



Neuroimmunology
Balo concentric sclerosis
Jul. 02, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Dermatomyositis is one of the five main clinicopathologically distinct inflammatory myopathies, the other four being (1) polymyositis, (based on the historical classification, even if now seemingly extinct), (2) necrotizing autoimmune myositis, (3) overlap myositis and the anti-synthetase syndromes, and (4) inclusion-body myositis (24). Although it may have been recognized earlier by Wagner (135), the first definitive description of dermatomyositis was reported by Unverricht in 1891 (133). Before the extensive use of immunosuppressive drugs, dermatomyositis was the cause of considerable disability and mortality, especially in children. During the last 20 to 30 years, extensive use of these drugs and a better understanding of the immunopathology of the disease have significantly improved the prognosis. Dermatomyositis, a disease affecting skin and muscle, is cared for not only by neurologists but also by dermatologists and, currently, most often by rheumatologists. However, the role of the neurologist remains essential to exclude other neuromuscular diseases associated with skin abnormalities, to substantiate the presence and degree of muscle weakness, to confirm the immunopathology of the disease, and to supervise the immunotherapeutic interventions. In children with dermatomyositis with muscle weakness as the major symptom, the pediatric neurologist is often consulted early in the disease and usually becomes the primary physician caring for these children.
Dermatomyositis occurs in both children and adults. It is a distinct clinical entity identified by a characteristic rash, which accompanies or, more often, precedes the muscle weakness. The skin manifestations include a heliotrope rash (blue-purple discoloration) on the upper eyelids with edema, a flat red rash on the face and upper trunk, and erythema of the knuckles with a raised violaceous scaly eruption (Gottron rash) (27; 75; 50; 38; 22; 39; 24; 89).
The initial erythematous lesions may result in scaling of the skin accompanied by pigmentation and depigmentation, resulting at times in a shiny appearance. In contrast to systemic lupus erythematosus, in which the phalanges are involved and the knuckles are spared, the erythema of dermatomyositis spares the phalanges. The erythematous rash can also occur on other body surfaces, including the knees, elbows, malleoli, neck and anterior chest (often a V sign), or back and shoulders (a shawl sign), and it may be exacerbated after exposure to the sun. Dilated capillary loops at the base of the fingernails are also characteristic of dermatomyositis. The cuticles may be irregular, thickened, and distorted, and the lateral and palmar areas of the fingers may become rough and cracked, with irregular, "dirty" horizontal lines, resembling a mechanic's hands. Dermatomyositis in children resembles the adult disease, except for more frequent extramuscular manifestations, as discussed later. Tiptoe gait due to flexion contracture of the ankles is a common early abnormality in children. Another common early abnormality in children is “misery,” describing an irritable child who feels uncomfortable, has a red flush on the face, is fatigued, does not feel like socializing, and has various degrees of muscle weakness (27; 75; 50; 37; 38; 22; 47; 39; 24). When the weakness develops, it takes the form of a myopathy, with proximal more than distal involvement. The degree of weakness can be mild, moderate, or severe, leading to quadriparesis. Some patients with the classic skin lesions appear to have clinically normal strength even up to 3 to 5 years after onset. This form, referred to as "dermatomyositis sine myositis" or "amyopathic dermatomyositis" (122), has a better overall prognosis. Although in these cases the disease appears limited to the skin, the muscle biopsy shows significant perivascular and perimysial inflammation and immunopathological features identical to those seen in classic dermatomyositis (28), suggesting that amyopathic and myopathic forms are simply variations on the theme of dermatomyositis affecting skin and muscle to various degrees.
Dermatomyositis usually occurs alone, but it may overlap with systemic sclerosis and mixed connective tissue disease, as well as with the “overlap myositis and anti-synthetase syndrome” subset (39). Fasciitis and skin changes, similar to those found in dermatomyositis, have occurred in patients with the eosinophilia-myalgia syndrome associated with the ingestion of contaminated L-tryptophan (94; 27); the eosinophilic fasciitis, and the macrophagic myofasciitis (57).
Extramuscular manifestations. In addition to primary disturbances of skeletal muscles and skin, extramuscular manifestations may be prominent in patients with dermatomyositis. These include (a) dysphagia, similar to that of patients with scleroderma or other forms of myositis (21); (b) cardiac abnormalities consisting of atrioventricular conduction defects, tachyarrhythmia, low ejection fraction, and dilated cardiomyopathy (either due to the disease itself or, more often, to hypertension or fluid retention associated with long-term steroid use); (c) pulmonary involvement, resulting from primary weakness of the thoracic muscles, rare drug-induced pneumonitis (eg, from methotrexate), or, most often, interstitial lung disease. Interstitial lung disease may precede the myopathy or occur early in the course of the disease, especially in patients who have anti-Jo-1 antibodies or antibodies to other synthetases, as discussed later; (d) subcutaneous calcifications, sometimes opening onto the skin and causing ulcerations and infections, especially in children (29; 38); (e) gastrointestinal ulcerations, seen more often in the childhood form, due to vasculitis and infection; (f) contractures of the joints, especially in the childhood form; and (g) general systemic disturbances, such as fever, malaise, weight loss, arthralgia, and Raynaud phenomenon, especially when dermatomyositis is associated with a connective tissue disorder.
The natural history of dermatomyositis is unknown, as most patients nowadays are treated early with steroids and immunosuppressants. The mortality rates reported 20 to 30 years ago are outdated. Clinical experience indicates that dermatomyositis responds to therapy more readily than the other forms of inflammatory myopathies, but in some patients the response still may not be optimal. We have also seen late progression in some patients, including the development of inclusion-body myositis in four patients. The rare patients with acute fulminating disease seem to be more difficult to treat and more resistant to therapies. In children, dermatomyositis may be a monophasic disease with infrequent flares once the disease is under control. Patients with interstitial lung disease may have a high mortality rate, requiring more aggressive treatment. The presence of anti-melanoma differentiation-associated gene 5 (MDA-5) defines a distinct subset with rapidly progressive lung disease and poor survival in patients with amyopathic and myopathic dermatomyositis (97). When dermatomyositis occurs in a setting of malignancy, the mortality can be also high, as dictated by the underlying neoplasm at the time of diagnosis.
The subcutaneous calcifications can be a major problem in dermatomyositis because once formed, they are not easily dissolved and are resistant to all therapies. When they protrude to the skin, they result in ulcerations, infections, and permanent disfiguring scars. The pathogenic mechanism of these calcium deposits is obscure. Fortunately, today we do not see them as often as in the past owing to early initiation of better therapies. Other complications are related to gastrointestinal ulcerations, melena, hematemesis, or even infarctions affecting long stretches of the bowel. Before the use of steroids and other immunosuppressants, these complications were serious, leading to death. Although rare now, they still occur, especially in children.
Overlap myositis or overlap syndromes. "Overlap syndrome" defines a disorder that shares the characteristics of two usually distinct disorders. As such, dermatomyositis is the only disease among the inflammatory myopathies that truly overlaps with two connective tissue disorders, (1) scleroderma and (2) mixed connective tissue disease. For years we knew that specific signs of systemic sclerosis or mixed connective tissue disease such as sclerotic thickening of the dermis, contractures, esophageal dysmotility, and microangiopathy are present only in dermatomyositis but not in other inflammatory myopathies. Conversely, signs of rheumatoid arthritis, systemic lupus erythematosus, or Sjögren syndrome are rare in dermatomyositis.
In the last decade overlap myositis has been recognized as a clinicopathologically distinct entity, separate from dermatomyositis. These patients present with systemic sclerosis-like lesions, mild-to-moderate proximal muscle weakness, arthritis in the form of subluxation of the interphalangeal joints, “mechanic’s hands”, Raynaud phenomenon, and interstitial lung disease. The syndrome is highlighted by antibodies against anti-RNA synthetases, especially Jo-1 (“Jo-1 syndrome”). Histologically, there is necrotizing perimysial and perifascicular myositis with actin myonuclear inclusions. The entity predominantly affects the perimysium (95).
Patients with systemic sclerosis and mixed connective tissue disease may also develop a rare but distinct myositis subtype identified as brachio-cervical inflammatory myopathy, characterized by weakness of proximal muscles of the upper limbs and cervical flexor and extensor muscles (108; 55).
Viruses. Coxsackie viruses, influenza, vaccinations, or even retroviruses have been proposed as possible triggering causes of dermatomyositis. However, no signs indicate that they play a pathogenic role in the disease, and their association has been peripheral and circumstantial. All attempts to isolate viruses, including retroviruses, from the patients' muscle or to amplify viral products by polymerase chain reaction have been unsuccessful (86; 28). The same applies to SARS-CoV-2 (COVID-19) during the pandemic. Although autopsy studies from patients hospitalized with severe pulmonary and systemic complications have shown some nonspecific endomysial inflammatory features, no convincing signs of the SARS-CoV-2 virus have been detected in the muscle tissue, and there is no evidence that SARS-CoV-2 can infect the muscle (40; 42; 21).
Cancer and cancer immunotherapy. The incidence of malignancies is increased in patients with dermatomyositis (12; 66). In a large series cancer was seen in 21% among 349 patients with inflammatory myopathies; 48% of them had dermatomyositis with antibodies to transcriptional intermediary factor 1γ (anti-TIF1-γ); most of the other anti-TIF1-γ-negative patients had necrotizing myositis (65). Ovarian cancer is most frequent, followed by intestinal, breast, lung, and liver cancer. In Asian populations, nasopharyngeal cancer is more common. In general, however, the cancer sites correspond to those occurring more frequently at the patient's age (13). Because tumors are often uncovered at autopsy or on the basis of abnormal findings on medical history and physical examination, blind radiologic searches are rarely fruitful (39; 24). A complete annual physical examination, with breast, pelvic and rectal examinations (including colonoscopy in high-risk patients), urinalysis, complete blood-cell count, blood chemistry tests, and chest x-ray, is usually sufficient and is highly recommended, especially in the first 3 years after diagnosis. PET scan screening for subclinical cancer has not been systemically utilized in patients with dermatomyositis, but it might be considered in high-risk patient groups. Dermatomyositis can be also one of the several neuromuscular complications induced by immune check point inhibitors, such as ipilimumab, pembrolizumab, nivolumab, atezolizumab, avelumab, or durvalumab (41). Dermatomyositis, polymyositis, and especially, necrotizing autoimmune myositis are the most frequent autoimmune myopathies triggered by pembrolizumab, ipilimumab, and nivolumab (117; 74; 41; 71). These agents are directed against cytotoxic T-lymphocyte-associated protein (CTLA)-4, programmed cell death (PD)-1 or PD-L1, to prevent the CTLA-4 or PD-1 from binding to their respective receptors CD80/86 and PDL-1. Blocking these molecules, the “inhibitory” costimulatory interactions between T cells and tumor cells are also inhibited, resulting in positive costimulation and strong cell activation, which allows both the T cells to kill tumor cells and the uncontrolled T cell activation to disrupt immune tolerance like taking the “brakes off” the immune system, causing various inflammatory autoimmune myopathies (41).
Myotoxic drugs. There is no evidence that myotoxic drugs cause dermatomyositis. Some rare old reports that cholesterol lowering drugs and specifically pravastatin, simvastatin, and atorvastatin may be causally linked to a disorder resembling dermatomyositis are unverified or unconvincing (115; 76; 102).
Presence of autoantibodies. Various autoantibodies against nuclear (antinuclear antibodies) and cytoplasmic antigens are variably found in up to 60% of the patients with all the inflammatory myopathies (27; 1992b; 1995b; 2006; 49; 75; 50; 37; 47; 39; 24). Most common among them are the antibodies directed against ribonucleoproteins involved in translation and protein synthesis that include various synthetases and translation factors. The antibody directed against the histidyl-transfer RNA synthetase, called anti-Jo-1, accounts for 75% of all the anti-synthetases and is clinically useful because it is associated with overlap myositis and interstitial lung disease. Although the antibodies are useful markers in highlighting these conditions, referred now as the “anti-synthetase” or “anti-Jo-1” syndrome, they are directed against ubiquitous cytoplasmic antigens and have no pathogenic significance; further, they may occur in patients with interstitial lung disease even in the absence of myositis (52; 64; 38). As already mentioned, the “anti-synthetase” or “anti-Jo-1” syndrome is now a distinct clinicopathologic entity clinically characterized by myositis, interstitial lung disease, arthritis, Raynaud phenomenon, fever, and “mechanic's hands” (72; 78; 130; 112; ).
More specific dermatomyositis-associated antibodies include: a) anti-complex nucleosome remodeling histone deacetylase (Mi-2), a nuclear antigen, which highlights the typical skin lesions and myopathic features but has less evidence of association with malignancy; b) melanoma differentiation—associated protein-5 (MDA-5), which is mostly connected with amyopathic dermatomyositis or interstitial lung disease and, as mentioned earlier, carries poor prognosis and has also been connected with mucocutaneous ulceration, palmar papules, nonscarring alopecia, and panniculitis (24; 89; 97; Tanboon et al 2020; 128; 129); new data in a large series confirmed that anti-MDA5 antibody-positive patients had higher probability of interstitial lung disease and worse prognosis than the anti-MDA5-negative patients; further, the amyopathic patients had higher mortality rates, suggesting the need for earlier and more aggressive treatment strategies (70); c) anti-small ubiquitin-like modifier-activating enzyme (SAE), seen in patients with typical dermatomyositis skin rash; d) transcriptional intermediary factor-1 (TIF-1) and nuclear matrix protein NXP-2, which are highly connected with cancer-associated adult dermatomyositis and the least associated with skin lesion, although their presence is influenced by geographic, racial, or genetic factors; and e) two other autoantibodies, one against 140 kd, the TIF-1α (transcription intermediary factor α), and the other against anti-p155, identified as the transcriptional intermediary factor 1 γ (TIF1γ), have also been described. The TIF-1α, and especially the TIF1γ, seem to be cancer-specific with 89% specificity for cancer-associated dermatomyositis (53; 81; 132; 65). Data using sensitive and specific methodology suggested that antibodies to TIF1γ and the nuclear matrix protein NXP-2 identify 55% of cancer-associated dermatomyositis (51). In a series, in anti-TIF1γ-positive patients with dermatomyositis, cancers were detected within 1 year of the diagnosis of myositis in 97% of the patients (65). Several autoantibodies associated with the anti-TIF1γ not enriched for HLA-DRB1*03, either in isolation or in combination, were also found in patients with juvenile dermatomyositis who had less severe muscle disease; this observation likely suggests that these antibodies lack overall specificity (118).
Immunopathology. The primary antigenic targets in dermatomyositis are components of the endothelium of the endomysial blood vessels. Signs of an ongoing angiopathy in dermatomyositis were first shown by Dr. Betty Banker on the basis of light and electron-microscopic observations (09; 08). Engel and colleagues showed that the earliest pathological alterations in dermatomyositis are changes in the endothelial cells consisting of pale and swollen cytoplasm with microvacuoles (69). Carpenter and Karpati confirmed and expanded these findings by demonstrating active focal destruction of capillaries, with undulating tubules in the smooth endoplasmic reticulum of the endothelial cells, vascular necrosis, and thrombi (15).
The first evidence that such alterations in the microvasculature were caused by an immune-mediated process was reported by Whitaker and Engel, who immunolocalized immune complexes in the endomysial blood vessels (136). Subsequently, Kissel and colleagues showed that the microvascular injury was mediated by the C5b-9 membranolytic attack complex, the lytic component of the complement pathway (77). Emslie-Smith and Engel, using a double-immunolocalization method that employs antibodies to membranolytic attack complex and a lectin (Ulex europaeus) as a specific endothelial marker, confirmed that membranolytic attack complex is deposited on capillaries and that these deposits occur early in the disease and precede signs of inflammation or structural changes in the muscle fibers (48). We confirmed these observations (27; 30) and further investigated the role of the complement (10).
(A) Cross-section of an H&E-stained muscle biopsy demonstrates the classic perifascicular atrophy (layers of atrophic fibers at the periphery of the fascicle) with necrosis and some inflammatory infiltrates (H&E stain ×...
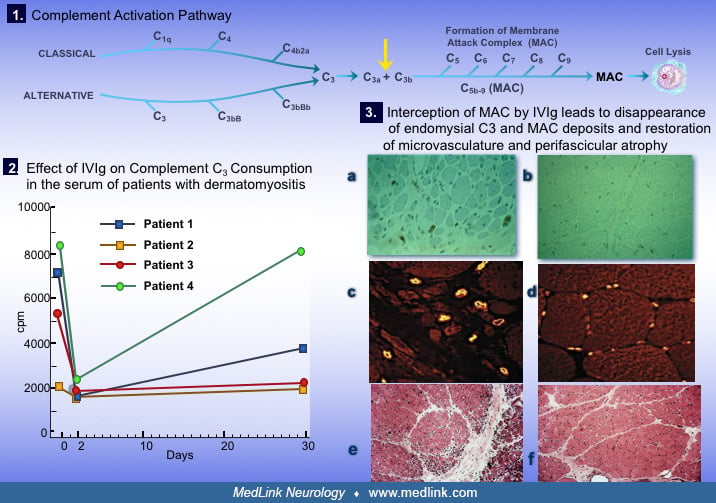
Utilizing an in vitro assay system, we found that the C3 uptake is high in the serum of patients with active dermatomyositis. Further, membranolytic attack complex and the active fragments of the early complement components C3b and C4b can be detected in the serum and appear to correlate with disease activity (10). In addition, the neoantigen C3bNEO, which is complex-specific because it gets exposed when the C3b is incorporated into an immune complex, is deposited on the muscle capillaries along with the membranolytic attack complex (10).
As discussed, the disease begins when putative antibodies or other factors activate complement C3, forming C3b and C4b fragments that lead to formation of C3bNEO and membranolytic attack complex, both of which are deposited in the endomysial microvasculature (29; 32; 33; 38; Dalakas 2011; 21). The membranolytic attack complex deposition on the intramuscular capillaries leads to osmotic lysis of the endothelial cells and necrosis of the capillaries. This results in marked reduction in the number of capillaries per muscle fiber and dilatation of the remaining capillaries in an effort to compensate for impaired perfusion. Larger intramuscular blood vessels are also affected in the same pattern, leading to muscle fiber destruction (often resembling microinfarcts) and inflammation. The perifascicular atrophy often seen in more chronic stages is a reflection of the endofascicular hypoperfusion and subsequent hypoxia that is prominent distally (47; 39; 24).
(A) Cross-section of an H&E-stained muscle biopsy demonstrates the classic perifascicular atrophy (layers of atrophic fibers at the periphery of the fascicle) with necrosis and some inflammatory infiltrates (H&E stain ×...
Data confirm the above by demonstrating that the perifascicular atrophy is consistently associated with focal microvascular depletion involving not only capillaries but also transverse vessels (83). Further, the microvascular C5b-9 membrane attack complex deposits result from activation of the classical complement pathway triggered by direct binding of C1q to injured endothelial cells (83). The finding that IFNy coordinates the local RNA overexpression of complement genes in all types of autoimmune inflammatory myopathies is interesting but of unclear significance because it lacks specificity for dermatomyositis, which is the main complement-mediated myopathy among all the autoimmune inflammatory myopathies (16).
Specific pathogenic autoantibodies against endothelial cells have not yet been identified despite some early efforts (17; 125). The activation of complement induces the release of cytokines, which, in turn, upregulate the expression VCAM-I and ICAM-I on the endothelial cells (124). These molecules serve as ligands for the integrins VLA-4, LFA-I, Mac-I expressed on T cells, and facilitate their exit through the blood vessel wall to the perimysial and endomysial spaces. Various chemokines are also upregulated at the protein and mRNA level (20; 111). Immunophenotypic analysis of the lymphocytic infiltrates demonstrates B cells, CD4+ cells, and plasmacytoid dendritic cells in the perimysial and perivascular regions, supporting the view that a humoral-mediated mechanism plays the major role in the disease (see figure below). In the perifascicular areas, if there are lymphoid infiltrates, they consist mostly of CD8+ cells and macrophages, which invade MHC-I-antigen-expressing muscle fibers. After successful therapy with IVIg, these abnormalities are not any more prominent in the repeat biopsies, as described later.
Gene arrays show that a number of adhesion molecules, cytokines, and chemokine genes are upregulated in the muscles of dermatomyositis patients. Most notable are genes induced by α/β-interferon (60), genes associated with ischemia and degeneration (131), and the KAL-1 adhesion molecule gene (110). The latter appears biologically relevant because it is significantly downregulated in dermatomyositis patients who improved after therapy (110). The KAL-I protein is upregulated in vitro by TGF-β and may have a role in inducing fibrosis. One of the proteins induced by α/β-interferon is the myxovirus resistance MxA protein, which is particularly abundant in the perifascicular regions of dermatomyositis muscles (60). The perifascicular regions of dermatomyositis contain many regenerating or degenerating fibers as they are in a stage of continuous remodeling (22; Dalakas 2011; 39; 24). As a result, they stain with alkaline phosphatase, desmin, neural cell adhesion molecule, with the autoantibody against chromatin remodeler Mi-2 (92), and with a variety of antibodies against immune or stressor molecules, including TGF-β, MHC-I, αB-crystallin, cathepsins, amyloid precursor protein, STAT-1 (triggered by interferon-γ), myxovirus resistance MxA protein (triggered by α/β-interferon), and a B-cell activating factor of the tumor necrosis factor family (BAFF) that plays a crucial role in B-cell survival and maturation (05; 54; 60; 07; 39). Therefore, the perifascicular myofibers display all the markers of inflammation, regeneration, and tissue remodeling (Dalakas 2011; 39). Based on the chronic overproduction of α/β-interferon-inducible proteins such as MxA, innate immunity also plays a role (60). Although some feel that MxA may be a primary event, classic studies have shown that this is a secondary innate immunity molecule and in distinct contrast to the primary process of complement-mediated microvasculopathy, as highlighted above, which: a) precedes all pathologic events, b) explains the noted ischemia and perifascicular atrophy, and c) explains the impressive improvement after complement inhibition by IVIg based on two controlled trials (34). We view the interferon factors as secondary playing a role mainly in enhancing or perpetuating the disease process (39; 24; 34). Sequentially, in dermatomyositis, the complement-mediated ischemic and hypoxic-damage is the primary pathogenic event preceding inflammation or fiber damage; this is sensed by the retinoic acid-inducible gene-1 signaling, which is part of innate immunity receptors, leading to activation of β-interferon, overexpression of MxA, and auto-amplification of perifascicular inflammation (126; 39; 34).
This scenario was also supported by the transcriptional analyses of genes involved in hypoxia and innate immunity; a hypoxia-driven pathology in perifascicular small fibers and by macrophages expressing markers of hypoxia predominated in juvenile dermatomyositis with no role of innate immunity or activation of type-1 interferon (IFN)-associated pathways (109). There was, however, a dichotomy that requires further study with the adult disease where perifascicular atrophy was prominently associated with molecules driving innate immunity, whereas hypoxia-related mechanisms were less relevant (109). Equally controversial is the contention that the sarcoplasmic MxA expression in non-necrotic/regenerating fibers associated with dermatomyositis-specific antibodies might be regarded as diagnostic of dermatomyositis regardless of the pattern or the distribution of MxA-positive fibers (129). Although perifascicular MxA expression is considered by some as diagnostic of dermatomyositis, a number of experts express skepticism. A new study showed that MxA is equally expressed in both lupus myositis and dermatomyositis, and it is also expressed in up to 20% of anti-synthetase syndrome myositis cases, supporting the aforementioned view that its expression is secondary (140).
The histological picture of skin lesions in dermatomyositis is characterized by dermal perivascular infiltrates consisting mainly of CD4+ cells, followed by macrophages (62). B lymphocytes are sparse and CD1+ Langerhans cells are diminished in the epidermis but increased in dermal papillae. Skin biopsies of the Gottron papules demonstrate cellular infiltrates consisting of activated CD4+ lymphocytes (expressing HLA-DR and CD40L molecules) located in the perivascular areas of the upper dermis and subepidermally (14). The CD8+ T cells are few. This finding is consistent with the humoral mediated process seen in the muscle of patients with dermatomyositis, as described earlier. Because the CD40 molecule was present on the basal keratinocytes while the neighboring CD4+ T cells expressed CD40L, it was suggested that the CD40-CD40-L system might be involved in the cutaneous manifestation of DM, probably via the upregulation of cytokines and chemokines, in a pattern similar to that described for the muscle (127). Upregulation of cytokines, chemokines and deposits of C5b-9 have been also observed in the skin lesions of patients with dermatomyositis (Magro and Crowson 1997). An overwhelming presence of IFN signature genes has also been observed (137). Although the immunopathology of the skin has not been studied as thoroughly as that of muscle, on the basis of these limited studies, it appears that in dermatomyositis a complement mediated endotheliopathy is a common pathway for both skin and muscle. The role of dendritic cells, plasmacytoid cells, ΜxA, and the APC function of keratinocytes or Langerhans cells in the manifestation of skin lesions remains to be determined.
The cause of calcifications, which are more prominent in juvenile dermatomyositis, also remains unclear. In two cases, the milk of calcium extracted from the subcutaneous collections was found to contain macrophages, IL6, IL1 and TNF- suggesting activation of macrophages. These cases have also responded to alendronate (98).
The exact incidence of dermatomyositis is unknown. Along with the other forms of inflammatory myopathies (polymyositis, per old classification; necrotizing autoimmune myositis; overlap myositis; and inclusion-body myositis), they occur in approximately 1 in 100,000 adults.
There is evidence that exposure to the sun may bring up the rash and potentially exacerbate the severity of muscle and skin disease. Therefore, sunbathing is not recommended.
The rash and the calcifications are so characteristic that the diagnosis of dermatomyositis is rarely in doubt. Sometimes muscle strength is normal (dermatomyositis sine myositis), in spite of clear evidence of subclinical muscle involvement in the muscle biopsy. At times, the rash may be barely detectable (especially in dark-skinned people), and the diagnosis can be made only in retrospect with the muscle biopsy based on perifascicular atrophy or on the basis of subcutaneous calcifications found accidentally (27). The skin changes in the phalanges may be distinguished from those due to systemic lupus erythematosus because the phalanges are involved and the knuckles are spared in the latter whereas the reverse is true in dermatomyositis. The only disorder that histologically resembles dermatomyositis is overlap myositis with Jo-1 antibodies, which has prominent perifascicular pathology, but in contrast to dermatomyositis, the fibers are not only atrophic but are also necrotic along with signs of abundant necrosis in the perimysium (95).
Calcinosis universalis (the abundant subcutaneous calcifications seen in rare juvenile dermatomyositis patients) should not be confused with myositis ossificans, a rare disease associated with skeletal deformities, such as short great toe or curved fingers. This disease begins in the second year of life and involves bone and subcutaneous tissue rather than muscle. The bone is deposited beneath the skin or within the muscle in the limbs or paraspinal regions, and this is often preceded by soft localized swellings (27).
Another, but now extinct, disease that potentially shared clinical features with dermatomyositis was the eosinophilia myalgia syndrome originally described in patients exposed to ingestion of contaminated L-tryptophan (94). In the eosinophilia myalgia syndrome, the skin can be tight and shiny but is not erythematous. Joint contractures were common. Although the inflammatory process was mostly confined to the subcutaneous tissue, inflammation can spread to the muscle and cause myopathic muscle weakness (68). The eosinophilia myalgia syndrome also resembled the Spanish toxic oil syndrome, but it has not yet been seen with other contaminants.
Dermatomyositis may rarely have some overlapping similarities with Shulman syndrome (eosinophilic fasciitis), especially joint contractures and thickening of the skin and subcutaneous tissue (119).
Another disorder that may have some apparent similarities with dermatomyositis is the macrophagic myofasciitis identified in French people at the areas of local intramuscular injection with aluminum-containing vaccines. This entity can cause systemic symptoms of fatigue, myalgia, and mild muscle weakness years after the vaccinations. Although it has no skin manifestations, the pathology is in the perimysial and perifascicular regions as in dermatomyositis. In contrast to dermatomyositis, however, the inflammatory cells are exclusively macrophages, and the capillaries are normal (58).
The clinical diagnosis of dermatomyositis is confirmed by serum muscle enzymes, electromyographic findings, and muscle biopsy. In classic cases, however, the typical skin manifestations in combination with muscle weakness are almost sure indicators of dermatomyositis, even on clinical grounds alone.
Serum muscle enzymes. The most sensitive enzyme is creatine kinase, which in the presence of active disease can be elevated to 50 times the normal level. Although creatine kinase activity usually parallels disease severity, it can be normal in some patients with untreated dermatomyositis and in others with dermatomyositis and connective tissue disease, probably reflecting the predominant involvement of the intramuscular vessels and the perimysium. Along with creatine kinase, serum glutamic-oxaloacetic transaminase, serum glutamic-pyruvic transaminase, glutamic pyruvic transaminase, lactate dehydrogenase, and aldolase may also be elevated.
Electromyography. Needle electromyography shows myopathic potentials characterized by short-duration, low-amplitude polyphasic units on voluntary activation and increased spontaneous activity with fibrillations, complex repetitive discharges, and positive sharp waves. This electromyographic pattern occurs in a variety of acute, toxic, and active myopathic processes and should not be considered diagnostic for the inflammatory myopathies. Mixed myopathic and neurogenic potentials (polyphasic units of short and long duration) can occasionally be seen in dermatomyositis as a consequence of muscle fiber regeneration and indicate chronicity of the disease. Electromyographic studies are generally useful to exclude neurogenic disorders and confirm active or inactive myopathy.
Muscle biopsy. Muscle biopsy is the definitive test to exclude other neuromuscular diseases and assess severity of involvement. Unique histological features are characteristic of dermatomyositis (38; 24).
The endomysial inflammation is predominantly perivascular or in the interfascicular septa and around rather than within the fascicles. The intramuscular blood vessels show endothelial hyperplasia with tubuloreticular profiles, fibrin thrombi (especially in children), and obliteration of capillaries. Necrosis, degeneration, and phagocytosis often affect groups of fibers within muscle fascicle in a wedge-like shape or at the periphery of the fascicle due to microinfarcts within the muscle. This results in perifascicular atrophy, characterized by two to 10 layers of atrophic fibers at the periphery of the fascicles. The presence of perifascicular atrophy is diagnostic of dermatomyositis, even in the absence of inflammation. It has been proposed that MxA stain may be of diagnostic value (134). Although doubtful, a large study showed promising specificity of the MxA pattern in connection with autoantibodies for classifying dermatomyositis subtypes (129).
The evidence that immunopathologic mechanisms are primarily involved in the pathogenesis of dermatomyositis justifies treating the disease with immunosuppressive therapies. Most of the treatment trials, however, have been empirical; large-scale control therapeutic studies in childhood or adult dermatomyositis have not been conducted (25; 26; 27; Dalakas 1994a; Dalakas 1994b; 37; 22; 47; 39; 24). As the specific target antigens are also unknown, these therapies are not selectively directed to the autoreactive T cells or to the complement-mediated process in the intramuscular blood vessels; rather, they induce nonselective immunosuppression or immunomodulation (30).
The goal of therapy in dermatomyositis is to improve function in activities of daily living as the result of improvement in muscle strength and alleviate the skin rash. Although improvement in strength is usually accompanied by a fall in serum creatine kinase, decreases of serum creatine kinase have to be interpreted with caution because most immunosuppressive therapies result in a decrease in serum muscle enzymes without necessarily improving muscle strength. Unfortunately, this has been misinterpreted as "chemical improvement" and has formed the basis for the common habit of "chasing" or "treating" the creatine kinase level instead of monitoring muscle weakness, a practice that has led to prolonged use of unnecessary immunosuppressive drugs and erroneous assessment of their efficacy (36). The importance of discontinuing these drugs if, after adequate trials, they have caused only reduction in creatine kinase and not objective improvement in muscle strength and skin rash has been repeatedly emphasized (25; 26; 27; Dalakas 2009). In dermatomyositis, the usefulness of creatine kinase levels in monitoring disease activity is insignificant because patients with dermatomyositis may have normal creatine kinase levels even from the outset when the disease is active. For patients with disease limited to the skin, our preference is to start with low doses of steroids or hydroxychloroquine sulfate and to avoid immunosuppressants until weakness develops.
Corticosteroids. Prednisone is the first-line drug of this empirical treatment. Its mechanism of action is unclear, but it may involve inhibiting recruitment and migration of lymphocytes to the areas of muscle inflammation and interfering with the production of lymphokines. Its effect on lymphokine IL1 may be important because IL1 is myotoxic and is secreted by the activated macrophages that invade the muscle fibers. Steroid-induced suppression of ICAM-I may also be relevant because downregulation of ICAM-I can prevent the movement of lymphocytes across the endothelial cell wall toward the muscle fibers.
Because early response to prednisone determines whether or not stronger immunosuppressive drugs will be needed, our preference has been to start with high-dose prednisone early in the disease (22a; 36). Others, however, prefer to start prednisone in combination with azathioprine or methotrexate in an effort to reduce the prednisone faster. A high dose of 1 gr/kg or between 60 to 100 mg/day as a single daily morning dose for an initial period of 3 to 4 weeks has been preferable. In patients with aggressive disease, methylprednisolone 1 gm IV every day for 3 days may be considered first, followed by the oral steroid dose. Prednisone is then tapered to a single every other day dose by gradually reducing an alternate "off-day" dose by 10 mg per week, or faster if necessitated by side effects. If there is evidence of efficacy and no serious side effects, the dosage is gradually reduced until the lowest possible dose that controls the disease is reached. If by the time the dosage has been reduced to 60 to 100 mg every other day and there is no objective benefit (defined as increased muscle strength and improved rash and not as decreased serum creatine kinase or subjective feeling of increased energy), the patient should be considered unresponsive to prednisone and tapering accelerated while another immunomodulating drug is considered (25; 26; 22a).
The merits of a single-dose, alternate-day program in minimizing side effects (cushingoid appearance, diabetes, obesity, high blood pressure, osteoporosis, avascular necrosis of the hip, retarded growth in children) while adequately controlling the underlying disease have been previously discussed (25; 26; 27).
Prednisone failures and nonsteroidal immunosuppressive therapies. Although in our experience almost all patients with dermatomyositis respond to steroids to some degree and for some period of time, some patients fail to adequately become steroid-resistant. The rationale for starting another immunosuppressive drug in dermatomyositis patients is based on the following factors: (a) need for a "steroid-sparing" drug when, despite steroid responsiveness, the patient is in danger of developing complications; (b) repeated relapses after attempts to lower a high steroid dosage; (c) ineffectiveness of an adequate high-dose prednisone for at least a 6-week period; and (d) rapidly progressive disease with evolving severe weakness and respiratory failure. The preference for selecting the next-in-line immunotherapy has been, however, empirical. The choice is usually based on a physician's own bias, personal experience with each drug, and assessment of the relative efficacy/safety ratio. The following therapies have been traditionally used in the treatment of patients with dermatomyositis:
Although lower doses (1.5 to 2 mg/kg) of azathioprine are commonly used, we prefer doses up to 3 mg/kg for effective immunosuppression. This drug is well tolerated, has fewer side effects, and, empirically, appears to be helpful for long-term or steroid-sparing therapy. Because azathioprine is usually effective after 3 to 6 months of treatment, patience is required before concluding that the drug is ineffective. Azathioprine, which is metabolized by xanthine oxidase, can be severely toxic to the liver or bone marrow if given concurrently with allopurinol, so combined use is not recommended.
Mycophenolate mofetil is now preferable to azathioprine because it works faster, although no control or comparable studies have been conducted, and seems also to help the skin lesions (56). Doses of 2 to 3 grams per day in two divided doses (1000 to 1500 twice daily) are the standard regimen.
Methotrexate, an antagonist of folate metabolism, is also preferred among some practitioners because it acts faster than azathioprine. It can be given orally starting at 7.5 mg weekly for the first 3 weeks (given in a total of three doses, 2.5 mg every 12 hours), increasing it gradually by 2.5 mg/week up to a total of 25 mg/week. An important but very rare side effect is methotrexate-pneumonitis, which can be difficult to distinguish from the interstitial lung disease accompanying dermatomyositis and often associated with Jo-1 antibodies.
Cyclophosphamide, preferably intravenously at doses of 0.5 to 1 gm/m2, has shown promising results in some patients (11). Although in our hands it was ineffective in patients with severe disease, the drug may be helpful in a subset of patients with interstitial lung disease.
Chlorambucil, an antimetabolite, has been tried in some patients with variable results. This drug was found safe and effective in five patients with dermatomyositis to the point that prednisone was reduced or discontinued (120).
Cyclosporine has been used with limited success. Low doses of cyclosporine can benefit children with dermatomyositis (63). Cyclosporine was tried as a first-line therapy in dermatomyositis patients with promising results (59).
In children with new-onset juvenile dermatomyositis, a controlled study comparing prednisone alone (47 patients) with a combined treatment of prednisone with either cyclosporine (46 patients) or methotrexate (another 46 patients) showed that combined therapy was more effective than prednisone alone; the safety profile and steroid-sparing effect, however, favored the combination of prednisone plus methotrexate (114).
Tacrolimus, acting as a calcineurin inhibitor, has shown promise in the treatment of difficult cases (138). However, the experience with this drug is limited.
Rituximab, an anti-CD20 monoclonal antibody that causes depletion of B cells, seems very promising in some patients with dermatomyositis (88). Rituximab was effective in adult dermatomyositis, showing an increase in muscle strength and improvement in scores of disease activity, general health, functional ability, and health-related quality of life with sustained effect during a median of 27.1 months of follow-up (91). The same effect was noted in nine patients with juvenile dermatomyositis (06). A placebo-controlled study conducted in 200 patients (76 with adult dermatomyositis, 48 with juvenile dermatomyositis, and 76 with polymyositis) did not meet the primary endpoint largely because of a study design. Patients were randomized to a rituximab-early group receiving the drug or placebo at weeks 0 and 1, and to a rituximab-late group receiving placebo or rituximab in weeks 8 and 9. There was no difference between placebo and rituximab at week 8, but at week 44, when all patients had received the drug, 83% of them had met the definition of improvement (103). Patients with autoantibodies, especially against Jo-1, Mi-2, or SRP, were more likely to improve in a follow-up multivariate analysis (02). Rituximab is now the preferred treatment in patients poorly responding to immunosuppressants and intravenous immunoglobulin.
Rapamycin, an immunosuppressant that binds to FKB protein, inhibits the IL2 synthesis by autoreactive T cells. It was shown to improve skin lesions in a patient with dermatomyositis unresponsive to available agents (101).
Plasmapheresis was not helpful in a double-blind, placebo-controlled study that we conducted (96).
The TNF-a inhibitors, including etanercept and infliximab, have been disappointing in the treatment of dermatomyositis. There is also evidence that they may even worsen or trigger the disease. A small, semi-controlled study of 16 patients, 11 randomized to etanercept and five to placebo, showed that five etanercept-treated patients were weaned off steroids; the rash, however, worsened in five etanercept-treated patients (100). Additional studies have clearly failed to show any benefit of etanercept and confirm the observation that this category of drugs can worsen the disease (113) and is not recommended for the treatment of dermatomyositis. A published, very small, dose-escalation, double-blind, placebo- controlled trial that was conducted 10 years ago assessed the effect of infliximab in 12 patients with dermatomyositis and polymyositis and showed no significant differences during a 16-week study period; the authors claim, however, that three patients experienced a benefit (116).
Intravenous immunoglobulin was effective in two small, uncontrolled studies (19; 85); its efficacy was, however, established with a controlled trial. In the first ever double-blind study, intravenous immunoglobulin was shown to be impressively effective in patients with refractory dermatomyositis, not only improving the strength and the skin rash but also clearing the underlying immunopathology (46; 31; 22b; 43; 34). The improvement begins after the first intravenous immunoglobulin infusion and is clearly evident by the second monthly infusion, often showing first in the skin.
Effect of IVIg on the skin manifestations (Gottron rash) and muscle strength of patients with dermatomyositis based on the first and pivotal placebo-controlled study. (Dalakas MC, Illa I, Dambrosia JM, et al. A controlled trial...
The benefit is short-lived (not more than 8 weeks), requiring repeated infusions every 6 to 8 weeks to maintain improvement. A long-term effect of IVIg was also demonstrated in adults (73) and in a large number of children (84). Long-term subcutaneous immunoglobulin is gaining popularity as an alternative to IVIg in patients with difficult venous access or those preferring a home care setting (18). Evidence reconfirms the efficacy of subcutaneous immunoglobulin with improvement in skin lesions and muscle strength (93). A prospective, double-blind, randomized, placebo-controlled phase 3 study evaluating the efficacy and safety of IVIg (octagam 10%) in patients with dermatomyositis (“ProDERM Study”) has now been published (03). Despite the many study limitations with inadequate scales, no immunology, and no immunopathology on repeated muscle biopsies (34), the results confirmed the original pivotal study (46). Most importantly, this study led to the FDA-approved indication. The safety and tolerability of IVIg in patients with dermatomyositis from the ProDERM study was also documented in the open-label extension period in which all patients received IVIg for six additional cycles (04). A total of 113 treatment-emergent adverse events were possibly related to treatment in 30 of 52 patients (57.7%) receiving IVIg and 38 events in 11 patients (22.9%) on placebo; eight patients discontinued therapy due to IVIg-related adverse events with eight thromboembolic events in six patients who exhibited more baseline risk factors. Lowering the infusion rate reduced the rate of these events, and none occurred at the lower IVIg dose. No hemolytic transfusion reactions or deaths were observed. The authors concluded that IVIg has a favorable safety profile in adult dermatomyositis, confirming many prior observations (43).
The mechanism of action of intravenous immunoglobulin in dermatomyositis is well established, even if not acknowledged in the study (03). The beneficial effect of IVIg in dermatomyositis is based on inhibiting the deposition of activated complement fragment on the capillaries (10) and subsequent suppression of cytokines and adhesion molecules, as has been convincingly shown with immunohistopathological studies on the repeated muscle biopsies of improved patients (46; 10; 23). IVIg inhibits complement activation at the C3a level, leading to interception of MAC formation, disappearance of MAC deposits on the endomysial capillaries, and restoration of microvasculature and perifascicular atrophy. IVIg also exerts an effect by saturating the Fc receptors and interfering with the action of macrophages invading muscle fibers (46; 23).
IVIg inhibits complement at the C3a level (1, arrow) 2 days after the infusion (2) and intercepts the formation of MAC. As a result, MAC is not detected in the repeated muscle biopsies of the improved patients anymore (3a, 3b)....
New biologicals may become promising in the future. Two JAK kinase inhibitors, tofacitinib in three patients (80) and ruxolitinib in one patient (67) seemed effective, probably by affecting circulating cytokine levels and regulating the activation of dendritic cells and T lymphocytes, which are known to be activated in the disease (104). Additional studies (139), including an open-label study in 10 patients, confirmed the efficacy of tofacitinib (106). A long-term extension study indicates that tofacitinib is safe and clinically beneficial in refractory dermatomyositis for up to 96 weeks (107). The effect of JAK inhibitors is now supported by additional dermatomyositis cases who responded to ruxolitinib including a juvenile case (01), and patients with refractory dermatomyositis with anti-MDA5 antibodies and interstitial lung disease (79). Ruxolitinib, in vitro, abolished the disruption of vascular disorganization of endothelial cells induced by type I interferon and clinically improved four patients with refractory dermatomyositis (82). Eculizumab, a monoclonal antibody directed against C5 that blocks the generation of C5a and the membrane attack complex assembly (40; 42), is now being tested in an ongoing a double-blind study in patients with dermatomyositis. For patients with amyopathic dermatomyositis, lenabasum, a cannabinoid receptor type 2 agonist that activates resolution of innate immune responses to reduce tissue inflammation and fibrotic processes, has been promising in a 16-week phase 2 double-blinded, randomized, placebo-controlled study in 22 patients (121). New biologics against complement FcRn inhibitors and CD19-targeting-chimeric antigen receptor (CAR)-T cells are now being explored in ongoing trials not only in dermatomyositis but also in other autoimmune inflammatory myopathy subsets (35).
An exciting new therapy is with anti-CD19 CAR-T cell, which is currently planned and being investigated in several autoimmune diseases, such as systemic lupus erythematosus, myasthenia gravis, stiff person syndrome, and myositis. CAR T-cell therapy resulted in clinical improvement and stable remission for up to 2 years in 15 patients with severe systemic lupus erythematosus, three patients with myositis, and four patients with systemic sclerosis (99). It appears to be safe and with great potential for achieving a lasting drug-free remission, especially since adverse events, such as infections, high-grade cytokine release syndrome, or ICANS, appear to be very low in contrast to the high incidence of these events seen in patients with cancer.
Collectively, our step-by-step approach in the treatment of dermatomyositis is as follows:
|
• Step 1: High-dose prednisone (oral or intermittent intravenous in acute cases). | |
|
• Step 2: A mild immunosuppressant, such as azathioprine, methotrexate, or mycophenolate only for steroid-sparing effect in steroid-responsive patients. | |
|
• Step 3: If step 1 fails or is not sufficiently effective, high-dose intravenous immunoglobulin. | |
|
• Step 4: If step 3 fails, or IVIg is not sufficiently effective, rituximab, or other anti-B cell agents. The new biologicals, especially JAK inhibitors, seem to be additional options. Results from new ongoing trials may, however, change the future therapeutic algorithm, as seems to be the case in myasthenia gravis (35). |
Treatment for calcinosis remains difficult. Attempts with alendronate (98), probenecid (61), or diltiazem thought to be promising, offer limited help.
Dermatomyositis can occur in the last trimester of pregnancy or during the puerperium, but it is not known whether pregnancy is responsible for triggering the disease. Pregnant women with dermatomyositis have been treated with steroids and have delivered normal but small babies (28), although miscarriages and stillborn babies have been reported.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Marinos C Dalakas MD
Dr. Dalakas of the National and Kapodistrian University of Athens Medical School in Greece and Thomas Jefferson University, Philadelphia, Pennsylvania, received speaker honoraria and consultancy fees from Argenx and Grifols.
See Profile
Nicholas E Johnson MD MSCI FAAN
Dr. Johnson of Virginia Commonwealth University received consulting fees and/or research grants from AMO Pharma, Avidity, Dyne, Novartis, Pepgen, Sanofi Genzyme, Sarepta Therapeutics, Takeda, and Vertex, consulting fees and stock options from Juvena, and honorariums from Biogen Idec and Fulcrum Therapeutics as a drug safety monitoring board member.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuroimmunology
Jul. 02, 2026
Neuromuscular Disorders
Jun. 16, 2026
Neuromuscular Disorders
May. 27, 2026
Neuromuscular Disorders
May. 21, 2026
Neuropharmacology & Neurotherapeutics
May. 14, 2026
Developmental Malformations
May. 08, 2026
General Child Neurology
Apr. 24, 2026
Neuromuscular Disorders
Apr. 23, 2026