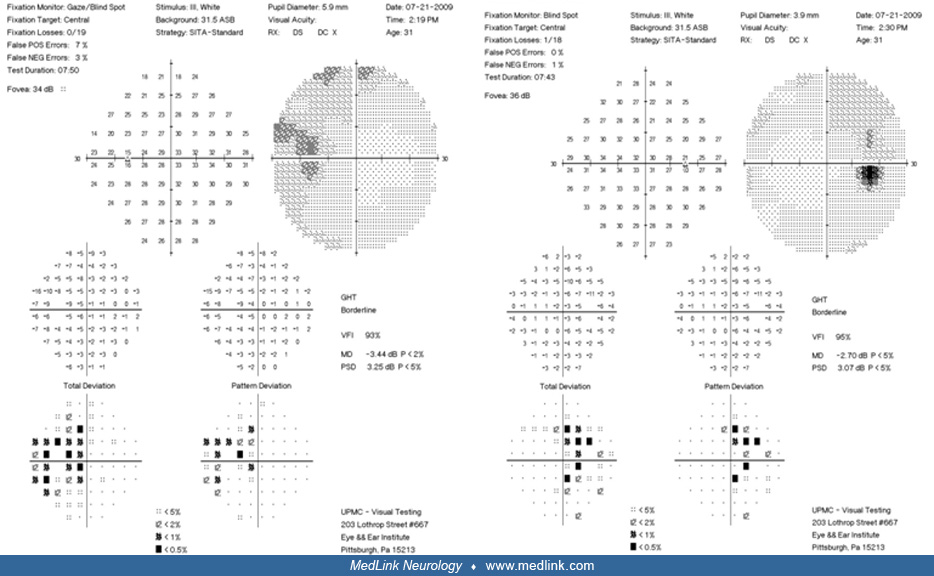
Case 2. A 31-year-old man presented with peripheral vision loss. Physical examination showed bitemporal hemianopia.
The patient had radiographic demonstration of a large sellar mass consistent with a solid and cystic craniopharyngioma.
The patient was offered surgical resection. After the risks and benefits of surgery versus conservative management were discussed, the patient elected to proceed with surgery. An endoscopic endonasal transplanum approach was used throughout resection of the tumor; the patient tolerated the procedure well. Postoperatively, he had one seizure, which was controlled, and he was started on antiseizure medication. The patient's neurologic examination on the day of discharge showed that he was alert, oriented to person, place, time, and situation, and able to follow commands in all four extremities. Cranial nerves III through XII were grossly intact. Although he had residual visual field deficits, as indicated by his formal visual field testing, his vision improved. Postoperatively, his pituitary function was intact with normal diuresis. A postoperative MRI was performed, demonstrating complete resection of the tumor with preservation of the pituitary stalk.
Visual field tests were repeated 4 months later, showing almost complete visual deficit resolution, and a complete pituitary panel proved normal pituitary function.
During early development, the primitive brain and the heart are separated by stomodeum, an ectoderm-lined structure that gives rise to the anterior lobe of the pituitary gland and the ectoderm-derived portion of the mouth. By the third week of development, Rathke’s pouch appears as a dorsal evagination of the stomodeum, creating the “craniopharyngeal” duct. The term “pharyngeal” is a misnomer because the endoderm-derived pharyngeal component of the mouth is located posterior to the buccopharyngeal membrane. The cells in the anterior wall of the pouch actively expand in number to form the adenohypophysis (anterior pituitary). The neurohypophysis is established from the caudal extension of the diencephalon, the infundibulum. Continuously proliferating cells in the anterior wall of the pouch extend to surround the stalk of the neurohypophysis to form the pars tuberalis. It is believed that the Rathke’s pouch may persist for unclear developmental reasons and transform into a craniopharyngioma. Observation of a congenital craniopharyngioma demonstrating ameloblastic and adenohypophyseal tissue specification supports a common tissue origin (156).
Although the tissue of origin is believed to be a component of the Rathke’s pouch and the craniopharyngeal duct, the exact cell of origin is unclear. The two histological subtypes of craniopharyngioma, adamantinomatous and papillary, are believed to represent products of distinct development, although the identification of a tumor demonstrating coexistence of both subtypes further complicates the cell of origin issue (102).
The adamantinomatous craniopharyngioma is a calcified multicystic tumor that may derive from teeth primordia.
Such origin is supported by the expression of enamel proteins and LEF1 (transcription factor critical for tooth development), indicating odontogenic epithelial differentiation (136; 07). Regions of calcification in adamantinomatous craniopharyngioma may be driven by expression of osteogenic factors such as BMP2 released through autocrine or paracrine mechanisms (141). Induction by BMP2 has been shown to differentiate adamantinomatous craniopharyngioma into an osteoblastic lineage (157). Molecular profiling has demonstrated mutation and nuclear-cytosolic expression of β-catenin in adamantinomatous craniopharyngiomas (135; 18; 17). The β-catenin protein normally functions in cell-cell adhesion, but “free” cytosolic form is involved in the Wnt signaling pathway, a cascade implicated in a variety of cancers. Mutations in the gene coding for β-catenin, CTNNB1, have been found in adamantinomatous craniopharyngiomas, and reportedly with highly sensitive sequencing can be found in 100% of cases (08). This observation confirmed the suspected role of Wnt/β-catenin signaling pathway in the pathogenesis of adamantinomatous craniopharyngioma. Animal studies show that only undifferentiated pituitary embryonic precursors and stem cells are tumorigenic when subjected to mutant β-catenin (89). The mutant stem cells, in turn, generate bulk tumors through interaction with neighboring cells in a paracrine manner, implying a noncell autonomous function of pituitary stem cells in the pathogenesis of adamantinomatous craniopharyngioma. Activation of the wild-type epidermal growth factor receptor (EGFR) pathway may play a role in tumor cell migration (57; 152). Histopathologic studies reveal cords of stratified squamous epithelium with intervening “wet keratin” and areas of calcification. Cystic regions containing cholesterol and debris may show yellow-green “machine oil-like” appearance. The adamantinomatous variant exhibits a lower expression of the tight junction protein claudin-1 that correlates with an invasive behavior compared to the papillary type and Rathke’s cleft cyst (142). Interestingly, 10 cases of craniopharyngiomas associated with familial adenomatous polyposis (which also results in Wnt pathway activation) have been described, with six of them presenting in the cerebellopontine angle and not in the sellar region (115). Genetic mutations of PBRM1 and BAP1 have been implicated in the malignant transformation (144).
In contrast, the papillary craniopharyngiomas do not show nuclear β-catenin, and mutations in CTNNB1 are not found in this tumor subtype.
On routine microscopy, papillary craniopharyngiomas have well-differentiated monomorphic squamous epithelium, lacking surface maturation. There is an absence of calcification and wet keratin. Genotyping in papillary craniopharyngioma revealed BRAF V600E substitution in 95% of tumor specimens, which leads to activation of the MAPK/ERK pathway (14). The CTNNB1 and BRAF mutations in adamantinomatous and papillary craniopharyngiomas, respectively, are clonal and mutually exclusive (46). Therefore, it is now established that these tumor subtypes are distinct entities with exclusive molecular underpinnings. Further evidence of this distinction is demonstrated by a study that analyzed whole genome, RNA sequencing and proteomic and phosphoproteomic profiles in 199 children with seven histologic types of brain tumors (119). In children with craniopharyngioma, their profiles were split by the by presence of BRAF V600E mutation into two subgroups, where each subgroup clustered with low-grade gliomas with matching BRAF mutations. This not only confirms the distinct molecular signature of both types of craniopharyngiomas but also highlights shared molecular alterations across disparate histology that may have treatment implications (119).
There are two theories proposed to explain the pathogenesis of craniopharyngiomas. The embryonic theory suggests that craniopharyngiomas originate from the neoplastic transformation of ectopic remnants of craniopharyngeal duct and Rathke’s pouch. The metaplastic theory proposes that craniopharyngioma results from squamous epithelial metaplasia of the pituitary (102). The majority of adamantinomatous craniopharyngiomas are developmental tumors. The pathogenesis of adamantinomatous craniopharyngioma is directly related to the CTNNB1 gene and overactivation of WNT/β-catenin pathway. However, it remains to be discovered why the tumorigenic potential of mutant β-catenin is restricted only to Rathke’s pouch embryonic precursors and adult pituitary stem cells (90). The pathogenesis of papillary craniopharyngioma may involve the metaplastic transformation of anterior pituitary epithelial cells (102).
Craniopharyngioma is a rare intracranial neoplasm with an annual incidence rate of 0.16 per 100,000 persons in the United States (97). It constitutes 4% of all brain and CNS tumors in children (ages 0-14) and 2.5% in adolescents (ages 15-19) (110) and in the United States, they are the second most common pituitary tumor (18.1%) after pituitary adenomas (77.9%) (21). For unclear reasons, they are relatively common in Nigeria and Japan (63; 70; 85).
Craniopharyngiomas show a bimodal age distribution with peaks during childhood (5 to 9 years of age) and at physical maturity (55 to 69 years of age) with an increased risk of recurrence in adult-onset craniopharyngioma (163; 97). Adamantinomatous craniopharyngiomas are the most common subtype (104). Papillary craniopharyngiomas are found predominantly in adults between the ages of 40 to 53 (104). The highest incidence is noted on African American patients, whereas American Indians and Alaska natives are the least affected (97). Rare cases have been reported in neonatal and fetal patients (25). Papillary craniopharyngiomas only represent 5.5% of craniopharyngiomas in children, whereas in adults, they are about 30% (97). Both sexes are equally affected (114; 97).
There are no known risk factors that predispose development of craniopharyngiomas. Therefore, there are no preventive measures.
Numerous pathologic entities in the suprasellar region can cause visual, endocrinologic-hypothalamic, and increased intracranial pressure symptoms and signs. Imaging studies such as MR and CT with consideration of the patient’s age can be helpful. In general, the differential diagnoses to consider are: pituitary tumor, hypothalamic glioma, hypothalamic hamartoma, clival chordoma, Rathke’s cleft cyst, meningioma, epidermoid and dermoid tumors, optic pathway glioma, teratoma, germ cell tumor, epidermoid tumor, chordoid glioma, xanthogranuloma, thrombosis of arachnoid cyst, giant suprasellar carotid aneurysm, lymphocytic hypophysitis, and Langerhans cell histiocytosis, solid tumor metastasis, and cholesterol granuloma.
Any combination of symptoms of headache, visual disturbance, growth failure, polyuria, or polydipsia should raise suspicion of a tumor of the sellar region. Cranial imaging studies with CT and MRI are the most sensitive preoperative diagnostic tools. Identifying calcified areas is important for surgical planning and is easily done on CT as compared to MRI. Calcification in the suprasellar region is present in 80% to 90% of patients, whereas 75% have at least one intratumoral cyst. The presence of two of these three features--namely suprasellar location, presence of calcification, or cystic nature--is strongly suggestive of craniopharyngioma on CT. Based on these features and the distinct appearance of craniopharyngioma, a fairly correct preoperative diagnosis can usually be made on imaging alone. However, when atypical features (such as hemorrhage and massive extension) are present, they may be difficult to distinguish from pituitary tumors. MRI is helpful in delineating tumor extent and tissue heterogeneity (eg, solid versus cystic). The greater tissue resolution of MRI also provides valuable information about the relationship of the tumor to surrounding structures and information concerning displacement or invasion of surrounding regions (68). The solid and cystic membrane portions of craniopharyngioma appear isointense in T1-weighted portion of MRI. On postcontrast CT and MRI, the solid portion and the cyst walls of craniopharyngioma show contrast enhancement. A particularly useful sign on MRI that may suggest craniopharyngioma is edema along the optic tracts--the ‘‘moustache sign” (54; 105). Specific textures in both T1 and T2, as well as the use of 3D T2-FLAIR with contrast sequences, may help differentiate craniopharyngiomas from pituitary adenomas and Rathke cleft cysts (10; 165). Moreover, there is growing interest in developing prediction models that can differentiate papillary and adamantinomatous craniopharyngiomas, as neoadjuvant therapies may become an option in the future, and both clinical and artificial intelligence models have been reported (41; 124).
The study of craniopharyngiomas by proton MR spectroscopy has revealed prominent lipid peaks correlating to the high levels of cholesterol present in the cysts (137). Combining traditional MRI with MR spectroscopy may be a potential avenue to improve the preoperative diagnosis of craniopharyngioma. A study of 23 parasellar lesions demonstrated 100% sensitivity and 94% specificity for craniopharyngioma when a combination of MRI and MR spectroscopy was used (36). The heterogeneous appearance on MRI with proteinaceous signal characteristics is also helpful. One report suggests that fluorodeoxyglucose (FDG) positron emission tomography may be helpful in the diagnosis and subsequent detection of recurrence (106). Another report found that carbon-11 methionine PET prior to proton therapy had significantly higher intra-tumor uptake in comparison to background white matter, providing an advantage over FDG PET in the identification of active craniopharyngioma (73).
The intraoperative distinction between craniopharyngiomas and other suprasellar tumors, such as cystic pituitary adenomas, may be facilitated by cytologic examination (138). Cytologic crush preparations reveal the presence of clusters of cuboidal cells with a palisading appearance and squamous cells with intervening calcification, psammoma bodies, and cholesterol crystals (31).
Before surgery, all patients should have thorough endocrinologic and ophthalmologic evaluations. Preoperative MRI should be obtained to delineate the extent of the tumor and CT to look for calcification. Children in particular may benefit from baseline neuropsychological testing. Endocrinologic studies should consist of complete chemistry, growth hormone level, luteinizing and follicle-stimulating hormone levels, serum and urine osmolality, diurnal serum cortisone levels, and thyroid function tests. Such measurements will be critical in the assessment of operative morbidity. Ophthalmologic evaluation should include fundoscopy, visual acuity, and visual field testing.
BRAF V600E mutation detection is a potential molecular marker that can be used to differentiate papillary craniopharyngiomas from Rathke cleft cysts (91) or Rathke cleft cyst with squamous metaplasia, which has the potential to transform to ciliated craniopharyngioma (161). Approximately 95% of papillary craniopharyngiomas carry the V600E activating mutation of the BRAF gene (B-Raf proto-oncogene) (91). A CTNNB1 mutation indicates an adamantinomatous craniopharyngioma (58).
The optimal management plan for patients with craniopharyngiomas should be customized based on the clinical features. Gross total resection is associated with a decreased recurrence rate and increased survival. Gross total resection is recommended in cases without optic pathway or significant hypothalamic involvement (68; 120; 121).
If a gross total resection cannot be performed safely, consensus guidelines by the European Association of Neurosurgical Societies (EANS) recommend subtotal removal of the tumor followed by radiation therapy (27). This is particularly relevant for those cases with the potential for injury of the pituitary stalk, which is most common on tumors that arise from the hypothalamic stalk and when the hypothalamus is located on the middle third of the tumor (123; 160). The postoperative endocrinological outcome depends on tumor invasion into the center of the pituitary stalk (33).
Surgery. Consensus regarding neurosurgical treatment of craniopharyngioma remains controversial. Surgical resection remains the first treatment option in managing craniopharyngioma. However, the view on the optimal extent of surgical resection has changed over the decades due to a greater understanding of the effects of hypothalamic damage and the fact that no current effective treatment of hypothalamic syndromes is available. Patients presenting with hydrocephalus or chiasmatic compression necessitate emergent surgery (125). Ventriculoperitoneal shunts are not recommended due to malfunction risk. In addition, potential overdrainage can preclude a future transcallosal surgical approach (103). The transcallosal approach allows management of the intra-third ventricle portions while avoiding hypothalamic injury (120). Resection is complicated by calcification and adherence of the tumor to nearby neurovascular structures, such as the optic chiasm-tract, pituitary, hypothalamus, and the vessels of the circle of Willis. Possible adverse effects of surgery on children can affect academic performance, personality development, eating disorders, as well as morbid obesity (51; 132). Hypothalamic syndrome is responsible for these complications, and it affects the growing child more than an adult who has already acquired a certain level of sociability, academic achievement, and growth (30). Up to 50% mortality has been observed following resection (103). Therefore, surgeons are moving away from aggressive gross total resection with acceptance of treatment-related morbidity towards conservative resection that can be safely performed.
The optimal surgical approach depends on the location of the tumor, which can be classified as inferomedial (sellar), superomedial (suprasellar), lateral, posterior (prepontine/interpeduncular), or intraventricular (04). In general, there are two strategies to approach these tumors: transcranial and endonasal. As the tumor is often located centrally between the carotid and optic nerves, below the chiasm and under the hypothalamus, all of these structures must be transgressed to reach the lesion. To overcome this, an expanded endonasal approach, where an endoscope is placed through the nose to directly access the tumor from the midline, has become the favored approach in the last 15 years (03). The technique uses the nose and paranasal sinuses as a corridor to the tumor, leaving neurovascular structures intact and obviating the need for an incision. A systematic review of the technique found it to be comparable and better in some instances to traditional approaches and consensus guidelines by the EANS currently recommend it for tumors in adults that are midline as it leads to improved gross total resection as well as better endocrine and visual outcomes (27; 140). Although in children there are additional concerns with this technique given the smaller skull size, multiple case series and a metanalysis that included 554 cases have found expanded endonasal approach to be safe and effective (76). Though CSF leaks were initially a common complication from this approach, using a pedicled nasoseptal flap reconstruction has been shown to reduce their occurrence in adults and children (49; 72).
Transcranial approaches (including pterional approach from a frontotemporal craniotomy, bifrontal or subfrontal approach for tumors with rostral extension, supraorbital) involve coming above and require varying degrees of bone removal, brain retraction, and neurovascular manipulation. This approach is most useful for tumors that extend laterally or intraventricularly. A supraorbital trans-eyebrow keyhole approach is minimally invasive and has been shown success in resection of craniopharyngiomas located in the third ventricle or suprasellar region (20). Alternatively, a bifrontal interhemispheric approach can be used by drilling directly into the tuberculum sellae to improve visualization (60). A subfrontal approach is optimal for accessing concealed retrochiasmatic tumor and management of hydrocephalus (120).
Gross total resection is recommended whenever feasible. Recurrence following gross total resection is 0% to 50%, which is significantly lower than recurrence following incomplete resection (25% to 100% after 10 years) (99). However, recurrence has been associated with an increased number of surgeries (163).
Conventional external beam radiation therapy. In cases where complete resection cannot be attained, partial removal of the tumor followed by radiation therapy can prolong the time to recurrence. The postoperative surgical cavity as well as the remaining cysts should be treated. The standard dose is daily fractions of 180 cGy to a total dose of 5400 cGy. Higher dose may increase the risk of injury to the optic apparatus. In young children (younger than 5 years of age), radiation therapy to minimize long-term cognitive deficits may be delayed if close clinical observation is possible. However, the decision must be carefully balanced with the heightened risk for recurrence and the potential need to deliver radiation to a larger tissue volume. Radiation is associated with neurocognitive impairment in young children, the risk of stroke, and the higher risk of radiation-induced malignancy at younger age. Several retrospective case series have noted excellent outcomes with 10- and 20-year progression-free survival of 77% to 89% and 54% to 66%, respectively (127; 150; 116). Yang and colleagues compared gross total resection to subtotal resection plus irradiation and found comparable 5- and 10-year overall survival (159).
Current literature has not solved the debate over the radiation treatment sequence. It is unclear whether planned radiation following subtotal resection is superior to subtotal resection, followed by salvage radiation at time of recurrence. A retrospective case series showed that children who were treated with surgery and salvage radiation at the time of recurrence had a less pronounced decline in full-scale IQ scores as compared to children treated with limited resection followed by radiation therapy, suggesting that a second intervention after relapse had a prominent negative effect on neurocognitive function (95).
Stereotactic radiosurgery. Fractionated stereotactic radiotherapy is a technique that combines the accurate focal delivery of stereotactic radiosurgery with the radiobiological benefits of reassortment and reoxygenation that occur with fractionation. This modality allows optimal sparing of normal tissue surrounding the tumor with the advantage of a reduced safety margin as compared to conventional radiation. Patients with craniopharyngiomas treated with fractionated stereotactic radiotherapy showed a local control rate of 92% to 100% after 10 years and 88% after 20 years. Overall survival at 10 years was 82%, and after 20 years was 67% (52; 148). Limited results in several case series have been promising, with overall treatment being tolerated well with no cases of radionecrosis or secondary malignancy reported, although this may be limited due to the duration of follow-up.
Small, preferably solid lesions that are spatially removed from the optic chiasm-tract may be amenable to treatment by stereotactic radiosurgery using gamma knife. A report described the use of gamma knife radiosurgery in 100 consecutive patients (67). The mean volume of treated tumor was 3.5 cc with a dose of 11.5 Gy. Complete response was seen in 19% with a partial response of 67%. An experience of 137 patients treated with 162 sessions of Gamma Knife surgery with a median radiological follow-up period of 46 months found tumor control rates of 73%, 74%, and 66% for solid, cystic, and mixed tumor types (75). Five- and 10-year overall survival rates were reported to be 91% and 84%, respectively.
Proton beam radiotherapy can provide a superior dose distribution and target conformation as compared with standard radiation therapy due to the rapid dose falloff known as the Bragg peak, reducing collateral radiation to normal tissue (103). Also, the risk of secondary malignancy may be reduced. However, altering tumor cystic components and rapid patient weight loss or gain can lead to fluctuating target size and shape, affecting tissue path length and protein sensitivity; thus, interval CT scans midway through therapy may be helpful (103; 98). A series of 14 adults treated with proton therapy for either de novo disease or recurrence showed 3-year local control in 100% of patients (130). Survival, disease-control outcomes, and toxicities were equivalent for proton beam therapy as compared to intensity-modulated radiation therapy (12). A series of 15 patients showed an actuarial 10-year survival rate of 72% (37). Limited evidence supports the superiority or inferiority of proton therapy compared to conventional photon therapy in craniopharyngiomas. A systematic review on clinical outcomes in craniopharyngiomas treated with proton therapy showed very low level evidence that proton therapy does not result in significant differences in 3-year overall survival (78). Among different modalities of proton beam therapy, a study of 10 patients showed reduced irradiation to the temporal lobes when using pencil beam scanning compared to volumetric modulated arc therapy and double scattering therapy (147). The results translated to better-predicted memory outcomes, though changes in estimated IQ did not reveal differences. A prospective study in a cohort of 94 patients with a median age of approximately 9 years showed improvement in cognitive outcomes after proton therapy, as compared to photon therapy (94).
Brachytherapy. Damage to the cyst wall epithelium by intracavitary delivery of radionuclides can help limit the production of cyst fluid. Preferred agents are all β-particle emitting isotopes such as yttrium-90, rhenium-186, gold-198, and phosphorus-32 because of limited penetration into the surrounding brain (19; 102). Brachytherapy may effectively manage cystic fluid production, but the exact calculation of an appropriate dose is often unclear. In the United States, only phosphorus-32 is available. A retrospective review of pediatric patients treated with phosphorus-32 brachytherapy after surgery determined that brachytherapy alone would not prevent the need for future intervention (06). Brachytherapy may be considered as an alternative treatment option only for recurrent monocystic craniopharyngioma. Complications such as hemorrhage, infection, damage to the visual pathway, and neurologic damage due to leakage of radioisotope may occur. Therefore, this option should be considered only after postoperative recurrence and conventional irradiation has been tried.
Chemotherapy. Systemic chemotherapy has no proven benefit in the treatment of craniopharyngioma (164). Intracavitary administration of bleomycin may reduce the size of the cyst and permit more thorough surgical resection by decreasing adherence to adjacent brain tissue (101; 62). However, such procedures should be performed by experienced clinicians because of potential catastrophic complications resulting from leakage of the toxic chemotherapeutic agent into vital regions of the brain (71). Potential use of an implantable carmustine wafer was reported in one patient, but no follow-up studies are available (74). Craniopharyngioma cells exposed to all-trans retinoic acid showed growth inhibition, apoptosis, and attenuation of the NF-kB signaling pathway (80).
Cystic childhood craniopharyngiomas may be treated by intracystic interferon-α instillation, as it does not have the neurotoxicity associated with bleomycin. Interferons exert antiproliferative, cytotoxic, and maturational effects and are generally tolerable with no significant side effects that would require treatment discontinuation (22). Patients achieved 60% to 98% reduction in cyst volume at 6-months after the last treatment cycle (32). The use of intradermally administered pegylated interferon has also been described in two phase 2 clinical trials (154; 44). And although without overt evidence of radiographic response, progression-free survival (7.8 and 19.5 months) in the setting of therapeutic tolerability is encouraging when compared to other existing systemic therapeutic options.
Targeted therapies. The identification of inappropriate activation of Wnt/β-catenin and BRAF signaling pathways may allow for treatment with highly selective targeted inhibitors.
With the establishment of BRAF V600E substitution in papillary craniopharyngiomas, effectiveness of combining BRAF-MEK inhibition has been established (15). Of the 16 newly diagnosed subjects with papillary craniopharyngioma, 15 demonstrated partial response or better to the combined inhibition.
Other promising targets include tocilizumab for the blockade of the interleukin-6 receptor; it has been shown to be expressed in high levels in craniopharyngiomas. Two reported cases showed response a primarily in the cystic components (48). Craniopharyngiomas also appear to express receptors for growth hormone, progesterone, and estrogen (56). Further, it has been reported that tamoxifen may inhibit craniopharyngioma growth in vitro (79). However, a meta-analysis did not find craniopharyngioma occurrence to be associated with growth hormone supplementation (05). If EGFR signaling indeed promotes in vivo migration of craniopharyngioma cells, small molecule inhibitors of EGFR may become an option (57). The B7 family ligand pathway, which includes CTLA-4/B7-1/B7-2 and PD-1/PD-L1, may also be a promising target as high expression of B7 proteins has been described in both adamantinomatous and papillary craniopharyngiomas (24; 153).
Several decades ago, the treatment of craniopharyngioma was based on the paradigm of aggressive resection strategies, even at the expense of postoperative morbidity. We now know that patients with craniopharyngiomas can have a high 20-year survival rate of 87% to 95%. Therefore, minimizing treatment-related morbidity is a priority. There is a delicate balance between the need to achieve complete resection and inflicting iatrogenic morbidity. Complete surgical resection should be performed when the tumor does not involve hypothalamus or optic structures. When the tumor is enmeshed with optic pathway or hypothalamus, it is unclear if complete surgical resection should be attempted. This is because despite macroscopic total resection, recurrence rates vary between 5% and 50% and there is a greater risk of injury to vital structures (62; 134). Surgical complications range from 0% to 15%. This underscores the importance of thorough preoperative planning to include postoperative radiation therapy in cases where complete resection is not feasible. Pituitary stalk preservation reduces postoperative endocrinopathy (diabetes insipidus and anterior pituitary insufficiency) by approximately 50% (109).
Advances in surgical technique, including expanded endonasal approach, as well as in radiotherapy have led to remarkable changes in the outcome of craniopharyngioma. One study found that although only 4% of patients between 1960 and 1984 had a total resection, 43% of patients did between 1985 and 2017 (38). This was accompanied by a reduction in diabetes insipidus and panhypopituitarism rates from 86% to 53%. Of note, patients treated at high-volume centers have better outcomes than those treated elsewhere (84).
Established risk factors for mortality include female gender, childhood-onset, tumor recurrence, hydrocephalus, higher BMI, and panhypopituitarism (126). Surgical complications depend on the particular approach and extent of resection into adjacent brain, with up to one third of patients with craniopharnygiomas who undergo craniotomy developing a complication (128). Development of a glioblastoma 15 years after delivery of radiotherapy has been observed (162).
Patients with craniopharyngiomas suffer long term morbidity due to endocrine, hypothalamic, visual, cognitive, and cardiovascular factors. Cardiovascular disease can occur as a result of the metabolic profile of these patients, which is thought to be induced by pituitary hormone insufficiencies as well as hypothalamic damage (133). The risk of developing cardiovascular disease in adults was found to be 3 to 19 times greater than that of the general population, and these cardiovascular risk factors must be managed (35). One study found vision was improved in 46% of patients after surgery, remained stable in 44%, and deteriorated in 10% (69). In a single-center study, 100% of childhood onset craniopharyngiomas and 93% of adult onset had hormone deficiencies at median follow-up of 12.8 years (64). Patients who undergo gross total resection have a lower likelihood of recurrence but with an increased rate of long-term endocrine deficits as compared to patients who have subtotal resection and subtotal resection with radiation therapy (134; 02; 47). Though some series report up to 83% patients developing panhypopituitarism and 73% diabetes insipidus, others describe lower rates of transient (40%) and permanent (20%) panhypopituitarism when using an endonasal surgical approach (16; 131; 77; 92). A 40-year metadata analysis of 185 patients showed that a shift to more conservative surgery reduced the prevalence of hormone deficiencies. Unfortunately, it was not associated with reduced hypothalamic and visual morbidities (145).
Hypothalamic damage may present as hypothermia, somnolence, or loss or circadian rhythm. In one study, 80% of patients after surgical resection had hypersomnolence and 35% had narcolepsy. Patients who were overweight or obese, as well as those with grade 2 hypothalamic involvement at diagnosis, were more likely to be diagnosed with narcolepsy (86). Hypothalamic obesity is now a well-known morbidity in patients with craniopharyngiomas, which impairs quality of life in survivors. Up to 80% of childhood-onset and 68% of adult-onset patients will have metabolic syndrome at follow-up and novel predictive scoring systems have been developed to detect patients at risk (64; 40). Weight gain results from damage to the ventromedial hypothalamus, and rates of obesity and short stature are positively correlated with the extent of hypothalamic damage (53; 61). Though hypothalamic obesity occurs despite an adequate replacement of pituitary hormones, early evidence suggests metilphenydate may be helpful (55; 59). GLP-1 analogs have been used in some randomized controlled trials in hypothalamic obesity (129; 42). Also, setmelanotide, a melatonin type 4 receptor agonist, has shown encouraging results in hypothalamic obesity (50). Visual deficits have been recorded in 40% to 48% of patients treated with surgery alone or in combination with radiation therapy. Visual outcome is adversely associated with the presence and duration of visual dysfunction at initial presentation and after radiation therapy (34).
Patients are at an increased cardiac and cerebrovascular risk (163). Pituitary deficiency and its treatment may contribute to enhanced morbidity due to the development of diabetes mellitus and radiotherapy confers an increased risk of developing new intracranial tumors (163).
Long-term neurocognitive complications in craniopharyngioma patients include executive dysfunction, inattention, and poor episodic memory (112). Neurocognitive complications are due to the hypothalamic location of the lesion, mammillary body involvement, as well as treatment-related effects (112). Radiation doses to the left hippocampus greater than 30 Gy have been correlated with a decline in some neurocognitive domains in this patient population (43). Up to 50% of patients have psychosocial impairment on long-term follow-up and up to 25% of patients are unable to work in their previous occupation or are behind expected school status (65; 111). Delayed radiation effects may manifest as cognitive dysfunction and the development of secondary intracranial tumors (125). However, in patients with gross total resection a study of 22 subjects found a quality of life similar to the general population at a median follow-up of 19 years (53).
There is no causal link between pregnancy and the development of craniopharyngioma. It is exceedingly rare for craniopharyngioma to present during pregnancy. However, pregnancy has been linked to symptomatic exacerbation of the underlying disorder (09). Rare cases of acute intratumoral hemorrhage during pregnancy have been reported. In a case series of eight pregnant women who presented with craniopharyngiomas, therapeutic abortion was necessary in two. The remaining six women had delivery between the 33rd and 40th weeks. Symptoms improved in seven of eight women postpartum (166). Such observations, although through largely isolated cases, require close monitoring of patients during pregnancy.
Anesthesia is not contraindicated in patients with craniopharyngiomas, and no special precautions are required. In patients with a history suggestive of diabetes insipidus, particular attention should be devoted to managing fluids and electrolytes. Evaluation of the hypothalamic-pituitary-adrenal axis should be performed to evaluate for subclinical hypocortisolism.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Deric M Park MD FACP
Dr. Park of Hackensack Meridian School of Medicine has no relevant financial relationships to disclose.
See ProfileHeather A Meller APN
Dr. Meller of Hackensack University Medical Center has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuro-Oncology
May. 27, 2026
Neuro-Oncology
May. 27, 2026
Neuro-Oncology
May. 23, 2026
Neuropharmacology & Neurotherapeutics
May. 11, 2026
Stroke & Vascular Disorders
May. 03, 2026
Neuro-Oncology
Apr. 30, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026