

Epilepsy & Seizures
Febrile seizures
Jun. 02, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Dravet syndrome is a severe developmental and epileptic encephalopathy caused by pathogenic variants in the neuronal sodium-channel α1-subunit gene (SCN1A) and is the most common monogenic cause of epilepsy. Dravet syndrome is characterized by an early onset in the first year of life (3 to 15 months), with the occurrence of febrile and afebrile, hemiclonic or generalized, and convulsive seizures in apparently normal infants. Dravet syndrome is later followed by other seizure types (myoclonic and atypical absence seizures, focal seizures, and obtundation status epilepticus) persisting into adulthood. Borderline forms have historically been described as lacking one of the above features of myoclonic seizures, atypical absences, or onset in the first year of life; however, they are now considered part of the Dravet syndrome spectrum. Developmental slowing becomes apparent within the second year of life and is followed by the emergence of significant intellectual disability and associated comorbidities, including behavioral difficulties and features of autism spectrum disorder. The EEGs show both generalized and multifocal abnormalities, without a specific pattern, but initial interictal EEGs may be normal. Pharmacoresistance is one of the main features, and episodes of status epilepticus are frequent. Treatment options are currently expanding and include the emergence of new gene-specific, disease-modifying therapies.
Dravet syndrome is a genetic disorder with 80% to 90% of cases being caused by pathogenic SCN1A variants, 90% of which occur de novo. Haploinsufficiency resulting in varying degrees of loss of protein function is thought to be the mechanism underlying most cases, with modifying factors, including the genetic and environmental background contributing to the variable phenotype of patients with pathogenic SCN1A mutations.
Experimental studies on animal models continue to provide new insights into the pathogenesis and possible treatments of this severe sodium ion channelopathy. There is no underlying brain lesion, and neuroimaging is normal. The long-term prognosis is unfavorable and includes significant mortality due to sudden unexpected death in epilepsy (SUDEP) or status epilepticus.
|
• Dravet syndrome is one of the most severe epilepsies in infancy, leading to a developmental and epileptic encephalopathy, with significant long-term neurocognitive and motor comorbidities. | |
|
• The onset is between 3 to 15 months of age with recurrent and often prolonged febrile and afebrile seizures in apparently normal infants. | |
|
• It is caused by pathogenic variants; the neuronal sodium-channel α1-subunit gene (SCNA1) is in 80% to 90% of affected individuals. | |
|
• Pathogenic variants are de novo in most cases, but some inherited variants have been reported in a minority of patients. | |
|
• The epilepsy often remains treatment-resistant despite evidence-based treatments, including sodium valproate, clobazam, and stiripentol, as well as the use of cannabidiol and fenfluramine. Topiramate, bromide, and the ketogenic diet are alternative options. | |
|
• Use of sodium channel blockers such as carbamazepine or lamotrigine is contraindicated as they may worsen seizures and developmental outcome. | |
|
• Prevention of status epilepticus with regular medication and emergency protocols is important to reduced mortality. | |
|
• Gene-specific, disease-modifying therapies are currently being developed and offer promising new treatment options. |
The history of Dravet syndrome has been detailed by Charlotte Dravet herself (46). Dravet syndrome was first described as “severe myoclonic epilepsy in infancy” by Charlotte Dravet in 1978 in a French medical journal from the Centre Saint-Paul, Marseille. She reported several very severe cases of epilepsy beginning early in life which, despite certain similarities, could not be categorized as Lennox-Gastaut syndrome for several reasons, especially their stereotyped mode of early onset, the frequent myoclonic seizures, and the absence of axial tonic seizures (45). Dalla Bernardina, who worked simultaneously in Verona, Italy, and the Centre Saint-Paul, also observed the same electroclinical features in 20 of his Italian patients (35). Subsequently, Dravet, Roger, Bureau, and Dalla Bernardina presented these 42 patients at the XIII International Epilepsy Congress in Kyoto (52).
In 1989, the revised classification of the International League Against Epilepsy placed this syndrome under “epilepsies and syndromes undetermined as to whether they are focal or generalized,” as the syndrome shows both generalized and localized seizure types and EEG paroxysms (31). It was defined as follows:
|
"Severe myoclonic epilepsy in infancy is a recently defined syndrome. The characteristics include a family history of epilepsy or febrile convulsions, normal development before onset, seizures beginning during the first year of life in the form of generalised or unilateral febrile clonic seizures, secondary appearance of myoclonic jerks, and often partial seizures. EEGs show generalised spike-waves and polyspike-waves, early photosensitivity, and focal abnormalities. Psychomotor development is retarded from the second year of life on, and ataxia, pyramidal signs, and interictal myoclonus appear. This type of epilepsy is very resistant to all forms of treatment." |
A landmark discovery was made in 2001 by Claes and colleagues who found the genetic etiology of Dravet syndrome with de novo pathogenic variants in the sodium-channel gene SCN1A in all of the seven studied probands with Dravet syndrome (29). Since then, research has expanded in documenting Dravet syndrome as a channelopathy at the severe end of the spectrum of SCN1A-related disorders (18; 88). Thus, the name “Dravet syndrome” is designated to include not only the classic “severe myoclonic epilepsy of infancy” but a spectrum comprising other phenotypic variants or borderline forms, including those without myoclonic seizures, onset in the second year, or without generalized spike and wave on EEG (61; 118; 88).
In the most recent ILAE proposals, Dravet syndrome is classified as a genetic epilepsy syndrome and a developmental and epileptic encephalopathy (08; 111). The complete description from the ILAE epilepsy manual is detailed below (30):
Dravet syndrome. Dravet syndrome (previously known as severe myoclonic epilepsy of infancy, SMEI) typically presents in the first year of life in a normal child with prolonged, febrile and afebrile, focal (usually hemiclonic), or generalized convulsive seizures. Other seizure types including myoclonic and atypical absence seizures appear between the age of 1 and 4 years. Seizures are usually intractable, and from the second year of life, children demonstrate cognitive and behavioral impairments. The clinical diagnosis is supported by the presence of abnormalities in the sodium channel gene SCN1A (found in 75% of cases).
|
Note. The term Dravet syndrome is now also used to encompass atypical or borderline cases, previously known as severe myoclonic epilepsy of infancy - borderland (SMEB). | |
|
Note. Dravet syndrome may be considered an “epileptic encephalopathy.” This term denotes the concept that the epileptic activity itself might directly contribute additional cognitive and behavioral impairments over those expected from the underlying etiology alone, and that suppression of epileptic activity might minimize this additional impairment. |
Clinical context. This syndrome is characterized by onset of seizures typically around 6 months of age. Most have had seizure onset less than 15 months of age; however, a small minority of cases have onset in the second year of life. Both sexes are affected. Antecedent, birth, and neonatal history are normal. The first seizure is associated with a fever in about 60% of cases. Not all patients start with febrile convulsions. Immunization may be a nonspecific trigger to the first seizure leading to an earlier age of seizure onset, but cases with onset with a vaccine proximate seizure have the same outcome as other children with Dravet syndrome. Sensitivity of seizures to fever may persist throughout life. Head size and neurologic examination are usually normal initially; over time ataxia and pyramidal signs may develop. Development is typically normal in the first year of life, with plateauing or regression in later years.
|
Caution. Antiseizure medications that have sodium channel blocking properties may aggravate seizures in this syndrome. | |
|
Caution. Tonic seizures and epileptic spasms are not expected; if present, consider other epilepsy syndromes. |
Mandatory seizures. Focal and generalized seizure types occur in this syndrome. A clonic-tonic-clonic sequence to the convulsive seizures may occur. Hemiclonic seizures may involve different sides of the body in different seizures.
Patients may have atypical absence seizures, myoclonic seizures, atonic seizures, or nonconvulsive status epilepticus.
One quarter of patients have seizures induced by visual stimuli.
EEG background. The background EEG activity is typically normal in the first year of life. Postictal slowing may be seen initially, and diffuse slowing may appear over time.
Interictal EEG. By the second to fifth year of age, generalized spike-and-wave and multifocal discharges are seen.
Activation. Photic and pattern stimulation precipitate generalized spike-and-wave, with or without associated clinical events (atypical absence seizures or myoclonic seizures). Photosensitivity can be present in infancy and is seen at all ages. EEG abnormality is enhanced by sleep deprivation and by sleep.
Ictal EEG. The ictal EEG varies according to the type of seizure.
|
Caution. The presence of diffuse electrodecremental patterns or paroxysmal fast activity is not seen: then Lennox Gastaut syndrome should be considered. |
Imaging. Neuroimaging is usually normal at onset. Abnormalities may be found later in life in 10% of cases, including generalized atrophy or hippocampal sclerosis.
Genetics.
Pattern of inheritance. In Dravet syndrome patients with pathogenic SCN1A variants, 95% are de novo and 5% are inherited. Carrier relatives are either unaffected or mildly affected with genetic epilepsy with febrile seizures plus phenotypes. Germline and somatic mosaicism have been reported.
Known genes. Approximately 80% to 90% of patients with Dravet syndrome have pathogenic variants or copy number variants in SCN1A. A small percentage of females with a Dravet syndrome-like phenotype have pathogenic variants in the PCDH19 gene. These females usually have clusters of seizures with fever as opposed to the prolonged status epilepticus with fever that occurs in SCN1A-related Dravet syndrome.
Family history of seizures/epilepsy. A family history of epilepsy or febrile seizures is present in 30% to 50% of patients. In some children with Dravet syndrome, the family history is consistent with genetic epilepsy with febrile seizures plus.
Differential diagnosis.
|
• Febrile seizures plus, genetic epilepsy with febrile seizures plus |
Dravet syndrome has been detailed in a book (46), a supplement to Epilepsia (03), and many relevant chapters in epilepsy books (49; 51; 94). The Epilepsia Supplement 3 of 2019 is devoted to “Dravet syndrome and other sodium channel related encephalopathies.”
Dravet syndrome typically begins during the first year of life (3 to 15 months of age). Development is normal prior to the onset of seizures. Affected infants develop generalized or unilateral convulsive seizures, which are predominantly clonic and frequently febrile. Myoclonic jerks, psychomotor slowing, and other neurologic deficits appear later. Mortality is high. Dravet syndrome usually develops in three stages:
|
(a) From onset up to 1 to 1.5 years: with mainly febrile convulsive seizures and febrile status epilepticus but normal development | |
|
(b) From age 1.5 up to 6 to 10 years: with frequent seizures of varying types, developmental stagnation, behavioral, neurocognitive, sleep problems, and neurologic abnormalities | |
|
(c) After 10 years: improvement in seizures, deteriorating gait, and intellectual and psychobehavioral disability but some developmental gains |
Seizures. Seizure onset is typically in the first year of life (peak at 5 months) with hemiclonic or generalized clonic and tonic-clonic convulsions in previously healthy children. These are usually febrile (70%) and may be associated with vaccinations or hyperthermia, including a warm bath. Seizures are severe, frequent, polymorphic, convulsive or nonconvulsive, focal, multifocal or generalized, and intractable. The various types of seizures are well demonstrated in YouTube with homemade video recordings and can be accessed at the following sites: https://www.youtube.com/watch?v=3NVBWX5GVaM, and https://www.youtube.com/watch?v=zkXmSg7G32M.
Convulsive seizures. The first seizures are usually clonic, tonic-clonic, or unilateral hemiclonic convulsions often with alternating sides. Commonly, these seizures are longer than simple febrile seizures. Convulsive seizures are present throughout the evolution in all patients.
“Falsely generalized” or “unstable” convulsive seizures are terms coined by Dravet (46; 47; 49). The “falsely generalized seizures” are characterized by a complicated semiology with some degree of discrepancy between clinical and EEG phenomena. The description reported by observers seems to correspond to a generalized tonic-clonic seizure, even if the seizures are generally shorter, but accurate video-EEG observations demonstrate that they are not generalized, neither at the onset nor at the offset, and that they differ from one patient to another. They occur especially during NREM sleep. The ‘‘unstable seizures’’ are characterized by the topographic changes of the ictal EEG discharge in the same seizure.
Unilateral convulsive seizures are the most characteristic. In the youngest patients, they correspond to hemiclonic seizures and often evolve to status. In older patients, they are shorter and associated with contralateral muscle tone changes. In all cases, there are postictal asymmetric EEG signs, and there is often postictal transient hemiparesis. These seizures can be on either side in the same patient—this alternating pattern being a clue for the diagnosis of Dravet syndrome. These seizures can be prolonged or repeated, often constituting status epilepticus.
The existence of tonic seizures is debatable; if they occur, they resemble the axial tonic seizures of Lennox-Gastaut syndrome, and they are mainly detected during sleep EEG recordings (83).
Myoclonic seizures appear between the ages of 1 and 5 years but sometimes at the same age with the onset of the convulsive seizures. They affect facial, limb, and axial muscles, causing flexion or extension and falls. They are frequent, often occurring several times per day and may cluster in myoclonic status epilepticus. On other occasions, jerks occur only hours or days before a convulsive seizure. Myoclonic jerks are usually violent and frequently mixed with numerous asynchronous and arrhythmic distal jerks manifested as twitching of fingers, which are difficult to differentiate from nonepileptic myoclonus. They usually disappear during stages III and IV of sleep.
Myoclonic jerks are usually accompanied by generalized spike waves and polyspike waves in the EEG. However, sometimes myoclonic jerks begin focally and are limited to one limb or the head prior to becoming generalized. These myoclonic seizures are often associated with interictal segmental myoclonus.
Atypical absence seizures (40% to 93% of patients) appear at different ages, at 1 to 3 years with myoclonic jerks or later from 5 to 12 years. They are short (5 or 6 seconds) with moderate impairment of consciousness and often with myoclonic jerks.
Focal seizures (43% to 78% of patients) are simple focal motor seizures or complex focal seizures. They occasionally progress to unilateral or bilateral convulsive seizures. Focal motor seizures manifest either as versive seizures or clonic jerks limited to a limb or a hemiface, or a combination of the 2. Complex focal seizures are characterized by loss of consciousness, autonomic manifestations (pallor, cyanosis, respiratory changes, drooling, sweating), oral automatisms, hypotonia, and rarely, stiffness, sometimes with eyelid or distal myoclonus. When the symptomatology is mild, it is difficult to distinguish the seizures from atypical absences without concomitant EEG. The seizures occur in patients who have one or several foci in the posterior and frontal brain areas.
Status epilepticus. Myoclonic, atypical absence, complex focal, and convulsive status epilepticus, either alone or in combination, are common and frequent (46; 47; 49; 78). These various types of status epilepticus may last for hours or days and may be facilitated or precipitated by photic stimulation, eye-closure, or fixation on patterns. Complex focal and, rarely, simple focal status epilepticus occurs. Episodes of EEG generalized spike and wave discharges interspersed with erratic small myoclonic jerks may persist for hours or days.
Obtundation status epilepticus (40% of patients) is a term used by Dravet for a type of nonconvulsive status epilepticus, which is relatively characteristic of this syndrome. It manifests with fluctuating obtundation (reduced level of alertness or consciousness), with low-amplitude, fragmentary, segmental, and erratic myoclonic jerks of the face and limbs that are sometimes associated with drooling, ataxia, and tonic manifestations (46; 47; 49). Duration is prolonged for several hours or days. Ictal EEG shows a mixture of irregular, arrhythmic, diffuse, and focal spike-wave discharges. Convulsive seizures can initiate, occur during, or terminate these episodes of obtundation status epilepticus.
Precipitation of seizures. Seizure precipitants are common and include fever (often in the context of infection or following vaccination), photic and pattern sensitivity, and change in environmental temperature such as that seen with hot baths, physical exercise, noisy environments, and strong emotions. A study on seizure precipitants in Dravet syndrome showed that in 99% of patients at least one seizure precipitant is cited by their parents (127). Seizure precipitants that reported in more than half of the cohort with Dravet syndrome were as follows: having a fever (97%), having a cold (68%), taking a bath (61%), having acute moments of stress (58%), and engaging in physical exercise (56%). Seizure precipitants were often related to ambient warmth or cold-warmth shifts (41%) and to various visual stimuli (18%). Within the categories, including stress and physical exercise, physical exercise was more often reported to provoke seizures in stress-sensitive patients.
Cognitive, behavioral, and neurologic abnormalities. Dravet syndrome has a characteristic developmental trajectory by age. Cognitive, behavioral, and neurologic abnormalities appear a few months to years after the onset of seizures (102; 103; 12; 47; 58; 49; 104; 05; 26; 57; 37).
At the very onset, the infants appear to be normal; however, subtle signs of developmental slowing might already be apparent in the first year of life. More overt developmental delay becomes apparent within the second year of life and is followed by definite neurocognitive impairment and behavioral disorders. Language progresses very slowly, and many patients do not reach the stage of constructing elementary sentences. Patients’ fine motor abilities do not develop well. There is poor hand-eye coordination. Lack of attention is one of the major factors responsible for the learning disability, as well as for hyperactivity and unmanageable behavior. Affected children are restless and are not interested in playing with educational toys or participating in the usual activities of their age group. Autistic behavior is common (09; 10; 67). Neurocognitive and behavioral changes are usually severe, but occasionally, they are mild or moderate (103).
Neurologic signs. Neurologic abnormalities emerge progressively and simultaneously with the delay in development. Signs consist of hypotonia, ataxia (60%), pyramidal signs (20%), uncoordinated movements, and interictal myoclonus. The association of hypotonia, ataxia, and motor neuropathy leads to postural changes (flexion at the hips, knees, and trunk, giving a “hunched over” appearance) and a peculiar way of walking and running. Gait tends to deteriorate from about 9 years of age, when patients gradually develop a crouch gait pattern (59; 62). There appears to be a strong positive correlation between cognitive and motor development, and a later age of independent walking was associated with a lower cognitive-developmental quotient (128; 134). Kyphoscoliosis and club feet are frequent and worsen with age. A distinctive speech, language, and oral motor phenotype in children and adults with Dravet syndrome has been reported (122). More than 70% of patients with Dravet syndrome have sleep problems (81). Autonomic disturbances and peri-ictal respiratory dysfunction may explain the high rate of SUDEP (70).
The improvement of epilepsy throughout ages is not associated with improvement in intellectual abilities and motor skills, which indicates that the unfavorable outcome of Dravet syndrome is not a pure consequence of epilepsy, but due to the underlying sodium channelopathy (18; 37).
An up-to-date review on neuropsychological phenotypes in Dravet syndrome hypothesized an original neuropsychological phenotype consisting of a defect in sensorimotor integration, especially of visuoconstructive abilities. This is particularly evident in the less impaired patients and in the first several years of life (05).
Diagnostic criteria for Dravet syndrome. Dravet proposed the following diagnostic criteria (46):
|
• Family history of epilepsy or febrile convulsions | |
|
• No previous personal history of disease | |
|
• Seizures beginning in the first year of life in the form of generalized or unilateral febrile clonic seizures | |
|
• Secondary appearance of myoclonic jerks and often focal seizures | |
|
• EEG showing generalized spike-wave and polyspike- wave, early photosensitivity, and focal abnormalities |
However, Dravet also emphasized that (46):
|
• A family history of epilepsy or febrile convulsions is not constant but variable (25% to 71%) | |
|
• The initial seizures are not always generalized or unilateral clonic but may be focal or myoclonic; they are not always febrile, and the clonic seizures often evolve to status epilepticus | |
|
• Not only myoclonic jerks and focal seizures appear secondarily but also atypical absences and obtundation statuses | |
|
• Photosensitivity may be associated with pattern-sensitivity | |
|
• Neurologic signs are not always present but are frequently observed: ataxia (60%), pyramidal signs (20%), and interictal myoclonus (36% to 85%) | |
|
• The MRI is normal at the onset | |
|
• Cognitive deficiency and personality disorders are present in all affected children during the course of the disease, but they are of variable degrees, from slight to severe, and may be detected only at the age when entering elementary school |
The diagnostic criteria for Dravet syndrome were revised by the ILAE Task Force on Nosology and Definitions as follows (138).
|
Seizures |
Recurrent focal clonic (hemiclonic) febrile and afebrile seizures (which often alternate sides from seizure to seizure), focal to bilateral tonic-clonic, or generalized clonic seizures |
|
Age at onset |
1–20 months |
|
Course of illness |
Drug-resistant epilepsy and intellectual disability |
|
MRI/EEG |
An MRI is not required for diagnosis but is highly recommended to exclude other causes. An ictal EEG is not required for diagnosis. |
|
Possible evolving syndrome |
In a child younger than 12 months old who presents with a prolonged hemiclonic or bilateral tonic-clonic seizure with fever and no other underlying cause, the possibility of Dravet syndrome should be considered. Further convulsive seizures (often with fever, and if prolonged or hemiclonic) would allow for a more definitive diagnosis of Dravet syndrome. A diagnosis would be further supported by the finding of a pathogenic SCN1A variant. |
|
Syndrome without laboratory confirmation |
In resource-limited regions, Dravet syndrome can be diagnosed in children without atypical features who meet all other clinical mandatory and exclusionary criteria, without EEG, MRI, and genetic testing. |
The ILAE Task Force specifies a number of “Alerts,” which are defined as criteria that are absent in the vast majority of cases within a syndrome but rarely can be seen. Alerts alone would not exclude the syndrome but should cause the clinician to rethink the diagnosis and undertake further investigations to rule out other conditions. The more Alerts that are present, the less confident one can be about diagnosis of a specific syndrome.
For Dravet syndrome these Alerts include the following:
|
• No history of prolonged seizures (> 10 min) or a lack of fever sensitivity as a seizure trigger | |
|
• Normal EEG background without interictal discharges after 2 years of age | |
|
• Age at onset 1–2 or 15–20 months old | |
|
• Developmental delay at seizure onset | |
|
• Focal neurologic findings (other than Todd paresis) | |
|
• Lack of pathogenic SCN1A or other causal variant | |
|
• Good efficacy with prophylactic sodium-channel agents, including carbamazepine, oxcarbazepine, and phenytoin |
The following exclusionary criteria must be absent in order to diagnose Dravet syndrome:
|
• Epileptic spasms or early infantile SCN1A developmental and epileptic encephalopathy | |
|
• MRI showing a causal focal lesion |
In atypical or borderline forms of Dravet syndrome, myoclonic, focal, or absence seizures may not occur, onset may be with afebrile seizures or focal seizures, febrile seizures may be rare, neurologic signs may be absent, and there may be almost normal psychomotor development in the first 4 years (46; 61; 117).
The outcome of Dravet syndrome is often unfavorable. All but a few exceptional cases have a poor prognosis regarding seizures and neurocognitive development, though progression of symptoms usually ceases at around 11 to 12 years of age (47; 58; 14; 49; 51).
Epileptic seizures tend to become less frequent and less severe after childhood. Fever sensitivity (temperature variations) persists throughout the clinical course, but its impact on seizure frequency and severity is milder than in infancy. Photosensitivity shows a tendency to disappear before the age of 20. Focal seizures stop, myoclonic seizures improve or disappear, and generalized convulsive seizures become less frequent than in childhood and mostly nocturnal.
EEG also changes with age but is still multiple and heterogenous, interictal, and ictal. The earliest reported age of generalized discharges is 6 months; the earliest reported age of focal discharges, diffuse background slowing, and focal slowing is 4 months. In the first year of life, the EEG is abnormal in 43% of these patients, rising to 90% in 1- to 2-year-olds (92).
Nearly all patients remain neurologically and cognitively impaired to a varying degree from severe (more than half) to relatively moderate or exceptionally mild intellectual disability. Many patients also have behavioral disorders and comorbidities, including autism spectrum disorder and features of attention-deficit hyperactivity disorder (ADHD), as demonstrated in a prospective 10-year follow-up study (54). Gastrointestinal and eating problems in Dravet syndrome are frequently observed, and patients should be carefully evaluated in this regard to ensure optimal nutrition (93). Premature death is high mainly due to status epilepticus and sudden unexpected death in epilepsy, affecting even young children.
Because outcome is not predetermined by genetic factors only, early recognition and treatment that mitigates prolonged/repeated seizures in the first year of life and avoidance of contraindicated medication might also limit the progression to epileptic encephalopathy (24; 39). Data from a natural history study in Dravet syndrome suggest that in infants and young children with Dravet syndrome, language and communication delays emerge from 2 to 3 years onwards, emphasizing that the optimal therapeutic window to prevent language and communication delays is before 3 years of age (101).
Mortality. Dravet syndrome is characterized by high premature mortality and a marked young age at death (108; 32; 114). Sudden unexpected death in epilepsy is the leading reported cause of death in this syndrome, accounting for nearly half of all deaths and tends to occur at a younger age (73% before the age of 11) than in other epilepsy cohorts (3% to 9% before the age of 10). Status epilepticus is the second most common cause of death after sudden unexpected death in epilepsy in Dravet syndrome (around a third of the cases).
In a comprehensive literature review on mortality in Dravet syndrome, SUDEP was the likely cause of death in nearly half of 177 cases (49%), followed by status epilepticus (32%) (114). Drowning or accidental death was reported in 14 cases (8%), infections in nine (5%), other causes in six (3%), and unknown in five (3%). Age at death was at a mean age of 8.7 ± 9.8 years; 73% died before 10 years of age. Similar to these results is a study of a cohort of 100 consecutively recruited, unrelated patients with Dravet syndrome (87 had pathogenic SCN1A variants); living cases had a median follow-up of 17 years (32). Seventeen patients died at a median age of 7 years (interquartile range 3 to 11 years) with the following causes of death: 10 SUDEP, four status epilepticus, two drowning, and one asphyxia. The Dravet-specific mortality rate per 1000 person-years was 15.84 (98% CI 9.01-27.85). The Dravet-specific SUDEP rate was 9.32 per 1000 person-years (98% CI 4.46-19.45), which is considerably higher than the 5.1 SUDEP rate per 1000 person-years for adults with refractory epilepsy (32). An international Delphi consensus recommends that SUDEP should be discussed with affected individuals/families as early as feasible (17).
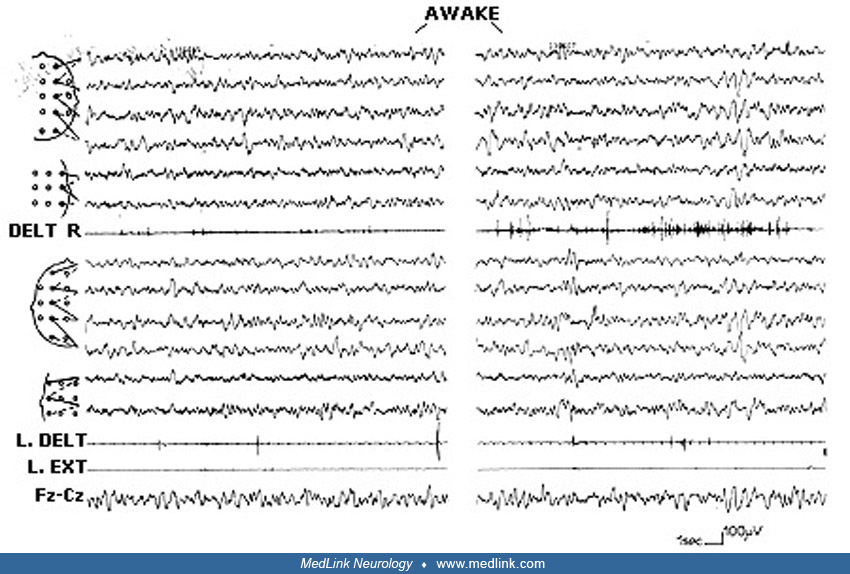
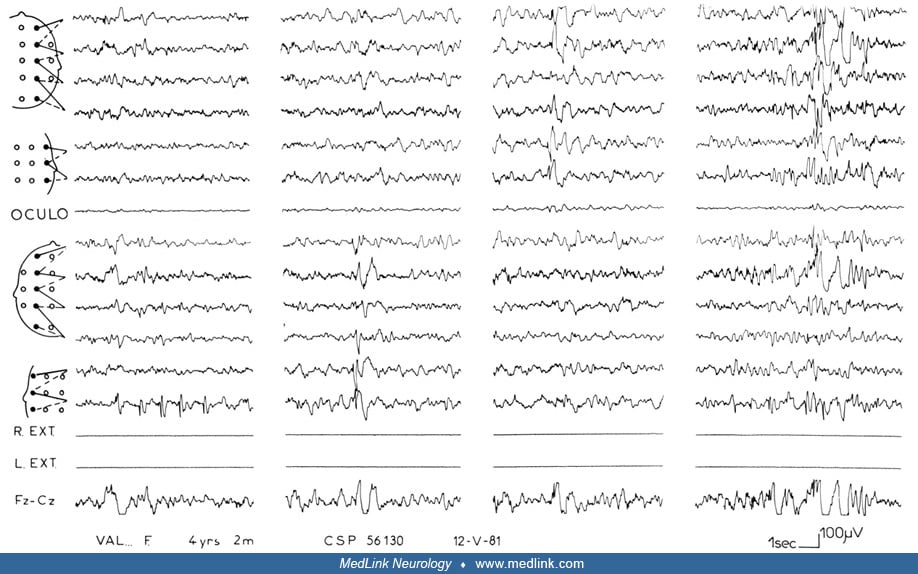
Case 1 (provided by Charlotte Dravet). The patient was born on March 1, 1994. There was no family history of epilepsy, febrile convulsions, or pathological antecedents. The patient demonstrated normal psychomotor development. The first unilateral febrile seizure occurred at 10 months on the left side for about 10 minutes. EEG was normal, and the patient was treated with phenobarbital. Other convulsive seizures, febrile and afebrile, occurred in the following months, either generalized or lateralized on the left. At 13 months, myoclonias and brief atypical absences with progressive and jerky head fall or complete fall appeared several times a day. The child then started to self-stimulate by pattern fixation. She began walking at 10 months and talking at 11 months. Psychomotor development began to slow and hyperkinetic behavior appeared. A pathogenic de novo SCNA1 was identified. CT, MRI scans, and numerous biological investigations were normal. The EEGs showed numerous generalized spike waves and polyspike waves. Different antiepileptic drugs were prescribed without success. Lamotrigine triggered a worsening of the seizures.
The child was referred to the Centre Saint Paul-Hospital Henri Gastaut in Marseille, France at 2 years 9 months of age. She had daily seizures consisting of atypical absences and clonic seizures, lateralized either on the left or on the right. She presented with a slight ataxia, diffuse hypotonia, and mild psychomotor retardation. She spoke in sentence fragments and played in a stereotyped manner but interacted well socially. EEGs showed background at 5 to 6 Hz, numerous generalized spike waves and polyspike waves that increased during sleep, associated with multifocal anomalies. No effect of intermittent photic stimulation. Numerous atypical absences were recorded that were spontaneous or elicited by patterns. One brief, right hemiclonic seizure was recorded, followed by a brief right motor deficit. This case is a typical example of Dravet syndrome. The slight degree of mental deficit at this age might be explained by the absence of status epilepticus as well as the absence of prolonged seizures during the course of the disease. However, at 10 years of age, she had the classical evolution with persistence of seizures, mainly nocturnal, and moderate mental retardation with an inability to read or write.
Case 2. A typical case of Dravet syndrome is a 5-year-old boy who had his first-ever seizure during a mild febrile viral illness at age 5 months. The seizure consisted of right-sided clonic convulsions of the face and the limbs probably interspersed with tonic manifestations. This lasted for 30 minutes. An EEG 2 days later was normal. However, a similar seizure progressing to generalized convulsions occurred 6 weeks later again during a mild febrile illness. Subsequently, he had hemi-clonic seizures and a convulsive status epilepticus again with fever every 4 to 7 weeks despite treatment with phenobarbitone and relevant precautions for febrile convulsions. At 9 months of age, he developed similar clonic seizures without fever, and some of them were preceded by bilateral or unilateral clusters of myoclonic jerks involving the head, body, or limbs. A video-EEG documented clusters of myoclonic seizures associated with EEG changes compromising generalized polyspikes and slow waves mainly of higher amplitude on the left. Mental and motor development appeared delayed. Myoclonic jerks became a daily routine. Various appropriate medications were ineffective. At age 16 months he also had frequent atypical absences and nonconvulsive status epilepticus. MRI was normal. Mental deterioration was rapid; he was ataxic, and there were signs of mild bilateral pyramidal paresis. At 2 years of age, a new form of epileptic seizures appeared. He would suddenly become unresponsive, his head turned to one side, he fumbled with his clothes and was pale. EEG showed generalized discharges of polyspikes and slow waves, 2 to 3 Hz spike and slow waves, and sharp and slow wave foci originating from any possible brain location. EEG background was very slow. At 5 years of age, he is severely retarded and ataxic, he can hardly make any meaningful communication, he is entirely dependent, and he continues to have mainly generalized tonic-clonic seizures. He was found to be SCN1A gene positive. From the family history, his father had three simple febrile seizures from 2 to 3 years of age.
|
• Dravet syndrome is caused by pathogenic variants in the gene encoding the alpha-1 subunit of the voltage-gated sodium channel (SCN1A). | |
|
• Evidence of SCN1A dysfunction in different neuronal networks across the brain points towards a channelopathy model, causing the neurologic features of Dravet syndrome that is beyond purely seizure-related damage. | |
|
• Seizure susceptibility is caused by impaired GABAergic firing in cortical interneurons, lowering the seizure threshold. | |
|
• Impaired firing of GABAergic cerebellar Purkinje cells results in motor disorder and ataxia. | |
|
• A genetic change will present according to its severity, the genetic background of the individual, and environmental factors, and it will affect a variety of neuronal networks according to channel distribution. | |
|
• This already-vulnerable system may be susceptible to secondary aggravating events such as status epilepticus (18). |
Dravet syndrome is purely genetic (29; 46; 86; 137; 49; 62; 24; 117; 82). The most important gene implicated is SCN1A, encoding the alpha subunit of the neuronal voltage‐gated sodium channel (Nav1.1). About 80% to 90% of patients with Dravet syndrome are shown to carry pathogenic SCN1A variants. These are mainly point mutations (90%), including missense, nonsense, and other pathogenic variants. The remaining changes consist of genomic rearrangements (10%), including single or multiple exon deletions, or chromosomal rearrangements involving both the entire gene or other contiguous genes (89). Most SCN1A abnormalities are de novo, but familial pathogenic variants occur in 5% to 10% of cases and are usually missense in nature (86). Two major classes of pathogenic SCN1A variants are associated with Dravet syndrome: those that result in haploinsufficiency (protein-truncating variants) and those that result in an amino acid substitution (pathogenic missense variants). Single nucleotide substitutions, small indels, and even whole gene deletions have been reported, and well over 1000 different pathogenic variants have been described to date. Although protein-truncating variants occur randomly throughout the gene, pathogenic missense variants are clustered in mutational hotspots. Approximately half of all encountered variants are protein-truncating variants, including frameshift and nonsense variants, which are predicted to cause a complete loss of protein function. The other half are pathogenic missense variants leading to varying degrees (full/partial) of loss of function (16). Compared to pathogenic missense truncating variants, protein-truncating variants are associated with an earlier mean onset of prolonged seizures, myoclonic seizures, and atypical absence seizures. Conversely, pathogenic missense variants occur most frequently in the voltage and ion-pore regions where changes in amino acid polarity are greater in the Dravet group compared to genetic epilepsy with febrile seizures plus group (137). In one report, significant differences were found in the distribution of truncating and missense variants across the SCN1A sequence among healthy individuals, patients with Dravet syndrome, and those with milder forms of SCN1A-variant positive epilepsy (66). Additionally, the rate of cognitive decline was found more rapid for patients with truncation variants regardless of the age at seizure onset, whereas age at onset is a predictor of the rate of cognitive decline for patients with missense variants (66).
Pathogenic variants in the SCN1A gene have been identified in epilepsy patients with widely variable phenotypes and modes of inheritance (18; 13). Mild impairment of Nav1.1 function predisposes to febrile seizures, intermediate impairment leads to generalized epilepsy with febrile seizures plus, and severe or complete loss of function leads to the intractable seizures and comorbidities of Dravet syndrome (16).
SCN1A haploinsufficiency producing NaV1.1 dysfunction mainly affects GABAergic neurons. Brain NaV1.1 is primarily localized in fast-spiking inhibitory interneurons; thus, the mechanism of epileptogenesis in Dravet syndrome is hypothesized to be reduced inhibitory neurotransmission leading to brain hyperexcitability (136; 105). In cortical interneurons it explains the epilepsy, in the cerebellum the ataxia, in basal ganglia and motor neurons the crouching gait, in hypothalamus the thermodysregulation and sleep difficulties, and dysfunction in all these structures contributes to the psychomotor delay (57). Furthermore, pathogenic SCN1A variants lead to changes in the dopamine system that may contribute to the behavioral abnormalities in Dravet syndrome (84).
Equally, GABAergic dysfunction in Dravet syndrome has been documented in a study of human GABAA-evoked currents using cortical brain tissue from patients with Dravet syndrome (107).
However, how do we explain the SCN1A-negative patients with Dravet syndrome? The 10% to 20% of SCN1A-negative patients may be false-negative cases that are carrying a pathogenic SCN1A variant that was missed on testing. Alternatively, affected individuals might carry variants that are undetectable with current screening techniques, or less likely, they might harbor pathogenic variants elsewhere (43). Somatic mosaicism in SCN1A occurs in up to 7.5% of cases and should be considered in patients with Dravet syndrome when standard screening for pathogenic SCN1A variants is apparently negative (38; 97). There does not appear to be a specific epigenetic signature in SCN1A pathogenic variants (72). In SCN1A-negative patients with Dravet-like presentation, a number of alternative genes have been reported, including SCN1B, PCDH19, GABRA1, GABRG2, STXBP1, HCN1, CHD2, and KCNA2. Many of these genes, however, are associated with their own distinct clinical phenotype.
The most significant Dravet syndrome mimic is PCDH19-related epilepsy, which was discovered in a subset of female patients presenting with clinical features similar to those of Dravet syndrome. However, females with PCDH19-related epilepsy present with clusters of febrile seizures and long periods of seizure freedom with an overall more favorable outcome, compared to Dravet syndrome (121). Other candidate genes for Dravet syndrome have been reported, but there is currently an insufficient body of literature to support their causative role. These are probably imitators of Dravet syndrome (117). More information can be accessed at the following site:http://epilepsygenetics.net/.
The frequency of a family history of either epilepsy or febrile seizures is extremely variable, from 15% to more than 50%. At least 13 families with two affected children and seven pairs of affected twins (five monozygotic, one dizygotic, one unknown) have been reported (49).
Dravet syndrome is not associated with previous significant brain pathology. In adult postmortem cases of Dravet syndrome, there was remarkable preservation of neurons and interneurons in the neocortex and hippocampi (23).
Myoclonia in Dravet syndrome represents cortical myoclonus with features that are similar to those characterizing progressive myoclonus epilepsy but differs because it does not have a severely worsening course and is not commonly associated with increased somatosensory evoked potentials or enhanced long-loop reflexes (21).
Animal models of Dravet syndrome. There are two preclinical models for Dravet syndrome that are considered to have translational validity: mouse models and zebrafish models. Both preclinical models have established protocols for measuring a seizure-related outcome that corresponds to the seizure-related primary endpoints used in clinical trials. There are several mouse models that have been developed for Dravet syndrome, and all are based on the removal (knock-out) or mutation (knock-in) of the Scn1a gene. Zebrafish models of Dravet syndrome based on the reduction (knock-down) or mutation (knock-in) of the Scn1a gene ortholog in the fish have also proven to have translational value using seizure endpoints.
Gene targeted mouse models of Dravet syndrome associated pathogenic variants replicate patients' phenotype and show reduced gamma-aminobutyric acid (GABA)ergic inhibition (136; 100; 68). In one of them, studies demonstrated a similar striking temperature- and age-dependence of the onset and progression of generalized and myoclonic seizures as well as the appearance of ataxia (99). The authors have demonstrated that the hyperexcitability and seizure susceptibility in these models originates in a dysfunction of GABAergic inhibitory interneurons, caused by haploinsufficiency due to Scn1a pathogenic variants. Moreover, loss of these sodium channels in cerebellar Purkinje cells of mutant mice and patients with Dravet syndrome may be sufficient to also cause their ataxia (98).
In a move towards developing gene-specific, disease-modifying therapies, Han and colleagues were able to demonstrate in the F1:129S-Scn1a+/− mouse model of Dravet syndrome that antisense oligonucleotides (ASOs) increase Scn1a expression and reduce seizures and sudden unexpected death in epilepsy incidence (63). The authors used targeted augmentation of nuclear gene output (TANGO) technology, which modulates naturally occurring, nonproductive splicing events to increase target gene and protein expression and to ameliorate the disease phenotype. Han and colleagues identified antisense oligonucleotides that specifically increase the expression of productive Scn1a transcripts in human cell lines, as well as in mouse brain. A single intracerebroventricular dose of a lead antisense oligonucleotide at postnatal day 2 or 14 reduced the incidence of electrographic seizures and sudden unexpected death in epilepsy in their mouse model of Dravet syndrome. Increased expression of productive Scn1a transcript and NaV1.1 protein was confirmed in the brains of treated mice, suggesting that targeted augmentation of nuclear gene output may provide a gene-specific approach for the treatment of Dravet syndrome. The first in-human trial using this new technology commenced in the summer of 2020.
Work by Valassina and colleagues showed proof of concept, highlighting that disease phenotype reversibility can be achieved when Scn1a gene activity is efficiently reconstituted in brain cells (123). Using an Scn1a conditional knock-in mouse model (Scn1aStop/+) in which Scn1a expression could be reactivated on demand during the mouse lifetime, the authors showed that Scn1a gene reactivation applied even after symptom onset in a mouse model of Dravet syndrome, thereby providing evidence that disease-modifying treatments might also be effective in individuals who are already symptomatic.
A different gene-specific approach to treat Dravet syndrome has been the reduction of excitatory SCN8A voltage-gated sodium channel transcripts. Decreasing the abundance of Scn8a transcripts with an antisense oligonucleotide by 25% to 50% has been shown to delay seizure onset and prolong survival in a mouse model of SCN8A encephalopathy as well as Dravet syndrome (80).
Zebrafish with a pathogenic variant in Scn1a represent salient phenotypes associated with Dravet syndrome, including seizures, early fatality, and resistance to antiepileptic drugs (42; 116; 60). The zebrafish model of Dravet syndrome is also used for antiepileptic drug screening in Dravet syndrome. Griffin and colleagues showed that phenotypic screening of drug libraries in zebrafish scn1a mutants rapidly and successfully identifies new therapeutics (60). They demonstrated that clemizole binds to serotonin receptors, and its antiepileptic activity can be mimicked by drugs acting on serotonin signaling pathways, eg, trazodone and lorcaserin. Coincident with these zebrafish findings, five medically intractable patients with Dravet syndrome were treated with a clinically approved serotonin receptor agonist (lorcaserin), and some promising results in terms of reductions in seizure frequency or severity were observed. Sourbron and associates demonstrated serotonergic modulation as an effective treatment for Dravet syndrome in a zebrafish mutant model (116).
It has been shown that selective activation of NaV1.1 by venom peptide Hm1a restores the function of inhibitory interneurons from Dravet syndrome mice without affecting the firing of excitatory neurons. Intracerebroventricular infusion of Hm1a rescues Dravet syndrome mice from seizures and premature death (105).
Dravet syndrome is a rare disease with the following incidence by Countries:
|
• In the United States, 1 per 15,700 and 1 per 20,900 for de novo pathogenic SCN1A missense variants (133). | |
|
• In Scotland, the incidence of SCN1A-positive Dravet syndrome is at least 1 per 15,500 live births as established in a prospective population-based national cohort study (120; 119). | |
|
• In Denmark, based on a 6-year birth cohort from 2004 to 2009, the incidence of Dravet syndrome was estimated to be 1 per 22,000 (06). | |
|
• In France, the incidence was 1 per 20,000 or 30,000 (135). | |
|
• In Sweden, the estimated incidence was 1 in 33,000 live births (95% CI 1:20 400-1:56,200), and prevalence was 1 in 45,700 children aged less than 18 years (95% CI 1:33,800-1:63,400) (106). |
The prevalence of Dravet syndrome in epilepsy has been reported as 3% and 5% in patients with epilepsy onset in the first year of life, 7% and 6.1% in patients with epilepsy onset before the age of 3 years, and 1.4% in patients with epilepsy aged from 1 month to 1.5 years.
Whilst diagnostic pick-up in children has improved significantly over the years, there are still adult patients that remain undiagnosed, emphasizing the importance of considering a diagnosis of Dravet syndrome in adult patients with epilepsy and intellectual disability (115).
Dravet syndrome is classified as ORPHA 33069 in the Orphanet classification, which can be accessed at http://www.orpha.net.
It is not possible to prevent the occurrence of this epilepsy in a child because it is usually due to a de novo pathogenic variant. However, it should be possible to decrease its severity and achieve better seizure control by establishing the diagnosis early and managing the treatment in a rational way (see management).
Lennox-Gastaut syndrome is virtually excluded by a history of prolonged febrile, hemiclonic, and clonic seizures in the first year of life. Later, features such as drop attacks, axial tonic seizures, and specific EEG abnormalities with rapid, high-voltage rhythms during sleep are characteristic of Lennox-Gastaut syndrome. Distinguishing adult patients with Lennox-Gastaut syndrome from those with Dravet syndrome is challenging, but dystonia of the neck (antecollis) and parkinsonian gait are significantly more common in Dravet syndrome (01).
Myoclonic-atonic epilepsy. In some cases, myoclonic-atonic epilepsy may have onset at a similar age as in Dravet syndrome with febrile convulsions preceding by several months of nonfebrile tonic-clonic and myoclonic-atonic seizures, which are the hallmark of the disorder. During the course of the disease, there are neither focal seizures nor persistent focal EEG abnormalities. The main seizure type is myoclonic-atonic with sudden drop attacks, which are unusual in Dravet syndrome.
Progressive myoclonic epilepsies could be suspected and eliminated by biological, neurophysiological investigations and often by fundoscopy.
In spite of the knowledge of its genetic etiology, the diagnosis of Dravet syndrome remains a clinical diagnosis. Therefore, in an infant younger than 1 year, the occurrence of repeated and prolonged febrile, hemiclonic, and generalized clonic seizures must suggest the diagnosis of Dravet syndrome, which will be affirmed after the appearance of other manifestations such as myoclonic, atypical absences, focal seizures, early photosensitivity, and delay in development. The presence of a pathogenic SCN1A variant is highly suggestive of a Dravet syndrome diagnosis.
When a vaccination precedes the first seizure, the vaccination is only a triggering factor and not the cause of the subsequent epilepsy (07; 126; 132; 36). Vaccination-associated earlier seizure onset does not alter the disease course of Dravet syndrome, although the risk of subsequent vaccination-associated seizures is probably vaccine-specific (126).
Prediction tools. Brunklaus and colleagues developed a prediction model to objectively estimate at disease onset whether a child will develop Dravet syndrome versus genetic epilepsy with febrile seizures plus (GEFS+) (15). The authors used clinical (age at disease onset) and genetic data (SCN1A genetic score) from 1018 participants to train and validate the model, which outperformed any previous or alternative models. The SCN1A epilepsy prediction model calculates a patient’s probability (%) of developing Dravet syndrome versus GEFS+ and is available online at no cost: http://scn1a-prediction-model.broadinstitute.org/. Importantly, the prediction model is not intended to replace clinical judgment but to inform and complement clinical decision-making based on objective and quantifiable data.
Hattori and colleagues proposed a scoring system to predict Dravet syndrome amongst infants with febrile seizures before the first birthday based on the clinical characteristics of infants and the SCN1A variant analysis (64). Age of onset of febrile seizure less than or equal to 7 months, a total number of seizures greater than or equal to 5, and prolonged seizures lasting more than 10 minutes were regarded as significant risk factors for Dravet syndrome. Other factors highly predictive of this syndrome were hemiconvulsions, focal seizures, myoclonic seizures, and hot water-induced seizures. Pathogenic SCN1A missense and truncating variants were detected significantly more often in the Dravet syndrome group than in the non-Dravet syndrome group. Gallagher and colleagues showed that status epilepticus as an initial seizure type is a highly specific, but not sensitive, early feature of Dravet syndrome (56).
Differentiation from febrile seizures. In Dravet syndrome, (1) the onset is earlier (before 1 year of age) than in febrile convulsions (18 to 22 months); (2) the seizure type is mainly clonic and often unilateral instead of generalized tonic-clonic or purely tonic; (3) the seizure episodes are more prolonged and frequent, even when treated; and (4) seizures can be precipitated by low fever (less than 38 degrees C). The diagnosis becomes more certain if other seizure types, photosensitivity, and neurocognitive deficits appear (49).
Febrile seizures plus (FS+). Afebrile and mild body temperature variation below 38 degrees centigrade, focal motor seizures or alternating hemiconvulsive seizures, and vaccination-related first seizure are in favor of Dravet syndrome (76).
Early infantile gain-of-function SCN1A developmental and epileptic encephalopathy. A new spectrum of gain-of-function SCN1A-related disorders has been described (15). Affected patients have distinct clinical presentations differing by age of onset and presence of arthrogryposis or movement disorders. The most severely affected infants present with congenital arthrogryposis, neonatal-onset epilepsy in the first 3 days of life, tonic seizures, and apneas accompanied by a significant movement disorder and profound intellectual disability. Others present later, between 2 weeks and 3 months of age, with a severe early infantile developmental and epileptic encephalopathy and a movement disorder. Milder forms manifesting with focal epilepsies with mild or no intellectual disability have been described (87). Clinically, the majority of gain-of-function variant carriers responded to sodium channel blocker treatment without evidence of symptom exacerbation.
SCN1A gain-of-function phenotypes have key clinical features that are different to typical SCN1A loss-of-function Dravet syndrome. The very early neonatal-onset seizures accompanied by arthrogryposis are highly specific for the newly described gain-of-function phenotype. Febrile seizures are a hallmark of Dravet syndrome but are not frequently encountered in gain-of-function–associated phenotypes. Early evidence of profound intellectual disability paired with the emergence of a prominent movement disorder in the first years of life is highly unusual for Dravet syndrome and more suggestive of a gain-of-function phenotype. The responsiveness to sodium channel blocking agents is equally atypical for Dravet syndrome.
The diagnosis is based on the clinical findings detailed in the clinical manifestations section of this article. Genetic testing and EEG are the most useful diagnostic tests in confirming the clinical diagnosis. All other laboratory tests are normal. Brain imaging (CT and MRI) is also normal except for a few cases with dilatation of the cisterna magna or slight diffuse cerebral or cerebellar atrophy and a few cases with hippocampal sclerosis during the course of the disease, particularly bilateral atrophic changes in the hippocampus, amygdala, and the temporo-limbic cortex (49; 79). The general consensus is that there is no metabolic abnormality. Tissue biopsies are normal. In the absence of a confirmative SCN1A genetic testing result, other causes of progressive myoclonus should be excluded.
EEG. In Dravet syndrome, there are no specific interictal EEG features, and ictal EEG of seizures vary by age and patients. However, both interictal and ictal features change progressively with the evolution, and it is this progressive modification that often suggests the diagnosis (49; 69; 77; 71). EEG findings seemed to be age-dependent, variable among different patients, and not influenced by the presence or absence of pathogenic SCN1A variants.
The role of EEG in the diagnosis and classification of epilepsy syndromes, including Dravet syndrome, has been detailed by the ILAE Neurophysiology Task Force (71).
The interictal EEG is normal at onset and has a tendency to deteriorate with time. Subsequently, focal, multifocal, and generalized paroxysmal abnormalities appear with photosensitivity occurring in more than 40% of patients (49; 71; 125).
EEG patterns are usually normal in sleep, but after the first year, there is usually a gradual slowing of the background activity, which is more marked if seizures are frequent. Focal and multifocal abnormalities often appear only during sleep. In some cases, during slow sleep there is an unusual pattern constituted by a subcontinuous sequence of diffuse slow bi-triphasic spikes predominating at the frontocentral regions (96). This pattern sometimes persists until adulthood and is not necessarily associated with tonic seizures. From the onset, throughout the evolution, the interictal pattern varies considerably according to the different treatments and probably because of differences in the seizure threshold among patients.
The background is variable, often with either a transitory or a permanent slowing. During the later stages of the disease, paroxysmal activities tend to disappear on awake EEGs but are prominent on sleep EEGs.
The ictal EEG abnormalities depend on the types of recorded seizures, which are convulsive or nonconvulsive, polymorphic, focal, multifocal, or generalized. These seizure types are described by the ILAE Neurophysiology Task as follows (71):
Unilateral convulsive seizures. The ictal discharge is characterized by rhythmic (two to three per second) bilateral slow waves of higher amplitude over the hemisphere contralateral to the clinical manifestations and intermixed with 10 Hz recruiting rhythms. In other focal unilateral seizures, the EEG pattern can be variable with onset over the frontal or frontal-central regions of one hemisphere, or with bilateral asymmetric onset, but always predominant over the frontal areas. The EEG onset consists of “pseudo-rhythmic” spikes and waves, contralateral to the clinical manifestations, and it may be periodically interrupted by a 1- to 2-second flattening of the EEG. In some cases, the unilateral seizure can be preceded by isolated massive jerks related to generalized spike-wave discharges starting several minutes before the unilateral seizure onset. These myoclonic seizures can be on either side in the same patient; this alternating pattern can be a clue for the diagnosis of Dravet syndrome.
Generalized and “falsely generalized” tonic-clonic seizures. These are probably tonic-clonic seizures of focal onset, and they differ from one patient to another. They occur especially during NREM sleep. The EEG discharge is always bilateral but of different modalities: (a) bilateral slow spike or spike-wave sometimes followed by a brief attenuation, and then fast activities intermixed with slow waves; (b) the abnormalities are initially bilateral but become and remain asymmetric during the seizure; and (c) bilateral but asymmetric from onset. The postictal EEG shows either a diffuse flattening or slowing. These seizures can be preceded several minutes by isolated massive jerks, increasing progressively in frequency until the onset of the convulsive event.
Unstable convulsive seizures are characterized by varying topographic changes of the ictal discharge. The seizure can start over one area of one hemisphere and then spread to another area of the same hemisphere or to the entire hemisphere, or asymmetrically to both hemispheres. The pattern of propagation is variable from one seizure to another in the same patient. In general, polygraphic video-EEG recordings of both these seizure types have documented complex ictal evolution and a degree of discrepancy between clinical and EEG manifestations.
Tonic seizures. These occur only exceptionally and are associated with diffuse discharges of polyspikes at 8 to 9 Hz (96).
Myoclonic seizures. These are accompanied by generalized spike- or polyspike-wave discharges at 3 Hz or more, lasting 1 to 3 seconds and of higher voltage over the central-parietal areas. The electromyography (EMG) can show postmyoclonic inhibition corresponding to a head drop.
Focal (complex focal) seizures. Ictal EEG consists of a rhythmic sequence of fast polyspikes intermixed with theta activity during the last part of the seizure involving, for the duration of the seizure, the temporal-parietal-occipital region of one hemisphere or, more rarely, a frontal region.
Atypical absences. These are associated with generalized regular or irregular slow spike-wave discharges (usually less than 2.5 Hz) lasting 3 to 10 seconds, and they are accompanied by impaired consciousness and often myoclonic features.
Nonconvulsive status epilepticus (obtundation status epilepticus). EEG background activity is replaced by diffuse delta slow waves, superimposed with multifocal spikes and spike-waves, sharp waves, and generalized spike-and-wave discharges predominating over frontal-central areas, associated with myoclonic jerks or without a clinical correlate (atypical absence status).
Recording protocols for Dravet syndrome proposed by the ILAE Neurophysiology Task Force are as follows (71):
|
Basic level. Planned recording during wakefulness and sleep with polygraphy (ECG, respiration, bilateral deltoid EMG) with hyperventilation and intermittent photic stimulation. Intermittent photic stimulation is important to record early photoparoxysmal responses. | |
|
Advanced level. Long-duration video-EEG with polygraphy, as above (with at least bilateral deltoid EMG). Include intermittent photic stimulation and hyperventilation, as in the basic level. Hospitalization of the child during a cluster of febrile or afebrile seizures provides a good opportunity. |
Sleep is important to enhance the probability of recording interictal discharges and unstable and falsely generalized seizures. More extensive EMG polygraphy will better demonstrate the ictal polymorphism of these seizure types. Repeat sleep EEGs when clinically indicated (ie, appearance of a new seizure type) to better document the evolution of the syndrome.
|
a. Worsening with suspicion of minor status |
The EEG report should highlight atypical EEG or video-EEG features that may cast doubts on a diagnosis of Dravet syndrome, such as persistent focal slowing of background activity associated with spike or polyspike-wave focus, which suggests focal structural epilepsy or frequent tonic seizures suggesting Lennox-Gastaut syndrome.
Levels of EEG diagnosis. The clinical diagnosis of Dravet syndrome is supported by serial (and as frequent as possible) video-EEG evidence that will document seizure polymorphism. Early EEG photosensitivity and video-recorded falsely generalized or unstable seizures provide pertinent diagnostic clues. In Dravet syndrome, there are no specific interictal features, and seizure polymorphism is unlikely to be shown by even an ictal EEG.
In the untreated child with suspected Dravet syndrome, the first EEG (both basic and advanced recording levels) can increase diagnostic certainty if it shows a combination of the following:
|
• Background showing of focal or diffuse slowing in postictal recordings, following repeated admissions for atypical febrile seizures or status epilepticus | |
|
• Early-onset photosensitivity | |
|
• Myoclonic and either unstable or falsely generalized convulsive seizures |
Genetic testing. When a diagnosis of Dravet syndrome is suspected, genetic testing must be performed on the patient and the parents (65). Not only does a positive SCN1A test result confirm the clinical diagnosis, it also helps treatment choice, especially in the very young, and it prevents additional investigations (11). However, the absence of a pathogenic SCN1A variant does not preclude this diagnosis because about 10% to 20% of patients with Dravet syndrome are found to be SCN1A-negative. Conversely, the presence of such a mutation does not allow the establishment of diagnosis if the clinical features do not fit the clinical criteria. Indeed, pathogenic SCN1A variants are present in patients with several other epilepsy types (see etiology).
Because many of the clinical criteria are not apparent in the first year of life and infants with Dravet syndrome initially experience a seemingly normal development, genetic testing should be considered in patients exhibiting any of the following:
|
• Two or more prolonged seizures by 1 year of age (often triggered by fever) | |
|
• One prolonged seizure and any hemiclonic (sustained, rhythmic jerking of one side of the body) seizure by 1 year of age (often triggered by fever) | |
|
• Two seizures of any length that seem to affect alternating sides of the body | |
|
• History of seizures prior to 18 months of age and later emergence of myoclonic or absence seizures. |
Traditionally, SCN1A genetic testing was done through bi-directional DNA sequencing and multiplex ligation-dependent probe amplification (MLPA) (118). Sequence analysis of the entire coding region of the SCN1A gene is performed first. If a pathogenic variant is not identified, deletion/duplication analysis via MLPA is performed. Seventy-three percent to 92% of all Dravet syndrome-associated genetic variants can be detected using sequencing of the SCN1A gene, and an additional 8% to 27% can be identified using deletion/duplication analysis. In recent years, the majority of SCN1A genetic testing is performed via next-generation sequencing epilepsy gene panels or whole exome/genome sequencing.
When SCN1A sequencing and copy number variant studies (MLPA) are negative, mosaicism of the SCN1A gene should be taken into account, and testing for variants in other genes that have been associated with Dravet-like phenotypes, such as GABRA1, STXBP1, SCN1B, and PCDH19, should be considered (110; 38). Evidence suggests that despite being considered a monogenic condition, there can be widespread genomic influences on the phenotype in Dravet syndrome, including an excess of rare variants in epilepsy-related genes causing blended phenotypes (34).
Following testing, consultation with a genetic counselor is recommended.
Dravet syndrome requires a multidisciplinary approach to management that is demanding and often challenging for the family and the treating health care professionals and is mainly supportive (49; 129; Wirrell 2016; 124; 131). A management strategy should include:
|
• Evidence-based treatment of epileptic seizures with appropriate medications and nonpharmacological therapies | |
|
• Prevention and emergency management of prolonged seizures/status epilepticus | |
|
• Avoidance (where possible) of precipitating factors such as febrile diseases, hyperthermia, and photic stimuli, which can trigger prolonged generalized or unilateral seizures | |
|
• Addressing behavioral, psychological, and cognitive difficulties | |
|
• Physiotherapy and mobility support for patients with motor disabilities | |
|
• Family support and signposting towards dedicated national and international Dravet syndrome support networks (for example, Dravet Syndrome Foundation U.S., Dravet Syndrome U.K., etc.). |
Antiseizure medication. Recommendations are based in part on expert opinion and on a limited number of evidence-based studies evaluating the best therapies for Dravet syndrome.
Although many medications are used to treat the seizures associated with Dravet syndrome, patients typically have medically refractory epilepsy, and polytherapy is often required. Traditionally, sodium valproate, benzodiazepines (clobazam or clonazepam), stiripentol, topiramate, and levetiracetam have been reported as the most useful antiseizure medications (27; 46; 47; 49; 14; 113; 129; Wirrell 2016; 131; 33; 109).
Long-term use of sodium channel blockers (carbamazepine, phenytoin, or lamotrigine) is contraindicated and should be avoided because they typically exacerbate seizures and can worsen the developmental outcome (14; 39).
In a parent-reported survey on antiseizure medication use in the European population with 274 patients with Dravet syndrome, most patients were between 4 and 8 years of age, and 90% had known pathogenic variants in SCN1A (04). Their epilepsy was characterized by multiple seizure types, although only 45% had more than four tonic-clonic seizures per month on average. The most common drug combination was sodium valproate, clobazam, and stiripentol, with 42% of the total population currently taking stiripentol. Over a third of patients with Dravet syndrome had taken sodium channel blockers in the past, and most had motor and behavioral comorbidities.
Newer guidance based on an international Delphi process concluded that valproic acid, clobazam, stiripentol, and fenfluramine may be considered as first- or second-line maintenance therapies for seizures due to Dravet syndrome (130). This guidance is likely to change over the coming years as several disease-modifying therapies are in clinical development. Provided these are safe and efficacious, their use may be recommended in Dravet syndrome.
Stiripentol. Stiripentol in combination with sodium valproate and clobazam has been shown effective in reducing seizures in Dravet syndrome in a randomized placebo-controlled trial, demonstrating that 15 out of 21 patients (71%) had more than 50% reduction in the frequency of clonic (or tonic-clonic) seizures in the treatment group versus 1 of 20 patients (5%) in the placebo group (P< 0.001) (28). Stiripentol, an allosteric modulator of the GABAA receptor, has been licensed by the European Medicines Agency for “use in conjunction with clobazam and sodium valproate as adjunctive therapy of refractory generalized tonic-clonic seizures in patients with severe myoclonic epilepsy in infancy (SMEI, Dravet syndrome) whose seizures are not adequately controlled with clobazam and valproate.” In 2018, stiripentol also received FDA approval. A report highlights the need for ammonia monitoring due to an observed risk of hyperammonemic encephalopathy in adult patients treated with stiripentol (139).
Cannabidiol. There is a growing interest in using cannabidiol to treat patients with Dravet syndrome (40; 41; 75; 91). In a double-blind, placebo-controlled trial, 43% of patients reported at least a 50% reduction in convulsive seizure frequency in the treatment group versus 27% of patients in the placebo group (P=0.08). There was a significant decrease in the median frequency of convulsive seizures per month in the treatment group versus the placebo group (-22.8 percentage points; P=0.01) (40; 41). In 2018, cannabidiol obtained marketing authorization from the FDA. It received marketing authorization from the EMA in 2019 for use as adjunctive therapy of seizures associated with Dravet syndrome, in conjunction with clobazam.
Fenfluramine. Modulating certain serotonergic receptors has offered a new avenue for developing future therapeutics in Dravet syndrome (42; 116). Fenfluramine, an agent that releases serotonin by disrupting vesicular storage and reversing serotonin transporter function, has historically been found very effective in seizure control of patients with Dravet syndrome (25; 112). In a randomized, double-blind, placebo-controlled trial, patients in the treatment group had a 62.3% greater reduction in mean monthly convulsive seizure frequency compared with patients in the placebo group (P< 0.001) (74). Echocardiography monitoring is mandatory because of the known risks of valvular heart disease and pulmonary arterial hypertension, though associated with higher doses of fenfluramine than those used for Dravet syndrome. Fenfluramine was approved by the U.S. Food and Drug Administration in 2020, and in December 2020, the European Medicines Agency (EMA) granted marketing authorization.
Potassium bromide can be very effective for treating seizures in Dravet syndrome. In a Japanese study, bromide was the most effective and well-tolerated antiseizure medication in Dravet syndrome, especially among SCN1A-negative patients (113).
Cenobamate. Cenobamate is a novel antiseizure medication that is being used in adult patients with focal-onset, drug-resistant epilepsy. Makridis and colleagues reported four adult patients with Dravet syndrome in whom the use of cenobamate resulted in a significant seizure reduction of more than 80%, with a follow-up of up to 542 days (85). The authors propose cenobamate as an antiseizure medication that may lead to a clinically meaningful reduction of seizure frequency in adult patients with Dravet syndrome. Use of cenobamate in pediatric patients has been associated with lack of efficacy and seizure worsening in a retrospective cohort of six patients (median age = 8.5 years), highlighting the need for further investigation and functional studies to confirm effectiveness of cenobamate in adult and pediatric patients (19).
This retrospective and single-center study analyzed the therapeutic response to cenobamate in six pediatric patients (median age = 8.5 years) with Dravet syndrome and SCN1A LoF variants. Patients were treated with cenobamate for a median follow-up of 7 months. Baseline seizure frequency was assessed over 3 months prior to treatment initiation. Response was defined as a greater than or equal to 50% reduction in seizure frequency and worsening as a greater than or equal to 25% increase compared to baseline. No patient met the responder criteria. Three patients experienced seizure worsening, including one patient who started presenting status epilepticus after a long status epilepticus-free period, and the remaining three showed no improvement. Adverse effects included sleepiness, restlessness, and loss of appetite, and cenobamate was discontinued in all patients due to lack of effectiveness or seizure worsening.
Ketogenic diet. The ketogenic diet has been efficacious in several studies (22; 53). In one study, the efficacy and tolerability of the ketogenic diet in Dravet syndrome was compared with various standard antiseizure medication regimens and vagus nerve stimulation (53). Thirty-two children with genetically confirmed Dravet syndrome treated since 1999 were analyzed retrospectively. Overall response to the ketogenic diet was 70% at 3 months and 60% at 12 months. No status epilepticus occurred while patients were on the diet, and the frequencies of prolonged generalized and myoclonic seizures were reduced. No severe side effects requiring withdrawal of the ketogenic diet were observed. Although the effect of the ketogenic diet was independent of age at initiation, it had to be withdrawn due to noncompliance more frequently in older children that were fed solid food compared with infants treated with the liquid ketogenic formula. The ketogenic diet was not significantly inferior to the current gold standard antiepileptic drug triple combination of stiripentol plus valproate plus clobazam (89%), bromides (78%), sodium valproate alone (48%), topiramate (35%), and vagus nerve stimulation (37%), but it was significantly more effective than levetiracetam (30%; p=0.037).
Vagus nerve stimulation, neurosurgical intervention. Vagal nerve stimulation or corpus callosotomy have limited success (44).
Status epilepticus. Prevention of status epilepticus by avoiding precipitating factors (febrile illness, hyperthermia, photic stimulation) and by providing the best possible prophylactic antiseizure medication treatment and emergency protocols is important and may influence developmental outcome. Carers of patients with Dravet syndrome should be provided with home emergency medication (diazepam or midazolam) as well as an individualized status epilepticus protocol, which can be carried out at their local emergency room. Beyond the initial treatment with benzodiazepines and use of sodium valproate, there is no consensus on the optimal in-hospital management of convulsive status epilepticus for this syndrome (131).
Management of nonepileptic manifestations. Multidisciplinary team involvement—including neuropsychology, physio, occupational and speech therapy, and social work—is essential to maximize the quality of life and well-being of patients with Dravet syndrome and their families. Behavioral, psychological, and social problems and autism spectrum disorder are common. Early identification of behavioral and psychological disorders and targeted psychological intervention are essential components of holistic care in Dravet syndrome (12; 10; 67).
Pharmacologic treatment of nonepileptic manifestations of the disease is often necessary. Attention-deficit hyperactivity disorder is often encountered in patients with Dravet syndrome, and psychostimulants can be helpful for this indication. Other psychoactive drugs are less studied in this context.
Repeated cognitive evaluations are recommended to better understand the factors responsible for the cognitive disability, the respective roles of the seizures, and antiseizure medications and to adjust the educational support. The management of the behavioral disturbances is problematic, and psychological support can help the family.
Targeting SCN1A haploinsufficiency and NaV1.1 dysfunction could improve nonepileptic manifestations of this disease as well (18; 57).
Family support. The psychosocial impact of severe epilepsy on patients and families is often complex due to medical, psychological, economic, educational, personal, and social repercussions (124; 73; 95). As families of individuals with Dravet syndrome are severely affected by this, helping these families cope with the stress and psychosocial and financial problems is challenging and absolutely necessary (20; 55; 90). It is important to provide families with contact information from dedicated national and international Dravet syndrome support networks that can provide further assistance (for example, Dravet Syndrome Foundation U.S., Dravet Syndrome U.K., etc.).
Several agents targeted for Dravet syndrome are currently in the developmental stage: TAK935, lorcaserin, clemizole, huperzine analog, ataluren, selective sodium channel modulators and activators, antisense oligonucleotide therapy, and adenoviral vector therapy (109).
There is a growing pipeline of gene-specific, disease-modifying therapies with the potential to address the underlying channelopathy beyond the epilepsy. These include programs in the late-preclinical or clinical stage that target the disease biology at the protein or gene level. For example, restoration of SCN1A protein levels can be achieved by supplying the cell with an extra copy of the gene using a viral vector, or by increasing expression of the SCN1A protein using the healthy copy of the gene. The latter can be achieved by using antisense oligonucleotide (ASO) treatment to boost Nav1.1 protein expression or by upregulating gene expression of the healthy SCN1A copy. A phase 1/2 study using an antisense oligonucleotide approach started in August 2020, the results of which are expected to be published later in 2025, and a clinical trial upregulating SCN1A expression commenced in 2024, with results expected to be published within the next 2 years.
Overall, the Dravet syndrome pipeline comprises three approved drugs and several additional treatments in development from late preclinical phase to registration. A report discussing current and future opportunities, including advances in gene therapy, can be found at the following site: https://www.draccon.com/s/Dracaena-Dravet-syndrome-pipeline-review-2024.pdf.
Transition. The transition from pediatric to adult care is often a daunting prospect for families. Dravet syndrome is still seen as a primarily pediatric disease. Care delivery in the pediatric setting is often very comprehensive whilst it appears that care is less than optimal in adult health care systems (02). A review addresses this issue, flagging the main barriers in caring for adults with Dravet syndrome, and provides a useful two-page transition summary guide based on the authors’ personal expertise and literature review (02).
Outcome is unfavorable with regards to seizure control and neurocognitive development as illustrated in the prognosis and complications section.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Andreas Brunklaus MD
Dr. Brunklaus of the Royal Hospital for Children, Glasgow, UK has received consultancy and speaker honorariums from work for Biocodex, Encoded Therapeutics, Jazz Pharma, Servier, Stoke Therapeutics, and UCB.
See Profile
Solomon L Moshé MD
Dr. Moshé of Albert Einstein College of Medicine has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Epilepsy & Seizures
Jun. 02, 2026
Neuropharmacology & Neurotherapeutics
May. 11, 2026
Epilepsy & Seizures
May. 08, 2026
Epilepsy & Seizures
May. 01, 2026
Epilepsy & Seizures
Apr. 30, 2026
Epilepsy & Seizures
Apr. 17, 2026
Epilepsy & Seizures
Apr. 13, 2026
Epilepsy & Seizures
Apr. 08, 2026