

Sleep Disorders
Sudden infant death syndrome
Jul. 05, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Typical absence seizures (previously known as petit mal) are brief (a few seconds to half a minute) generalized epileptic seizures of abrupt onset and abrupt termination. They have two essential components: (1) clinically, the impairment of consciousness (absence) and (2) EEG generalized 2.5 to 4 Hz (not less than 2.5 Hz) spike-and-slow wave discharges. The impairment of consciousness may be conspicuous or inconspicuous, severe or less severe. Impairment of consciousness is a prerequisite of any definition of absence seizures. Typical absences are clusters of clinico-EEG manifestations that may be syndrome related. Absences may be the only type of seizures occurring in the patient, as in childhood absence epilepsy, or may be part of a phenotype in which other seizures eg, myoclonic, are the predominant type, as in juvenile myoclonic epilepsy. Typical absences are fundamentally different and pharmacologically unique compared to any other type of seizure, which also makes their treatment different. Antiseizure medications effective for focal seizures are contraindicated for absence seizures. In this article, the author details developments in the pathophysiology, genetics, and pharmacological treatment of absence seizures and related epileptic syndromes.
• Typical absence seizures (previously known as petit mal) are brief (few seconds to half a minute) generalized epileptic seizures of abrupt onset and abrupt termination. | |
• The defining manifestations of typical absence seizures are impairment of consciousness, conspicuous or inconspicuous, and generalized 2.5 to 4 Hz spike-wave discharges. | |
• Typical absences are fundamentally different and pharmacologically unique compared to any other type of seizures, which also makes their treatment different. | |
• The clinico-EEG manifestations of absence seizures are syndrome-related. Childhood and juvenile absence epilepsy are the archetypal syndromes of typical absences. | |
• Typical absence seizures are more frequent (pyknolepsy) in childhood absence epilepsy and less frequent (spaniolepsy) in Juvenile absence epilepsy. Briefer or even inconspicuous typical absence seizures are seen in juvenile myoclonic epilepsy concomitant or not with myoclonic jerks. | |
• Typical absences, particularly in adults, are frequently misdiagnosed as focal seizures with detrimental effect on patient management. | |
• Antiseizure medications effective for focal seizures are contraindicated for absence seizures. | |
• Genetic and nonpharmacologic strategies are emerging topics in the treatment of typical absences. |
Poupart, in 1705, was the first to describe absences (156). Tissot described a girl with absences “avec un tres leger movement dans les yeux” and frequent generalized tonic-clonic seizures (162). The term “epileptic absence” was first used by Calmeil (22). Shortly thereafter, Esquirol coined “petit mal” (48). Gowers gave a most accurate description of the absence seizures “without conspicuous convulsions” (64). Friedman reported a long-term favorable prognosis but believed that these absences were not epileptic (53). Sauer coined the name “pyknolepsy” (from the Greek word pyknos, meaning closely packed, dense, or aggregated) (142). Adie defined pyknolepsy as follows (02):
…a disease with an explosive onset between the ages of 4 and 12 years, of frequent short, very slight, monotonous minor epileptiform seizures of uniform severity, which recur almost daily for weeks, months, or years, are uninfluenced by anti-epileptic remedies, do not impede normal and psychical development, and ultimately cease spontaneously never to return. At most, the eyeballs may roll upwards, the lids may flicker, and the arms may be raised by a feeble tonic spasm. Clonic movements, however slight, obvious vasomotor disturbances, palpitations, and lassitude or confusion after the attacks are equivocal symptoms strongly suggestive of oncoming grave epilepsy, and for the present they should be considered as foreign to the more favorable disease. I shall be well satisfied if I have made it appear probable to you that there does exist a form of epilepsy in children which is distinguishable by its clinical features and in which the prognosis is always good (02). |
Gibbs and colleagues described the clinico-EEG characteristics of absences (55). Lennox in 1945 referred to the petit mal triad as absence, myoclonic, and akinetic seizures, and the introduction of trimethadione revolutionized the treatment of absence seizures (91). The petit mal triad of Lennox, which was misused and misunderstood, was clarified by the Commission on the International League Against Epilepsy with the differentiation of typical from atypical absences (54). Both Penry and colleagues and Stefan and colleagues studied absences with video-EEG (128; 150). Panayiotopoulos and colleagues described syndrome-related characterization of typical absence seizures with video-EEG analysis (125; 123; 124; 121).
A more detailed history of typical absence seizures can be found in a review publication (20).
Typical absences are brief, generalized epileptic seizures of sudden onset and termination. They have two essential components: (1) clinically the impairment of consciousness (absence) and (2) EEG generalized 2.5 to 4 Hz spike-and-slow wave discharges (38; 121; 144; 15).
Typical absences are a cluster of clinico-EEG manifestations that occur in idiopathic generalized epilepsies and may be syndrome related.
Impairment of consciousness may be conspicuous (severe or less severe) or inconspicuous (requiring a meticulous video-EEG assessment). Impairment of consciousness is prerequisite of any definition of absence seizures. Typical absence seizures are often associated with other symptoms, such as automatisms, autonomic disturbances, or regional (mouth or eyes) or widespread (head, limbs, and trunk) rhythmic or random myoclonia (15). The clinical expression of absence seizures is variable among children and during different absence seizures in the same child.
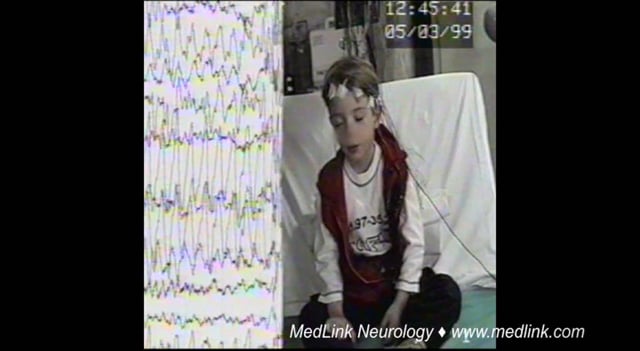
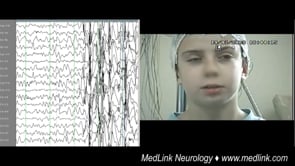
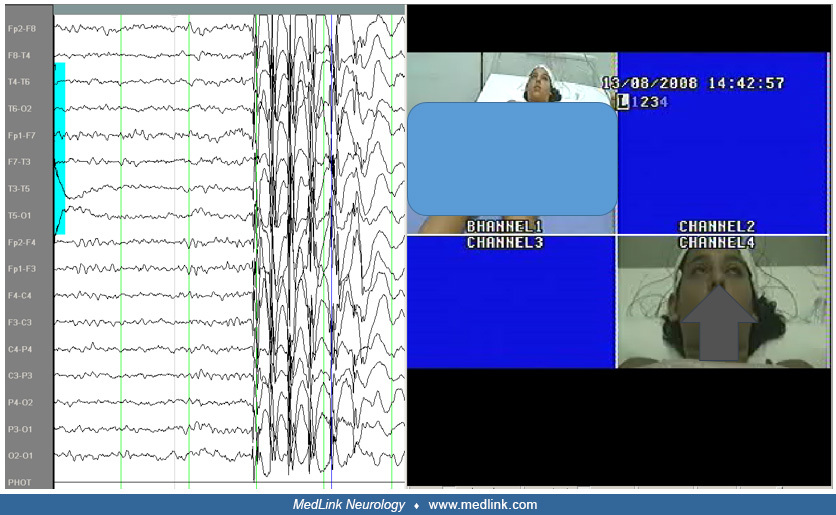
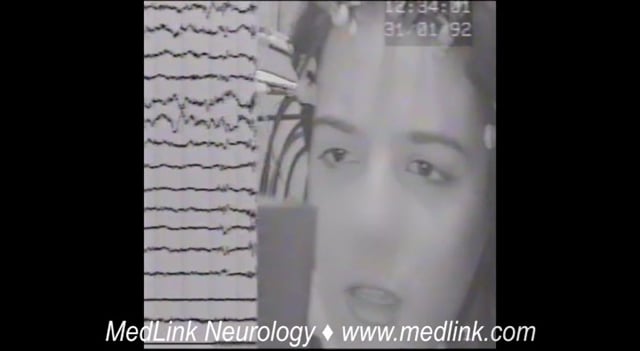
This 5.8-year-old boy was assessed following two generalized seizures a few days prior to admission. The first event occurred while sitting. He suddenly lifted his right upper limb, his head turned left, his eyes turned upwards...
The EEG discharge is rarely longer than 30 seconds (89). It may be continuous or fragmented; and it may be composed of single or multiple spikes, which may or may not be consistent with the slow wave.
The typical absence seizures may remit with age or be part of a phenotype with aberrant electroclinical features and variable evolution, requiring continuous treatment. Thus, the clinico-EEG manifestations of typical absences are, by definition, widespread and often not as classical as in their archetype, childhood absence epilepsy. The prefix “typical” is not to characterize them as “classical,” but to differentiate them from “atypical” absence seizures occurring mainly in symptomatic epilepsies.
Atypical absences differ from typical absences in the following ways (38; 37; 118; 119; 120; 06):
• Atypical absences occur only in the context of mainly severe symptomatic or unknown (previously known as cryptogenic) epilepsies of children with learning difficulties, who also suffer from frequent seizures of other types such as atonic, tonic, and myoclonic seizures. | |
• In atypical absences, onset and termination is not as abrupt as in typical absences, and changes in tone are more pronounced, slower, and last longer than in typical absence seizures. | |
• Ictal EEG of atypical absence is of slow, less than 2.5 Hz, spike-and-slow wave. The discharge is heterogeneous, bilateral, often asymmetrical, and may include irregular spike wave and slow wave complexes and fast or other paroxysmal activity. Background interictal EEG is usually abnormal with widespread slow activity. | |
• The final distinguishing characteristic involves the neural circuitry involved in the spike-wave discharge. In typical absence seizures, the epileptiform activity is constrained within thalamocortical circuitry. In contrast, there are experimental, clinical, behavioral, and neuroimaging data for the involvement of both thalamocortical and limbic circuitry in atypical absence seizures. Thus, the progression of ictal events and the mechanisms by which these recruit several brain areas may provide an explanation for the differing characteristics of typical versus atypical absence seizures (165). See atypical absence seizures. |
In brief, typical absence seizures differ from atypical absence seizures in terms of network circuitry involved, clinical manifestations, morphology, frequency of spike-and-wave discharges, and cognitive outcome.
The latest ILAE position paper of the operational classification of seizure types recognizes typical absences, atypical absences, absences with eyelid myoclonia, and myoclonic absences as types of generalized epileptic seizures (15). In the 2017 ILAE classification, absences were classified as "generalized nonmotor (absence) seizures." This definition was revised in 2025 to remove “nonmotor,” recognizing that typical absences often have motor phenomena, including automatisms, head movements, and blinking, particularly in eyelid myoclonia with absence and myoclonic absences (144; 15). The updated definition is as follows (15):
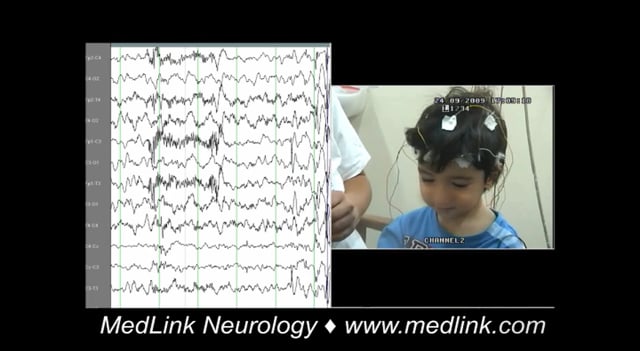
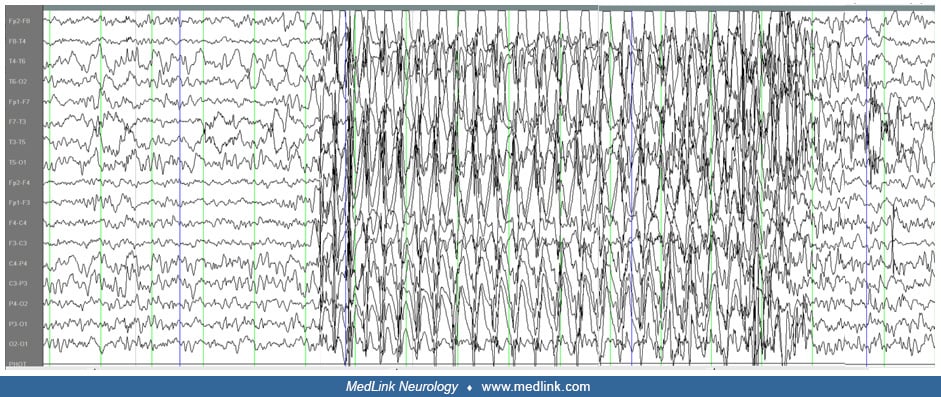
Typical absence seizure (3.1.1) is a generalized epileptic seizure characterized by sudden onset, interruption of ongoing activities, a blank stare (loss of facial expression), and possibly a brief upward deviation of the eyes. The patient is usually unresponsive; in most cases, awareness is impaired as well. However, occasionally, patients may recall the ictal events after the seizure (eg, test words given during the seizure). Oral or manual automatisms occur in 86% of patients, and eye involvement with blinking, eye opening, or subtle eyelid or perioral myoclonus occurs in 76.5% of patients. There is immediate return to normal activity, although children may be momentarily confused as they reorient themselves. Duration is a few seconds to half a minute (median 7 seconds; range: 2 to 26 secons*), but rarely they may last more than 30 seconds. Ictal EEG is characterized by regular, bilateral-synchronous (“generalized”) spike-waves. In the first seconds of seizure onset, the frequency of the spike-waves is around 3 Hz (range: 2.5 to 4 Hz in childhood absence epilepsy, 3 to 5.5 Hz in juvenile absence epilepsy). Disorganized discharges, defined by brief (less than 1 second) or transient interruptions in the ictal rhythm, or waveforms of different frequency or morphology are significantly less common in childhood absence epilepsy than in juvenile absence epilepsy. The seizures are typically provoked by hyperventilation in most untreated patients with childhood absence epilepsy. They may be provoked by intermittent photic stimulation as well. In childhood absence epilepsy, seizures typically occur multiple times per day but are often under-recognized. In juvenile absence epilepsy, typical absence seizures occur less than daily in the untreated state.
Caution. Individual absence seizure longer than 45 seconds or with a post-ictal phase, then consider focal seizure. | |
Caution. Onset of absence seizures less than 4 years, then consider glucose transporter disorders. |
EEG. Background/interictal/activation: Please refer to specific syndromes and etiologies in which this seizure type occurs.
Ictal EEG. Generalized spike-and-wave is mandatory. Regular 3 Hz generalized spike-and-wave occurs with typical absence seizures in childhood absence epilepsy. In absence seizures beginning in adolescence, faster irregular 3.5 to 6 Hz generalized spike-and-wave and polyspike-and-wave occurs.
Caution. Slow spike-and-wave (less than 2.5 Hz), then consider atypical absence seizures. |
• Absence with eyelid myoclonia: repetitive, rhythmic, fast (greater than 4 Hz) jerks of the eyelids, with upward deviation of the eyeballs and with head extension; often very frequent and provoked on eye closure, voluntary or on command, and by photic stimulation. | |
• Myoclonic absence: 3 Hz myoclonic jerks of upper limbs with tonic abduction. | |
• Atypical absence: more prolonged subtle altered awareness often seen in individuals with intellectual disability. | |
• Focal impaired awareness seizures | |
• Daydreaming or inattention or other. |
• Childhood absence epilepsy |
Transient loss of consciousness without conspicuous convulsions. In 1881 Gowers described transient loss of consciousness without conspicuous convulsions as follows (64):
|
A patient stops for a moment whatever he or she is doing, very often turns pale, may drop whatever is in the hand…There may be a slight stoop forward, or a slight quivering of the eyelids…The attack usually lasts only a few seconds. The return of the consciousness may be sudden and the patient, after the momentary lapse, may be in just the same state as before the attack, may even continue a sentence or action which was commenced before it came on, and suspended during the occurrence. |
The clinical manifestations of typical absence seizures vary significantly among patients (128; 34; 24; 76; 120; 06). Behavioral arrest and impairment of consciousness may be the only clinical symptoms, but this may be combined with other manifestations, such as motor automatisms and eye involvement, and absences may occur in clusters (84; 06).
Absence with impairment of consciousness only. The hallmark of the absence attack is a sudden onset and interruption of ongoing activities, often with a blank stare conspicuous or inconspicuous concomitant with brief generalized spike-and-wave discharges (06).
If the patient is speaking, speech is slowed or interrupted; if walking, he or she stands transfixed. Usually, the patient will be unresponsive when spoken to. The attack lasts from a few seconds to (rarely) half a minute and terminates as rapidly as it commenced.
In less severe absences, the patient may not stop his or her activities, though reaction time and speech may slow down. In their mildest form, absences may be inconspicuous to the patient and imperceptible to the observer, as disclosed on video-EEG recordings with errors and delays during breath-counting or other cognitive testing during hyperventilation. Typical absence seizures that are related to brief generalized spike-and-wave discharges are impossible to identify while the patient is counting or responding to a given world. This is particularly so in early childhood. Only when carefully assessing a video-EEG recording can we identify very brief loss of contacts. These brief typical absence seizures are seen in cases where myoclonic seizures and typical absence seizures coexist in the same phenotype of an idiopathic generalized epilepsy or syndrome.
This 5.8-year-old boy was assessed following two generalized seizures a few days prior to admission. The first event occurred while sitting. He suddenly lifted his right upper limb, his head turned left, his eyes turned upwards...
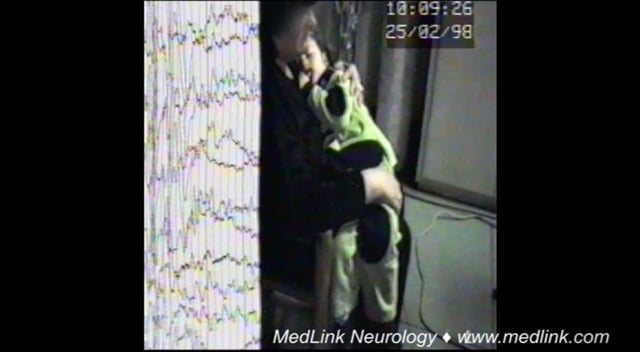
Absence with clonic components. During the absence as above, clonic motor manifestations, rhythmic or arrhythmic and singular or repetitive, are particularly frequent at the onset. They may also occur at any other stage of the seizure. The most common are clonic jerking of the eyelids, eyebrows, and eyeballs, together or independently, as well as random or repetitive eye closures. Fast eyelid flickering is probably the most common ictal clinical manifestation and may occur during brief generalized discharges without discernible impairment of consciousness. Myoclonias at the corner of the mouth and jerking of the jaw are less common. Myoclonic jerks of the head, body, and limbs may be singular or rhythmical and repetitive, and they may be mild or violent.
Absence with atonic components. Diminution of muscle tone is not unusual and may lead to drooping of the head and, occasionally, slumping of the trunk, dropping of the arms, and relaxation of the grip. Rarely, tone is sufficiently diminished to cause falls.
Absence with tonic components. Tonic muscular contraction may affect the extensor or the flexor muscles symmetrically or asymmetrically. The head may be drawn backwards (retropulsion) or to one side, and the trunk may arch.
Absence with automatisms. Automatisms are common in typical absences when consciousness is sufficiently impaired, and they are more likely to occur 4 to 6 seconds after onset.
Automatisms are more or less coordinated, adapted (eupractic or dyspractic), involuntary movements that may be an unconscious continuation of the preservative automatisms, de novo automatisms, or both. They vary in location and character from seizure to seizure, the same patient having both simple and complex absences. Perioral automatisms such as lip licking, smacking, swallowing, or mute speech movements are the most common. Scratching, fumbling with the clothes, and other limb automatisms are also common. Automatisms can be evoked, and their pattern and distribution can be changed by passive movements, postural repositioning, or other ictal stimulations.
Absence with autonomic components. Autonomic components consist of pallor and, less frequently, flushing, sweating, dilatation of pupils, and incontinence of urine. Mixed forms of absence are the rule rather than the exception.
Visual hallucinations and other focal symptoms. Exceptionally, patients may have visual hallucinations or visual illusions during the absence or absence status. Epigastric sensations may also be an unusual manifestation of adult absence epilepsy (173).
Absence status epilepticus. Absence status epilepticus may occur in 5% to 16% of cases (44). Absence status occurs in patients who have idiopathic generalized epilepsy with typical absences and consists of generalized, nonconvulsive seizures characterized by impairment of consciousness. Other intermittent manifestations include automatisms or subtle myoclonic, tonic, atonic, or autonomic phenomena. Persistent confusion or stupor are associated with characteristic 1- to 4-Hz spike-and-wave or polyspike-and-wave discharges on EEG, usually without other typical clinical manifestations of absence seizures. An absence seizure becomes absence status when it lasts longer than 10 to 15 minutes (time point t1). The time point t2 that may cause long-term consequences has not as yet been defined (160).
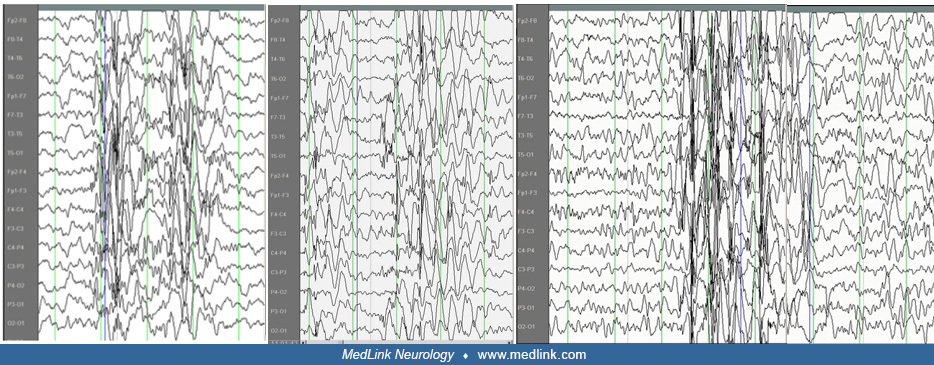
Clinical and EEG correlations. The ictal EEG is characteristic with usually regular and symmetrical generalized discharges of 3 to 4 Hz spike wave and slow wave complexes at a duration of 3 to 30 seconds. The discharge spike-wave frequency varies from onset to termination. It is usually faster and unstable in the opening phase (first second), becomes more regular and stable in the initial phase (first 3 seconds), and slows down towards the terminal phase (last 3 seconds). The intradischarge frequency and the relation of spike or multiple spike wave and slow wave frequently vary. Discharges are often of higher amplitude in the anterior regions.
In a large, structured database of 11,919 consecutive, routine video-EEG recordings, 2742 seizures were extracted and analyzed to determine the duration of epileptic seizure types in patients who did not undergo antiseizure medication withdrawal (89). The duration of typical absence seizures was rarely longer than 30 seconds (range: 2.75–26.5 s) compared to atypical absences, which lasted 2 to 100 seconds.
Although interictal background is not slowed in typical absences, a cohort study found paroxysmal interictal findings to occur frequently in children with typical absences, including those diagnosed with childhood absence epilepsy. These findings included occipital intermittent rhythmic delta activity, focal interictal discharges, and generalized spike-wave discharges, all of which were not found to influence prognosis in childhood absence epilepsy in this study (69). Another study similarly found frequent interictal focal spike-wave discharges in childhood absence patients during wakefulness and sleep, most commonly in the frontal regions (115).
Variability in clinical and EEG characteristics are seen in childhood absence epilepsies with aberrant electroclinical features and in those idiopathic generalized epilepsies or syndromes where typical absence seizures are part of the phenotypic expression.
There are reports of a few adult patents with idiopathic typical absence seizures and generalized paroxysmal fast activity in their EEG; these patients are usually pharmaco-resistant with bad prognosis (49; 68; 11; 14).
The diagnosis and management decision in adults is mainly based on self-reporting, but people often misreport their seizures (180). In a study, the misreporting rates in adults with absence seizures were assessed with inpatient video-EEG telemetry or outpatient ambulatory EEG; only 24% of 759 reported seizures to have an EEG correlate (131). Age, sex, time of epilepsy, video-EEG telemetry versus ambulatory EEG, epilepsy syndrome, or medication were not significant predictors of over-reporting, and patients were predominantly over-reporters or under-reporters, not both. Use of zonisamide or valproate was associated with under- reporting, possibly through an impact on attention. These findings indicate that self-reported absence seizures are a poor measure for treatment decisions due to both over- and under-reporting.
The prognosis and complications are syndrome related. Childhood absence epilepsy, if diagnosed with strict inclusion and exclusion criteria, usually remits within 2 to 5 years from onset (121; 120), with long-term seizure freedom often occurring between 10 and 14 years of age (134; 06). Although seizure freedom outcomes remain promising, studies are importantly recognizing the prevalence of significant behavioral and attention problems in children with absence epilepsy (8% compared to an expected 2.1% among healthy controls) (145; 18). Studies of absence medication effects on behavior have found valproate to have consistently poorer outcomes on behavior and cognition (145). Poor prognostic factors in childhood absence epilepsy include occurrence of motor seizures or generalized tonic-clonic seizures, absence persistence after 6 months of treatment, myoclonus, and neuropsychiatric comorbidities, such as attention difficulties (69). A cohort study found no relationship between interictal EEG discharges and long-term seizure-free outcome in childhood absence epilepsy (70). In all other syndromes, such as epilepsy with myoclonic absences or epilepsy with absence myoclonia, there is probably a lifelong liability to absences, myoclonic jerks, and generalized tonic-clonic seizures and a higher prevalence of cognitive comorbidities (40; 149); 64% to 80% of patients with epilepsy with eyelid myoclonia demonstrate pharmacoresistance (69). Generalized tonic-clonic seizures have also been described as a poor prognostic indicator for juvenile absence epilepsy. Patients with absence seizures in the context of juvenile myoclonic epilepsy are likely to require lifelong seizure therapy, with 78% recurrence rates among those who withdraw therapy (69).
For clinical vignettes of classical typical absence seizures, see childhood absence epilepsy and juvenile absence epilepsy.
• Calcium channel and GABA receptor mutations are known to be associated with absence seizures. However, genetic causes are likely polygenic and heterogeneous. | |
• Absence seizures originate in likely focal areas of the cortex and spread to the thalamic nuclei, with spike-and-wave discharges propagating via mutual connectivity. |
Absences are classified as an idiopathic generalized epilepsy by the ILAE (15), meaning there is no known etiology other than genetic. Calcium channel– and GABA receptor–associated mutations have long been documented as associated with absence seizures. Evidence supports polygenic and heterogeneous genetics underlying the disease. The pathophysiology of absences is generally understood to involve spike-and-wave discharges within the thalamocortical neuronal pathways.
Idiopathic generalized epilepsies with typical absences are genetically determined, as indicated by the high incidence of similar disorders among families. Genes associated with calcium channels (specifically T-type) and GABA A and B receptors are implicated in absence seizures (52; 30; 97; 134). However, inheritance is thought to be multifactorial and polygenic, and precise inheritance patterns are not known (06).
Feucht and colleagues found a significant association between a polymorphism in GABRB 3 in chromosome 15q11 and in 50 families with childhood absence epilepsy (52). Marini and colleagues found GABA-A receptor gamma2 subunit (GABRG2) gene mutations on chromosome 5 in a large family with childhood absence epilepsy and febrile seizures (97). Chen and colleagues found 68 variations, including 12 missense mutations in the calcium channel CACNA1H gene in patients with childhood absence epilepsy (30). The identified missense mutations occurred in the highly conserved residues of the T-type calcium channel gene. However, another study of 33 nuclear families, each with two or more individuals with childhood absence epilepsy, provided conclusive evidence that genes encoding GABA-A and GABA-B receptors, voltage-dependent calcium channels, and the ECA1 region on chromosome 8q do not independently account for the childhood absence trait in a majority of the families (135). More recently, Casillas-Espinosa and colleagues found that the R1584P gain-of-function mutation in the CaV3.2 T-type Ca2+ channel gene (Cacna1h) acted as a modulator of seizure development and also depressive-like behavior (28). Gokce-Samar and colleagues described the epilepsy phenotype of heterozygous variants in RAR-related orphan receptor B (RORB) as absence seizures with mild to moderate intellectual disability (63).
Several mutant mouse models that display behavioral arrest and spike-and-wave discharges have been created via genetic mutations that affect various voltage-gated calcium channels, the ubiquitous sodium/hydrogen exchanger on chromosome 4, and the AP-3delta protein on chromosome 10. Two genetic rat models of absence epilepsy, Genetic Absence Epilepsy Rat from Strasbourg (GAERS) and WAG/Rjj, carry polygenic mutations in calcium channels and various chromosomes and are thought to better represent human absence seizures, although no perfect animal model exists and more studies are needed to better identify implicated genes and their roles (80). A thalamocortical computer model demonstrated polygenic interaction; decreased GABAA conductance and increased T-type calcium channel conductance in concert were shown to convert spindle to spike-and-wave oscillations at a lower threshold than alone (87).
A study of 160 patients with confirmed genetic etiology and absence seizures showed high genetic heterogeneity with 56 different genes identified (most common were SLC2A1 (n=27), SLC6A1 (n=17), SYNGAP1 (n=15), CHD2 (n=11), and SCN1A (n=11); most mutations were de novo (13). Importantly, many of these patients were classified as having developmental and epileptic encephalopathies or epileptic encephalopathy, and only seven were diagnosed with childhood absence epilepsy. Most of the most common mutations were more commonly associated with atypical absences.
Of significant interest are reports documenting absence seizures mainly of early onset as a key feature of familial glucose transporter 1 (GLUT1) deficiency; these are discussed in detail in the Differential diagnosis section. GLUT1 facilitates the transport of glucose across the blood-brain barrier.
The pathophysiological mechanisms of absence seizures have been studied in humans and in various animal models with generalized spike-and-wave discharges associated with behavioral arrest. It appears that the generalized spike-and-wave discharges are generated and sustained by highly synchronized abnormal oscillatory rhythms in thalamocortical networks that mainly involve the neocortical pyramidal cells, the reticular nucleus, and the relay nuclei of the thalamus. Neither the cortex nor the thalamus alone can sustain these discharges, indicating that both structures are involved in their generation. Avoli reviewed the findings obtained over the past 70 years on the fundamental mechanisms underlying generalized spike-wave discharges associated with absence seizures (10). Studies in genetic animal models have demonstrated that spike-and-wave discharges are initiated in the primary somatosensory cortex and then rapidly propagate to motor cortices and thalamic nuclei. More specifically, deep layer pyramidal neurons of the somatosensory cortex are ictogenic neurons in absence seizures (42).
Despite much progress in our understanding of the neuropathological mechanisms, researchers still do not have a clear picture of the “widespread synchrony accounting for absence seizures” (77). Furthermore, following the traditional, strict dichotomy of “generalized” versus “focal,” generalized absence seizures may be counterproductive to the development of new ideas and treatment. In a 2022 publication, childhood absence seizures were discussed with respect to changing scientific concepts and newer findings (151). The data from semiology and structural and functional studies using quantitative electroencephalography, video-EEG monitoring, magnetoencephalography, magnetic resonance imaging, and positron emission tomography, as well as neuropathology, suggest a nosological spectrum from focal to generalized seizure-generating mechanisms. In fact, functional findings indicate that the frontal lobe, with its projections to other brain areas, may play an important role in generalized absence seizures and suggest new research approaches for diagnosis and treatment. Therefore, the distinction between generalized and focal epilepsy is, at times, imperfect. Frontal-onset absences constitute a specific subtype of childhood absence epilepsy, and the cortical initiating network is the major player in generating absence seizures (151).
Furthermore, typical absence seizures can be present with other seizure types and are commonly associated with being difficult to treat. Better understanding the underlying mechanisms of how absence seizures are generated and maintained may help to improve treatment outcomes. One study provides fMRI-informed magnetoencephalography multilayer network analyses showing beta/gamma cross-frequency and within-frequency coupling in the frontoparietal and frontofrontal regions during childhood absence seizures (159). These results provide a possible mechanistic explanation for previous fMRI and neurophysiology studies showing that focal areas of the precuneus and frontal lobe are critically important for the initiation and maintenance of generalized rhythmic spike-wave discharges characterizing absence seizures. In another study, the simultaneous changes in electrical (neuronal) and optical dynamics (hemodynamic, with changes in (Hb) and cerebral blood flow) of eight pediatric patients experiencing 25 typical childhood absence seizures during the transition from the interictal state to the absence seizure by simultaneously performing EEG, functional near-infrared spectroscopy (fNIRS), and diffuse correlation spectroscopy (DCS) approach were studied (112). About 20 seconds before the onset of the spike-and-wave discharge, they observed a transient direct current potential shift that correlated with alterations in fNIRS and DCS measurements of the cerebral hemodynamics detecting the preictal changes. Whether such information may ultimately be relevant for diagnostic and therapeutic approaches requires future confirmation.
Both inhibitory and excitatory neurotransmissions are involved in the genesis and control of absence seizures. This may be the result of an excessive cortical excitability due to an imbalance between inhibition and excitation or of excessive thalamic oscillations due to abnormal intrinsic neuronal properties under the control of inhibitory GABAergic mechanisms. It is likely that the generation of absences is due to a predominance of inhibitory activity, in contrast to generalized or focal convulsive seizures where an excess of excitatory activity is present (96).
Researchers have noted that there are complicated genetic mechanisms behind absence epilepsies, especially mutations in genes that code for GABA receptors, calcium channels, and ion channel proteins. These mutations impart an altered effect on the thalamocortical neuronal pathways. Simultaneous EEG and functional magnetic resonance imaging measurements document cortical deactivation and thalamic activation. Cortical deactivation is related to slow waves and disturbances of consciousness of varying degrees (101). Motor symptoms correspond to the spike component of the 3 Hz spike-and-wave-discharges, and thalamic activation can be interpreted as a response to overcome cortical deactivation (163).
The involvement of thalamus as the generator of the generalized spike-and-wave discharges is documented by the following: (1) stimulation of the medial thalamus induces a cortical generalized spike-and-wave discharge without leading to self-sustained activity and (2) thalamic neurons can intrinsically generate action potentials in both a tonic and a burst-firing mode. In a study of MRI diffusion and volumetry abnormalities in subcortical nuclei, 11 patients with absence seizures and 11 controls, patients had increased mean diffusivity values bilaterally in thalamus, putamen, and left caudate nucleus; increased fractional anisotropy value in bilateral caudate nuclei; and loss of volume in bilateral thalamus, putamen, and pallidum (95). These findings of microstructural changes provide further evidence for the involvement of thalamus and basal ganglia in propagation and modulation of spike-wave discharges in absence seizures. The thalamus has also been shown to contain pH-sensitive neurons that are activated in response to arterial carbon dioxide, providing a mechanism by which hyperventilation induces absence seizures (141).
The relative importance of the cortex in the initiation and synchronization of the generalized spike-and-wave discharges is mainly documented by the finding that following thalamectomy, instigation of generalized spike-and-wave discharges persists even though the thalamus is required to maintain rhythmicity once the discharges are established. Additional reports from animal models indicate that absence seizures arise from the somatosensory cortex, thus, providing support of a "focal hypothesis" of absence epilepsy (103; 132). During the first cycles of the seizure, the cortex drives the thalamus; thereafter, the cortex and thalamus drive each other, thus, amplifying and maintaining the rhythmic discharge. In this way, the "cortical focus" theory for generalized absence epilepsy bridges cortical and thalamic theories (104). Aarabi and colleagues applied linear and nonlinear synchronization measures to characterize the synchrony between cortical regions and detect cerebral epileptic states in scalp EEG recordings recorded prior to and during typical absence seizures (01). An overall rapid increase in the synchronization level between different cerebral regions was observed during the ictal state. During the interictal state, the degree of interdependence between EEG channels was significantly less than that observed in the ictal state (p< 0.05), although some patients experienced a preictal increase of synchronization. The authors concluded that their findings support the hypothesis of having a focal susceptibility of the cerebral cortex prior to absence seizures and further underlie that this susceptibility is reproducible and patient specific.
Various studies have suggested focal origins of absences. MEG studies of connectivity and cortical activations preceding spike-and-wave discharges found consistent low frequency frontal cortical discharges prior to the first generalized spikes that were preceded by an occipital source. This suggests a pathologically predisposed state towards synchronous seizure networks with increasing connectivity from interictal to preictal and ictal state, whereas the occipital and frontal low frequency early preictal sources demonstrate that the spike-wave discharges are not suddenly arising but gradually build up in a dynamic network (67). Additional MEG and EEG epoch studies support source initiation in the frontal cortex that then becomes more widespread throughout the cortex with decreased thalamic localization (172; 157; 82), whereas others show more synchronous cortical and thalamic activation (79). Studies of EEG-fMRI show consistent thalamic activation in order to overcome a variety of cortical deactivation changes (105). These studies advocate for focal brain areas responsible for absences although they appear bilaterally symmetric and generalized on conventional EEG.
Carney and colleagues studied 13 subjects with absence seizures during EEG-fMRI (27), finding two subgroups that could be distinguished by positive versus negative signal change in the dorsolateral prefrontal cortex (DLPFC). When the DLPFC-POS group was compared to the DLPFC-NEG group, time-course analysis revealed a larger positive blood oxygenation level-dependent deflection following onset of the absence seizures in cortical and subcortical areas beyond the DLPFC. Masterton and associates examined absence epilepsy subnetworks in eight patients revealed by event-related independent components analysis (eICA) of fMRI (98). Six eICA components were identified in a number of brain regions, including the striatum, representing distinct generalized spike-wave-related subnetworks. These findings suggest that the earliest activity associated with generalized spike-wave may be in posterior cortical regions and provide new evidence that the thalamostriate network may play a more important role in the generation of generalized spike-wave than suggested by previous studies (98). Liao and associates studied the dynamical intrinsic functional architecture of the brain during absence seizures with simultaneous EEG in order to understand the network mechanisms of seizure initiation, maintenance, and termination in absence epilepsy (93). They measured dynamic connectivity maps of the thalamus network and the default mode network, as well as functional connectome topologies, during the three different preictal, ictal and postictal intervals. The analysis of dynamic changes of anticorrelation between the thalamus and the default mode network is consistent with an inhibitory effect of seizures on the default mode of brain function, which gradually fades out after seizure onset. Also, the authors observed complex transitions of functional network topology, implicating adaptive reconfiguration of functional brain networks (93).
T-type calcium channels and GABA receptors are known to be genetically implicated in absences.
Some of the neurons involved in the cortico-thalamic-cortical system include cortical glutamatergic neurons originating on cortical layer VI and projecting to the nucleus reticularis of the thalamus; thalamic relay neurons that have excitatory projections to cortical pyramidal neurons; and neurons from the thalamic nucleus reticularis containing inhibitory GABA-ergic projections that connect with other neurons from the same nucleus and with thalamic relay neurons. These neurons do not connect directly with the cortex (126). Neurons from the thalamic nucleus reticularis can fire in an oscillatory pattern (for example, rhythmic bursts involved in the generation of sleep spindles) or continuously in single spikes (tonic firing during wakefulness). Shifts between these two firing patterns in the thalamic nucleus reticularis are modulated by spikes present in thalamocortical networks and neurons from the thalamic nucleus reticularis. These are mediated through low-threshold, transient calcium channels known as T-type channels, which are present in high densities in thalamic neurons responsible for normal and pathologic thalamocortical rhythms, including spike-and-wave discharges (31). After depolarization, T-type channels briefly allow calcium inflow before becoming inactivated. Reactivation requires a relatively long hyperpolarization facilitated by GABA-B receptors. Therefore, abnormal oscillatory rhythms can originate from T-type channel abnormalities, or from the increased GABA-B activity (126).
Ethosuximide exerts its anti-absence effect either by reducing thalamic low-threshold calcium currents probably through a direct channel-blocking action that is voltage dependent (36) or through a potent inhibitory effect in the perioral region of the primary somatosensory cortex (96).
Of neurotransmitters, GABA-B receptors play the most prominent role by eliciting long-standing hyperpolarization required to drive low threshold calcium channels for the initiation of sustained burst firing. GABA-B agonists, such as baclofen aggravate, and GABA-B antagonists suppress typical absences. GABAergic drugs (such as vigabatrin and tiagabine) are pro-absence substances; they interfere with the degradation of, and the re-uptake of, GABA (96; 120). The only exception of GABAergic activation inhibiting absences is that of the reticular thalamic nucleus, with exclusively GABA-A receptors; it functions as a pacemaker to synchronize thalamocortical oscillations (56; 75). Enhanced activation of GABA-A receptors in this nucleus decreases the pacemaking capacity of these cells, therefore, decreasing the likelihood of generating absence seizures.
Functional imaging with positron emission tomography demonstrates normal cerebral glucose metabolism and benzodiazepine receptor density in absence epilepsies with diffuse hypermetabolism during 3 Hz spike-and-wave discharges (139; 46). There is no evidence of any interictal overall abnormality of opioid receptors in idiopathic generalized epilepsy, but typical absences have been found to displace 11C-diprenorphine from the association areas of the neocortex. In contrast, binding of 11C-flumazenil to central benzodiazepine receptors has been shown to be unaffected by serial absences (46).
Ictal single photon emission computed tomography shows an overall increase in the cerebral blood flow (179) and may be useful in detecting secondarily generalized cases (78).
During absence seizures, there are pronounced changes in cerebral Hb-oxygenation (21). These start several seconds after the EEG-defined absence onset and outlast the clinically defined event by 20 s and 30 s. The changes consist of decrease in [oxy-Hb] and an increase in [deoxy-Hb] during absence seizures indicating a reduction of cortical activity.
A significant reduction in the amplitude of P300 on the visual and auditory continuous performance test was reported in patients with absence seizures (45). According to the authors, this indicates the impaired capacity of absence patients to mobilize and sustain attentional resources.
Microdysgenesis and other cerebral structural changes were reported in some patients with childhood absence epilepsy and juvenile absence epilepsy from autopsy (102) and MRI (175) studies. These results were not replicated in a blinded study (114). Microdysgenesis may be inconceivable for a benign, age-dependent, and age-limited epileptic syndrome such as childhood absence epilepsy, though the current ion channel hypothesis for the pathogenesis of idiopathic generalized epilepsy does not preclude microscopic or ultramicroscopic abnormalities.
|
• Typical absences are most common in children and in female patients. | |
|
• Myoclonic absences and juvenile absence epilepsy are more prevalent in adults compared to childhood absence epilepsy. |
Typical absences are more common in children than in adults and have a female prevalence. The female-to-male ratio was reported as 1.3:1 for the age group 4 to 8 years, 2:1 for children under the age of 4 years, and 1:1 for those older than 8 years of age (38; 37; 06). Idiopathic generalized epilepsies, including childhood absence epilepsy, juvenile myoclonic epilepsy, juvenile absence epilepsy, generalized tonic-clonic seizures, and other absence syndromes account for 15% to 20% of all epilepsies (32). Childhood absence epilepsy is the most prevalent idiopathic generalized epilepsy, with an annual incidence rate between 6.3 and 8.0 out of 100,000 in children younger than 15 years of age (70; 06). The frequency of childhood absence epilepsy among children with epilepsies is estimated to be 10% to 17%, with peak onset between 4 and 10 years of age (18; 69).
Though more common in children, typical absences also occur in approximately 10% of adults with epilepsies often combined with other types of generalized seizure (123).
Absence status epilepticus may occur in 5% to 16% of cases, with typical absence seizures starting before the age of 10 years (44). Absence status is rare among children in population-based studies (0% to 3%) (41).
The prevalence of typical absence status appears to be syndrome related, ranging from as high as 57.1% in perioral myoclonia and absences and 46.2% in “phantom” absences with generalized tonic-clonic seizure to as low as 6.7% in juvenile myoclonic epilepsy (03).
The best way to prevent absence seizures is to appropriately diagnose and treat early. In the case of absence status epilepticus or absence clusters, an abortive medication may be recommended (08).
Absences may be the only seizure type for a patient, particularly in childhood absence epilepsy. However, in other syndromes, such as juvenile absence epilepsy, typical absences may be the predominant type amongst other coexistent seizures (eg, myoclonic jerks and generalized tonic-clonic seizures). They may be mild and nonpredominant with myoclonic jerks and generalized tonic-clonic seizures as the main seizure type, as in juvenile myoclonic epilepsy. Typical absence status epilepticus may occur in approximately 25% of patients with idiopathic generalized epilepsies and typical absences (03).
Typical absence seizures of idiopathic generalized epilepsies are also easy to differentiate from atypical absences that occur only in the context of mainly severe symptomatic or cryptogenic epilepsies in children with learning difficulties who also suffer from frequent seizures of other types, such as atonic, tonic, and myoclonic seizures. These seizure types are discussed in the Terminology and clarifications section.
Although the differential diagnosis of typical absence seizures should be straightforward, they are frequently misdiagnosed as focal impaired consciousness seizures, especially in adults (Table 1) (123; 120; 06). A typical absence seizure can be reproduced by the hyperventilation test, whereas a focal impaired consciousness seizure cannot. Further, typical absence seizures occur daily, are shorter than 30 seconds, are frequently associated with bilateral facial myoclonic jerks or eyelid fluttering, are of sudden onset and termination, are not associated with complex behavioral automatisms or complex hallucinations or illusions, and there are no postictal manifestations.
In general, a detailed clinical history will differentiate between absence seizures and nonepileptic staring spells in the majority of cases. Such paroxysmal nonepileptic events are observed in children with inattention, “daydreaming,” Munchausen by proxy–reported apneas, and nonepileptic absence episodes related to Rett syndrome. Hearing loss and intellectual disability may also cause perceived inattention and staring spells. During routine examination, typical absence seizures can be provoked by hyperventilation in almost 90% of the cases. The abrupt onset and termination of typical absence seizures and the provocation by hyperventilation makes the diagnosis easy, particularly when capturing an episode during a video-EEG recording.
|
Typical absences |
Focal impaired consciousness | |
|
Clinical criteria |
|
|
|
Duration for less than 30 seconds* |
As a rule |
Exceptional |
|
Daily in frequency |
As a rule |
Rare |
|
Reproduced by hyperventilation |
As a rule |
Exceptional |
|
EEG criteria | ||
|
Ictal generalized 3 to 4 Hz spike-and-wave |
Exclusive |
Never |
|
Normal EEG in untreated state |
Exceptional |
Frequent |
|
| ||
Childhood and juvenile absence epilepsy are the most pure syndromes of typical absence seizures, with ictal manifestations showing similar clinical and EEG findings. In childhood absence epilepsy, age-related typical absences are the only, the most disturbing, and the most characteristic seizure type. The impairment of consciousness is more conspicuous than in any other syndrome, and the EEG discharge is harmonious with no polyspikes or fragmentations.
In contrast, in juvenile absence epilepsy they are milder and less frequent. Furthermore, juvenile absence epilepsy often manifests with infrequent generalized tonic-clonic seizures and sporadic, infrequent myoclonic jerks.
Absence seizures of early onset (a few months to 4 years) are particularly demanding in their diagnosis and management (37; 73; 120). There are two main categories in this age group: the nonmyoclonic and the myoclonic. In the nonmyoclonic group, there are cases with spanioleptic and pyknoleptic absence seizures. The spanioleptic are reminiscent of juvenile absence epilepsy, whereas the pyknoleptic are either childhood absence epilepsy-like or an early expression of juvenile myoclonic epilepsy with or without positive response to intermittent photic stimulation (37). These are not a specific expression of a distinct syndrome. The only syndrome recognized easier in this age group is eyelid myoclonia and absences evoked with voluntary or on-command eye closure (37). Absence seizures of early onset may also herald more severe forms of idiopathic and symptomatic generalized epilepsies (25; 57; 169; 04; 58). In the myoclonic group, jerks may be the only seizure type or jerks may combine with typical absences or generalized tonic-clonic seizures or even with absence seizures and generalized tonic-clonic seizures. Conspicuous or inconspicuous absences may precede or follow a brief jerk.
If diagnosis of epilepsies by age at onset is to be followed, idiopathic generalized epilepsies exist in infancy and childhood, whereas typical absence seizures are part of the phenotype either as the only seizure type or combined with myoclonic jerks or generalized tonic-clonic seizures. In this respect, not all typical absence seizures occurring in childhood belong to the syndrome childhood absence epilepsy. Some of these epilepsies are well recognized or not yet recognized as syndromes by the ILAE Commission on Classification, and some other in infancy and early childhood remind us of syndromes we see in childhood and adolescence. The ILAE defined the Idiopathic Generalized Epilepsy Syndromes in 2022 (71).
The absence syndromes in childhood and adolescence comprise several groups and subgroups of syndromes with or without photosensitivity.
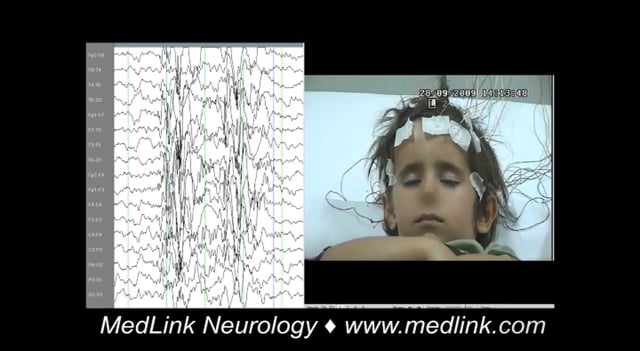
Childhood absence epilepsy (previously known as pyknolepsy). Childhood absence epilepsy is the archetypal syndrome of typical absence seizures (72; 120; 100). This is an idiopathic (genetic) generalized epilepsy with frequent (tens or hundreds per day) typical absence seizures, characterized by an abrupt onset and termination of a brief (4 to 30 seconds but usually around 10 seconds), complete or partial loss of consciousness that occur in otherwise normal children. The associated EEG 3 Hz generalized spike-and-wave discharge (range 2.5 to 4Hz) is bilateral, symmetrical, synchronous, and usually most prominent in frontocentral regions (37). Typical age of onset is before 10 years, with a peak at 5 years of age. Remission occurs in 75% to 90% of patients off antiseizure medications long-term (70). Clinically, there is abrupt and severe impairment of consciousness. The eyes spontaneously open, and overbreathing, speech, and other voluntary activity stop within the first 3 seconds of the discharge. Automatisms are frequent. The eyes stare or move slowly; random eyelid blinking (usually not sustained) may occur. The background EEG is normal.
Juvenile absence epilepsy. Juvenile absence epilepsy is an idiopathic generalized epilepsy mainly characterized by typical absences that are less frequent (spaniolepsy) and probably not as severe as childhood absence epilepsy (72; 120). Generalized tonic-clonic seizures and myoclonic jerks are part of the phenotype in 80% and 15% to 30% respectively, and absence status epilepticus occurs in about 20% of cases (37). Age at onset is between 7 and 16 years with a peak at 10 to 12 years. Juvenile absence epilepsy is a lifelong disorder, but absences tend to become less severe with age. Almost half of patients have poor seizure control with a high rate of pharmacoresistance (40). The ictal EEG shows generalized, spike or multiple spike-and-slow waves at 2.5 to 4 Hz.
Exclusion criteria for juvenile absence epilepsy. Mild impairment of consciousness, brief ictal discharges (less than 4 seconds), eyelid or perioral myoclonus, rhythmic limb jerking, and single or arrhythmic myoclonic jerks during the absence ictus are incompatible with juvenile absence epilepsy. Visual, photo-sensitive, and other sensory precipitation of absences may be against the diagnosis of juvenile absence epilepsy.
Myoclonic absence epilepsy. Myoclonic absence epilepsy is a rare generalized unknown (cryptogenic) or symptomatic epilepsy, where typical absence and myoclonic jerks are the predominant type of seizures in the phenotype (37). Severe bilateral rhythmical clonic jerks, often associated with a tonic contraction, occur during the absence concomitant with generalized spike-and-wave discharges in the EEG. Each spike correlates with a jerk and the slow wave with the tonic contraction. Two thirds are symptomatic and one third idiopathic or unknown. Approximately 45% have intellectual disability from start and 70% final. Almost 50% persist above the age of 20 years old.
Eyelid myoclonia and absences (formerly known as Jeavons syndrome). Eyelid myoclonia “and absences” is preferable to “with absences” for the reason that absence seizures provoked during photic stimulation are evoked independent to eye closure and with eyes open. Eyelid myoclonia and absences is an idiopathic generalized epilepsy manifested with eyelid myoclonia and typical absences as the predominant seizures in the phenotype (37; 149). Eyelid myoclonia consists of marked, rhythmic, and fast jerks of the eyelids immediately after voluntary or on-command eye closure in the light and is often associated with jerky upward deviation of the eyeballs and retropulsion of the head. The seizures are brief (3 to 6 seconds) and occur mainly after eye closure. Onset is usually in early childhood. All patients are highly photosensitive in childhood, but this declines with age. Infrequent generalized tonic-clonic seizures are inevitable in the long term, and they are likely to occur after sleep deprivation, fatigue, and alcohol indulgence. Myoclonic jerks of the limbs may occur but are infrequent and random. Eyelid myoclonia and absences may be resistant to treatment and lifelong. The EEG ictal manifestations consist mainly of generalized polyspikes and slow waves at 3 to 6 Hz, though these are more likely to occur immediately after eye closure in an illuminated room. Total darkness abolishes the abnormalities related to eye closure. Photoparoxysmal responses are recorded from all untreated young patients.
Juvenile myoclonic epilepsy. Juvenile myoclonic epilepsy is a genetically determined, common idiopathic generalized epilepsy. Prevalence is 5% to 11% among adult and adolescent patients with other epilepsies, and both sexes are equally affected. Juvenile myoclonic epilepsy is characterized by myoclonic jerks on awakening, generalized tonic-clonic seizures, and typical absences in more than one third of the patients. The seizures have an age-related onset with absences first appearing either in childhood or adolescence, followed by myoclonic jerks and generalized tonic-clonic seizures in the middle teens. Rarely, absence seizures concomitant with myoclonic seizures appearing before the age of 3 years may herald juvenile myoclonic epilepsy. In such cases, the final diagnosis is made at an older age when relapses occur after treatment is discontinued and the classical electroclinical features of juvenile myoclonic epilepsy are recorded. Seizure-precipitating factors like sleep deprivation and fatigue, alcohol, photosensitivity, and mental and psychological arousal are prominent. All seizures are probably lifelong, although absences may become less severe with age; jerks and generalized tonic-clonic seizures commonly improve after the fourth decade of life. Typical absences are usually highly mild and simple (with no automatisms or localized limb jerks), and impairment of consciousness is subtle. Generalized discharges of 3 to 6 Hz spike-and-waves have an unstable intradischarge frequency with fragmentations and multiple spikes. In the author’s experience, myoclonic jerks occur in all by definition and typical absences in at least 48%, generalized tonic-clonic in 60%, and positive response to intermittent photic stimulation in 75% of cases (37). Absence seizures (conspicuous or inconspicuous) are usually evoked during photic stimulation and may precede or follow a jerk.
The following are additional possible syndromes with typical absence seizures (118; 119; 120; 146; 176):
Facial (perioral, eyebrow, or mixed) myoclonia and absences. This is an idiopathic generalized epilepsy with onset in childhood or early adolescence (37; 120; 146; 176). Rhythmic myoclonus of the perioral or eyebrow muscles occurs during the absence, together with a variable impairment of consciousness. The absences are frequent and may be brief. Absence status is common. Generalized tonic-clonic seizures are common but infrequent and present in 93.3% of patients with perioral myoclonia (176) and 61.4% of patients with eyelid myoclonia (146). Clusters of absences or absence status usually precede generalized tonic-clonic seizures. Absences and generalized tonic-clonic seizures may be resistant to medication, unremitting, and possibly lifelong. Other patients may have a mild but long-standing course. Ictal EEG shows high-amplitude generalized discharges of typical but often irregular, rhythmic multiple spike waves and slow waves at 2 to 4.5 Hz. Interictal EEG in perioral myoclonia also shows generalized spike and wave. Photosensitivity is common in absences with eyelid myoclonia but may decrease with age or in response to medications (146).
GLUT1 deficiency syndrome. Of clinical importance is the diagnosis of absence seizures associated with glucose transporter-1 (GLUT1) deficiency syndrome (137; 153; 90; 106; 136; 57; 62; 168; 116) that responds to ketogenic diet (85). GLUT1 deficiency syndrome (OMIM 606777) is an autosomal dominant disorder due to SLC2A1 gene mutations that result in abnormal transport of glucose into the brain. GLUT1 deficiency syndrome has been associated with a variety of clinical phenotypes and severity (90; 109). GLUT1 deficiency syndrome is characterized by seizures mainly of early onset and refractory to antiepileptic drugs; deceleration of head growth; delays in mental and motor development; spasticity; ataxia; dystonia; dysarthria; opsoclonus; paroxysmal, exercise-induced dyskinesia; and other paroxysmal neurologic manifestations, often worse prior to meals. Affected infants appear normal at birth. Apneic episodes and abnormal episodic eye movements simulating opsoclonus may precede the onset of seizures by several months. Seizures begin between the ages of 1 and 4 months in 90% of cases. The frequency and severity of seizures vary among affected individuals. Typical or atypical absences, mainly of early onset, are the most prominent seizure types. Typical absences may imitate various syndromes of idiopathic generalized epilepsy with absences, such as epilepsy with myoclonic absences or childhood or juvenile absence epilepsy. Ketogenic diet is highly effective in controlling the seizures. An early diagnosis and early start of a ketogenic diet may prevent deterioration. Phenobarbital is contraindicated.
Clinical diagnostic clues for GLUT1 deficiency syndrome include early-onset drug-resistant epilepsy, delayed development, cognitive impairment, movement disorders such as dystonia or ataxia, and family history. Symptoms and EEG abnormalities often worsen during the fasted state and improve after eating. Cerebral imaging is normal. In GLUT1 deficiency there is (1) reduced CSF glucose concentration (hypoglycorrhachia) that seldomly exceeds 40 mg/dL; (2) low ratio of CSF glucose concentration to blood glucose concentration (approximately 0.33±0.01; normal ratio: 0.65±0.01); and (3) normal blood glucose concentration. Molecular genetic testing for SLC2A, which is the only gene known to be associated with GLUT1 deficiency, is now clinically available. Families with members manifesting various types of idiopathic generalized epilepsy with absences (early-onset absence seizures, childhood or juvenile absence epilepsy, juvenile myoclonic epilepsy) should be tested for SLC2A1 gene mutations (152). However, in a study of 84 children with rigorous diagnosis of early-onset absence seizures, no mutations in the SLC2A1 gene were detected (05). A study found 27 of 160 patients with absence seizures to have an SLC2A1 gene mutation (13).
The syndrome of phantom absences and generalized tonic-clonic seizures. “Phantom absences” denote typical absences that are so mild that they are inconspicuous to the patient and imperceptible to the observer. They are disclosed by video-EEG recording and breath-counting or other cognitive testing during hyperventilation with brief (usually 3 to 4 seconds) 3 to 4 Hz spike or multiple spike wave and slow wave discharges. The absences are simple, occasionally with eyelid blinking. They may be clinically unrecognized, but they usually manifest in adult life with generalized tonic-clonic seizures, and often with absence status epilepticus (124; 88).
A 31-year-old female with a 16-year history of paroxysmal convulsions and 26-year history of unrecognized “phantom absences” was reported (Jiang and Zhou 2023). She was finally diagnosed with idiopathic generalized epilepsy with phantom absences, absence status, and generalized tonic-clonic seizures. The authors stress the importance of video-EEG recording in identifying phantom absences as the mild and temporary impairment of cognitive function fails to attract the attention of patients, their families, and clinicians (124).
Typical absences with specific modes of precipitation (reflex absences). Absence seizures can be provoked with specific modes of precipitation (eg, photic, pattern, video games, thinking, reading, fixation-off).
Absences with single myoclonic jerks during the absence ictus. Typical absences with single, often violent jerks of the head, body, or limbs during the absences ictus may appear in early childhood and continue in adult life, often with other types of generalized seizures. They are frequently difficult to treat and may be associated with a bad prognosis.
Symptomatic and unknown (cryptogenic) absences. Although typical absences are considered the paradigm seizure type of idiopathic generalized epilepsy, they may occasionally be symptomatic, arising as a consequence of a known disorder of the central nervous system (51). Symptomatic and cryptogenic absences may be focal or diffuse, traumatic, metabolic, or inflammatory. In most cases an etiologic link is not proven, and it is likely that they are coincidental. The mesial surfaces of the frontal lobe are most likely to generate typical absences. “Brief blank spells” with 3 Hz spike-wave EEG paroxysms mainly due to subependymal heterotopia have been reported (133).
Increasing evidence from animal and clinical studies has indicated that focal changes may occur prior to absence seizures; however, the relationship of absence seizures with epileptogenic lesions remains unclear. Clinical, imaging, and electrophysiological data of 16 patients who had absence seizures and structural lesions were retrospectively collected and analyzed (154). Nine had Lennox-Gastaut syndrome and seven had non-Lennox-Gastaut syndrome. Three out of seven patients with non-Lennox-Gastaut syndrome had atypical absence seizures and four had typical absence seizures. All 16 patients had structural lesions on imaging. The lesions were located in the posterior cortex in nine patients (9/16), and in the four with typical absence seizures, the lesions were located in deep brain regions, including right thalamus, cingulate gyrus, deep parietal lobe, and insular lobe. All patients underwent lesion resection and had seizure-free outcomes during follow-up. The intelligence quotient also improved by 10.71 ± 3.90 one year after surgery.
The absence seizures in Dravet syndrome have been studied by Tsuda and colleagues (161). The mean age at their onset was 16.2 +/- 7.1 months, their duration ranged from 2 to 180 seconds, and the frequency of the EEG discharge was from 2 to 4 Hz (median=3 Hz). Clinical manifestations included eyelid-myoclonus (17%) and generalized myoclonus (44%). Absence seizures in Dravet syndrome are characterized by an early age of onset, a high incidence of irregular and disorganized 3 Hz generalized spike-and-wave morphology, and the frequent association of generalized myoclonic movement as well as the absence of automatism as compared to typical absence seizures.
An observational EEG study found comparable median seizure duration between typical absence (7.25 sec), myoclonic absence (7.5 sec) and absence with eyelid myoclonia (4.5 sec) (89). A separate study found absences associated with juvenile myoclonic epilepsy to be of shorter duration but that other syndromes were not associated with seizure duration; neither were eye opening, eyelid movements, and level of awareness (140).
In a commentary on diagnostic strategies for the diagnosis of typical absences and related syndromes, Scheffer and Berg rightly emphasized the following (143):
|
We should not strive to achieve a specific result but to use valid, accepted methods that lead us to the most reliable result, one we hope is as close as possible to an elusive truth, whatever that may be. As researchers, we need to remain dispassionate about our findings and accepting of new data that challenge or even refute our previously held views of epilepsy syndromes, constantly but methodically revising and refining our understanding to make it more robust. |
The diagnosis of typical absence seizures with conspicuous, profound impairment of consciousness in childhood is relatively straightforward. Their brief duration with abrupt onset and termination, the high frequency (as a rule), and nearly invariable provocation with hyperventilation makes them one of the easiest types of seizures to diagnose. Automatisms, such as lip smacking or licking, swallowing, fumbling, or aimless walking are common, and these should not be taken as evidence of focal impaired consciousness seizures. In focal seizures, the semiology of automatisms is variable, depending on the area of the cerebral cortex in which the seizures originate, and their treatment is entirely different. The detailed sleep-awake EEG, particularly video-EEG, will confirm the correlation of clinical events, unprovoked or provoked, with generalized spike-and-slow wave discharges. This may reveal features favoring a specific epileptic syndrome and, therefore, may determine long-term prognosis and management.
In practical terms, a child suspected of typical absences should be asked to overbreathe for 3 minutes, counting his or her breaths while standing with hands extended in front. Hyperventilation will provoke an absence in more than 90% of those who suffer. This procedure should preferably be videotaped for documentation of the clinical features. In infancy and childhood, hyperventilation is encouraged by blowing either a paper, a paper streamer, or a straw inside a cup with fluid causing bubbles (37). In addition, in children under the age of 3 or 4 years, the presenting symptom may be myoclonic jerks or a generalized tonic-clonic seizure or, rarely, the very brief inconspicuous absence seizures. This type of typical absence seizure will be discovered by careful observation of facial expression during the brief generalized spike-wave discharges. Rarely in this age group, absence seizures are the only seizure type, either pyknoleptic or spanioleptic, reminiscent of childhood or juvenile absence epilepsy, respectively.
The video-EEG documentation may be particularly useful if absences prove resistant to treatment, if other seizures develop, or for future genetic counsel. Focal spike abnormalities and asymmetrical onset of the ictal 2.5 to 4 Hz spike-wave discharges are common and may be a cause of misdiagnosis, particularly in resistant cases.
The EEG, preferably sleep-awake video-EEG and particularly in infancy and early childhood, is the single most important procedure in diagnosing typical absence seizures. Ictal EEG demonstrates high amplitude discharges of spike, multiple spike, and slow-wave discharges usually 3 to 4 Hz (range 2.5 to 6 Hz). These discharges can sometimes have maximum frontal or begin with unilateral spikes (172; 157; 163; 82).
Hyperventilation induces absence seizures in almost all (more than 90%) untreated or treated with inappropriate drug cases. Hyperventilation is preferably done while standing in order to identify concomitant unsteadiness or perseverative movements. They are best studied with video EEG. Ideally, all children with absence seizures should have video-EEG recordings in an untreated state, as this may reveal features favoring a specific epileptic syndrome and may, therefore, determine long-term prognosis and management. If this is not possible, the clinical manifestations of the seizures should be documented with video by the parents or the treating physicians. The absences are videotaped while the patient is holding hands in front of him or her and counting his or her breaths while overbreathing for 3 minutes. In a randomized multicenter controlled trial among 20 children (4 to 10 years old) with typical absence seizures, hyperventilation while sitting without backrest provoked 17 absences and while being in supine position 13 absences (p = 0.031) (138). All patients that had absence seizures during supine hyperventilation also had seizures during sitting hyperventilation and were of shorter duration (mean 8.69 seconds).
Breath-counting during hyperventilation (wherein the patient is asked to count his or her deep breaths) is an important, often neglected, method to detect impairment of consciousness during the discharge. Practical, easy to perform, and clinically relevant, this method may reflect impaired performance in daily life. Frequently, the patients are able to recall numbers or phrases told to them during the spike wave and slow wave discharge, whereas these may be associated with serious errors in breath-counting (120). Despite its practicality, breath-counting is rarely performed in EEG examinations of these patients; unfortunately, absences are often not apparent without this procedure. The significance of breath-counting may be appreciated by reviewing the video clips with the volume off.
Be aware that the ability to recall numbers or phrases or count may not be affected during brief spike-and-slow wave discharges. This diagnostic method is also difficult in early childhood and infancy. In addition, the given message or counting rate should be quicker than the brief discharge in order to identify concomitant impaired consciousness. Only by observing carefully the video-EEG during brief generalized spike-wave discharges, we may observe concomitant relevant transient impassive facial expression and a vague look. Furthermore, in the younger age group, hyperventilation is encouraged by blowing either a paper, a paper streamer, pinwheel, spinning wheel, or a straw inside a cup with fluid causing bubbles (37).
Sleep EEG patterns are normal. Generalized discharges of polyspike waves and slow waves are more likely to increase, but a reduction is also observed during sleep. The discharges are shorter, may be fragmented, and usually are devoid of discernible clinical manifestations, even in those patients who have numerous clinical seizures with motor manifestations during the alert state.
In the few cases for which an accurate and complete diagnosis of absence seizures is difficult, particularly in early childhood, the use of low-cost, portable EEG device monitoring may be useful in facilitating diagnosis. For example, EEG home monitoring is feasible in pediatric patients as long as an EEG wearable is easy to put on and is comfortable. Remote seizure monitoring will be one of the elements of personalized treatment for childhood absence epilepsy and juvenile absence epilepsy, similar to personalized drug titration and determining the duration of pharmacotherapy (59). EEG wearables have also been shown to be useful in managing the treatment of patients already diagnosed with absence epilepsy.
Twenty-four-hour EEG recording is the gold standard for assessment. It is often performed in the hospital and is expensive, obtrusive, and time-consuming to review. A study investigated the performance of an unobtrusive, two-channel, behind-the-ear EEG- based wearable, the Sensor Dot monitoring at home, to detect typical absences in adults and children (155). The authors concomitantly recorded 284 absences on video-EEG and Sensor Dot. Using the wearable Sensor Dot, epileptologists were able to reliably detect (F1 score = .73) typical absence seizures and reduced the review time of a 24-hour recording from 1 to 2 hours to around 5 to10 minutes. However, for research purposes, absence seizures were defined as having 3 Hz generalized spike-wave discharges for at least 3 seconds. Although the accuracy is not perfect, absences usually occur very frequently; therefore, it seems that this method will not significantly influence the outcome. This method can be useful for quantifying the absence seizures only in well-documented cases of the syndrome, with proper routine EEG recording methodology.
Children with absence seizures, particularly those with childhood absence epilepsy, eyelid myoclonia and absences, and facial myoclonia and absences should also be assessed neurophysiologically (149). Some children have deficits involving attention, executive function, visuospatial memory, and reading. Such an assessment should be periodically performed to provide appropriate supportive help. Depression, anxiety, and attention deficit hyperactivity disorder have also been reported in children with childhood absence epilepsy (100).
In a systematic literature search, 33 studies reporting on cognitive performance in children with absence epilepsy were considered (171). Neuropsychological tests were classified into the following domains: intelligence; executive function; attention; language; motor and sensory-perceptual examinations; visuoperceptual/visuospatial/visuoconstructional function; memory and learning; and achievement. In contrast to the common belief that cognition in absence epilepsy is generally undisturbed, lower than average neurocognitive performance was noted in multiple cognitive domains, which may influence academic and psychosocial development. Children with childhood absence epilepsy have elevated rates of adverse behavioral, psychiatric, language, and cognitive comorbidities, including attention problems, anxiety, depression, social isolation, and low self-esteem (158). These psychiatric and cognitive abnormalities were correlated with disease duration and interictal spike load in a publication (47). This, together with the failure of antiseizure drug monotherapy in children with absence epilepsy, the persistence of cognitive impairments even after apparent seizure suppression, the high probability of the development of generalized tonic-clonic seizures (158), and absence-associated comorbidities (anxiety and depression) (164), necessitates the identification of novel therapeutic targets.
In a Danish cohort study, school performance and psychiatric comorbidity in 114 children with childhood absence epilepsy were compared with both population controls and nonneurologic chronically ill children. Compared to both control groups, children with childhood absence epilepsy had increased psychiatric comorbidity, including ADHD, insomnia, and higher frequency of psychiatric visits. A considerable proportion of these children receive special needs education in primary/secondary school, albeit insufficient to normalize their considerably lower grade point average in the 9th grade by 1.7 grade points (18).
A systematic review and meta-analysis synthesized human and animal models that investigated mood disorders and absence seizures among 35 articles meeting inclusion criteria (66). Patients with absence seizures had greater odds of developing depression and anxiety when compared to controls (odds ratio: 4.93, 95% CI: 2.91–8.35, p < 0.01). A strong correlation between absence seizures, depression, and anxiety was also found in the form of a bidirectional relationship sharing underlying mechanisms, such as genetic predisposition, neurophysiology, and anatomical pathways. Furthermore, antiseizure and antidepressant drugs affect both absence seizures and mood disorders, potentially indicating a common psychopathology.
In general, it is known that genes coding for T-type calcium channels and for GABA receptors have been associated with the etiopathogenesis of childhood absence epilepsy, and abnormal oscillatory rhythms can originate either from T-type channel abnormalities or from the increased GABA-B activity (126). Therefore, antiseizure drugs that suppress T-type calcium channels (eg, ethosuximide, valproate) are effective anti-absence drugs, whereas those that increase GABA-B activity (eg, carbamazepine, vigabatrin), exacerbate the frequency of absence seizures. Furthermore, GABA-A agonists (eg, benzodiazepines), which preferentially enhance GABA-ergic activity in neurons from the thalamic nucleus reticularis, can suppress absence seizures (126).
Monotherapy if typical absences are the only type of seizures. Treatment is mainly with sodium valproate or ethosuximide, which are of equal efficacy controlling absences in around 85% of patients (38; National Guideline Alliance (UK) 2022; 32; 92). The rest (15%) responded to the combination of sodium valproate and ethosuximide or levetiracetam, lamotrigine, and benzodiazepines (38). A large meta-analysis using surface under the cumulative ranking found the efficacy ranking for monotherapy for absence to be as follows, from highest to lowest: ethosuximide, valproate, placebo, lamotrigine (32). Ethosuximide should be preferred as monotherapy in idiopathic generalized epilepsies with typical absence seizures only, whereas valproate should be added or used as monotherapy for those cases with aberrant electroclinical features, particularly generalized tonic-clonic seizures. Lamotrigine monotherapy is less effective with nearly half or less of the patients becoming seizure free (35; 74; 60; 178; 19; 148; 92). Glauser and colleagues performed a double-blind, randomized, controlled clinical trial to compare the efficacy, tolerability, and neuropsychological effects of ethosuximide, valproic acid, and lamotrigine in children with newly diagnosed childhood absence epilepsy (60). Drug doses were incrementally increased until the children were free of seizures, the maximal allowable or highest tolerable dose was reached, or a criterion indicating treatment failure was met. The primary outcome was freedom from treatment failure after 16 weeks of therapy; the secondary outcome was attentional dysfunction. Differential drug effects were determined by means of pairwise comparisons. The 453 children who were randomly assigned to treatment with ethosuximide (156), lamotrigine (149), or valproic acid (148) were similar with respect to their demographic characteristics. After 16 weeks of therapy, the freedom-from-failure rates for ethosuximide and valproic acid were similar (53% and 58%, respectively; odds ratio with valproic acid vs. ethosuximide, 1.26; 95% confidence interval [CI], 0.80 to 1.98; P=0.35) and were higher than the rate for lamotrigine (29%; odds ratio with ethosuximide vs. lamotrigine, 2.66; 95% CI, 1.65 to 4.28; odds ratio with valproic acid vs. lamotrigine, 3.34; 95% CI, 2.06 to 5.42; P< 0.001 for both comparisons). There were no significant differences among the three drugs with regard to discontinuation because of adverse events. Attentional dysfunction was more common with valproic acid than with ethosuximide (in 49% of the children vs. 33%; odds ratio, 1.95; 95% CI, 1.12 to 3.41; P=0.03) (60). A meta-analysis of 4282 patients found that ethosuximide had higher 3- and 6-month seizure-free outcome rates than valproate (odds ratio: 1.3, 95% CI: 0.66–2.8), and lamotrigine had a lower rate of seizure-free outcome than valproate (odds ratio: 0.40, 95% CI: 0.23–0.77) (32).
In a study, ethosuximide was associated with a higher rate of complete remission than valproate in childhood absence epilepsy, which may suggest that ethosuximide has a beneficial potential disease-modifying effect on this epileptic syndrome (16). Furthermore, in another study, class 1 evidence was provided that valproate is associated with more significant attentional dysfunction than ethosuximide or lamotrigine in children with newly diagnosed childhood absence epilepsy (99).
Genetic variation plays a role in the differential drug response of childhood absence epilepsy. Specifically, polymorphisms in T-type calcium channels were associated with poor response to ethosuximide, whereas one T-type calcium channel variant was more common among responders to lamotrigine (61). An ABCB1 P glycoprotein variant was associated with poor response to lamotrigine. No variants were associated with response to valproate.
Meta-analysis found no significant differences between the safety profiles of any antiseizure medication as monotherapy or adjunctive therapy (32). The most frequent side effects of ethosuximide are abdominal pain, nausea, vomiting, and diarrhea (134). For this reason, it should be taken with meals. Valproate in particular has been associated with inattention and behavioral side effects (145) as well as weight gain (134). Lamotrigine is generally well tolerated but carries a risk of Stevens-Johnson syndrome. In a publication, two cases of childhood absence epilepsy were reported that showed seizure disappearance after ethosuximide was added to valproate and ethosuximide drug eruption occurred, suggesting the possibility that the epilepsy disappears due to immune responses to antiseizure medication (107).
If monotherapy fails or unacceptable adverse reactions appear, replacement of the antiepileptic drug by another of the three drugs is the alternative. As second monotherapy, ethosuximide and valproate demonstrate higher freedom from failure proportions and greater efficacy than lamotrigine; valproate is more often associated with attentional dysfunction (33). When valproate does not control typical absence seizures, ethosuximide is the drug of second choice.
The most effective combination treatment is valproate with small doses of lamotrigine, which is possibly due to a pharmacodynamic interaction between these two drugs (122; 86), or levetiracetam slow titration when absence and myoclonic seizures co-exist. Inhaling medical carbogen containing 5% CO2 and 95% O2 can suppress hyperventilation-induced absence seizures and spike-and-wave discharges and, therefore, it could be a therapeutic approach for hyperventilation-related absence seizures (177).
In 20 patients with childhood absence epilepsy and incomplete seizure control in therapy with first antiseizure medication (seven ethosuximide, five valproate, five levetiracetam, and three lamotrigine), perampanel was added as first add-on antiseizure drug (113). None of the patients had generalized tonic-clonic seizures. Fifteen cases became seizure-free, and video EEG was negative for epileptic abnormalities (five with valproate, five with levetiracetam, three with ethosuximide, and two with lamotrigine). The retention rate after 12 months of follow-up was 75%. The tolerability profile was favorable. The patients who were seizure-free with adjunctive perampanel were switched to perampanel monotherapy (mean follow-up on perampanel monotherapy = 5.56 ± 1.59 months). Nine of 15 remained seizure free in monotherapy with perampanel, whereas the other six relapsed and the double antiseizure therapy was restored (113).
Novel T-type calcium channel blockers are under investigation in animal models, some as analgesics; they may emerge as novel treatment options for absences (94).
Monotherapy in syndromes of idiopathic generalized epilepsy with typical absence seizures and other types of generalized seizures such as myoclonic jerks, generalized tonic-clonic seizures, or both. Established first-line monotherapy is with sodium valproate, which controls absence seizures in 75% to 85%, generalized tonic-clonic seizures in 70% and myoclonic jerks in 75% (38; 37; 117; 120; National Guideline Alliance (UK) 2022). A major problem is that sodium valproate is undesirable in women because of teratogenic effects, and polycystic ovarian syndrome. Therefore, it should not be used in females of childbearing age, particularly if contraception is not being used.
The adverse effects of valproate and its lack of efficacy in 25% of patients have prompted the search for alternatives among newer AEDs. Levetiracetam appears to be most promising of all because it is highly effective in controlling generalized tonic-clonic seizures, myoclonic jerks, photosensitivity (17; 166; 110), and probably absence seizures (29; 43; 167; 50; 134). In a report, aggravation of absences with levetiracetam has been reported in six patients resistant to other AEDs (09). These six patients were retrospectively found from an unspecified number of probably a large population of children with absence epilepsy from three pediatric neurology departments in France; how many patients benefitted from this drug has not been reported. In another study, only about one-quarter of children with absence epilepsy became seizure free with levetiracetam (111). When effective, this occurred at a relatively low dose.
Lamotrigine is effective in controlling generalized tonic-clonic seizures and absence seizures, but it exaggerates myoclonic jerks (120). Topiramate is ineffective as monotherapy in absence seizures of childhood absence epilepsy (130) and often has serious cognitive and other adverse reactions.
Monotherapy should not be abandoned before making sure that maximum tolerated dose has been achieved if smaller doses have failed. If monotherapy fails or unacceptable adverse reactions appear, then replacement of one by the other is the alternative.
Polytherapy in syndromes of idiopathic generalized epilepsy with typical absence seizures and other types of generalized seizures such as myoclonic jerks, generalized tonic-clonic seizures, or both. For monotherapy failures, sodium valproate, topiramate, levetiracetam, lamotrigine, and probably zonisamide appear to be effective in polytherapy. Surface under the cumulative ranking score found the most effective adjunctive therapy to be topiramate, followed by levetiracetam, lacosamide, perampanel, and then lamotrigine (32). Small, minute doses of lamotrigine added to adequate doses of sodium valproate may have a dramatic beneficial effect in more than half of the patients (122). In persistent myoclonic jerks, the best add-on drug is clonazepam that in small doses (0.5 to 2 mg) prior to sleep may have a dramatic beneficial effect. Ethosuximide should be added only for uncontrolled absence seizures. Zonisamide is efficacious in approximately half of patients with absence seizures (127; 134). In 2005, a chart review of 45 patients (mean age 11.4 years; range 2.8 to 17.9 years) with absence seizures was conducted to assess the effectiveness and safety of zonisamide in absence epilepsy. Patients received a mean daily dose of zonisamide 343 mg or 9.0 mg/kg (range, 100 to 600 mg or 2 to 24 mg/kg). Twenty-three of 45 patients (51.1%) obtained seizure freedom, whereas 16 of 45 patients (35.6%) experienced 50% or greater seizure reduction. Five patients (11.1%) had less than 50% seizure reduction, whereas one patient (2.2%) had an increase in seizure frequency. Nineteen patients (42.2%) reported one or more unwanted effects, with decreased appetite (15.5%) and sleepiness (11.1%) being the most common. Two patients discontinued zonisamide therapy, one for an increase in seizure frequency and apparent inefficacy and sleepiness in the other (174).
Newer antiseizure medications, such as brivaracetam and perampanel, seem to be promising options. Cannabidiol was found to be likely ineffective and potentially exacerbating for absence seizures (12). Well-conducted clinical trials aimed at evaluating the efficacy of alternative monotherapy (beyond ethosuximide, valproic acid or lamotrigine) or the combination of antiseizure medications on difficult-to-treat typical absences, are warranted (134).
Amantadine may constitute an efficacious alternative treatment for refractory absence seizures (129). In one study, a response of at least 50% seizure reduction was reported in more than 50% of patients; among responders, a majority had greater than 90% reduction in seizure frequency (129).
Furthermore, chronic treatment with the sodium channel blocker, lacosamide, in genetic absence epilepsy rats modified the development of absence seizures, suggesting that lacosamide exerts beneficial effects on absence seizure epileptogenesis (26).
In summary, ethosuximide should be used as monotherapy in those cases that experience absence seizures only. Sodium valproate, or even lamotrigine, should be used in absence seizures with aberrant electroclinical features, alone or in combination. Alternative choices are levetiracetam, benzodiazepines, zonisamide, brivaracetam, perampanel, and topiramate. Any relevant trigger factors such as lack of sleep or noncompliance should also be avoided. Absence status emergency treatment includes lorazepam or diazepam, or even sodium valproate. As with any other seizure-type epilepsy, a basic neuropsychological assessment, follow-up, and appropriate intervention is advisable.
Be aware in adult patients, even for absence seizures, there is a self-reporting dependency on seizure reporting in order to determine seizure frequency for epilepsy management. Studies show that absence seizures in adults are highly misreported (180). Using inpatient video-EEG telemetry or outpatient ambulatory electroencephalography, only 24% and 26% of seizures were correctly identified in the over- and underreporting assessment groups, respectively (131).
Contraindicated drugs. The treatment of idiopathic generalized epilepsy is demanding because many antiepileptic drugs are either ineffective or exaggerate absences and myoclonic jerks (147; 120). An antiepileptic drug is contraindicated not only when it exaggerates seizures but also when it is ineffective in controlling the seizures that it is supposed to treat. It may cause unnecessary adverse reactions and deprives the patient of the therapeutic effect that could be provided by another antiepileptic drug. In general, GABA-ergic anticonvulsants, such as carbamazepine, vigabatrin, and tiagabine are contraindicated in the treatment of absence seizures, irrespective of cause and severity. This is based on clinical and experimental evidence. In particular, the GABA agonists vigabatrin and tiagabine are used to induce, not to treat, absence seizures and absence status epilepticus (96; 120). The error of prescribing these drugs in the treatment of absence seizures may be of the same magnitude as prescribing a gluten-rich diet in the treatment of celiac disease. Similarly, phenytoin, phenobarbital, gabapentin, and pregabalin should not be used in the treatment of absence seizures (120).
Nonpharmacologic treatment options. Animal models of absence seizures have identified potential excitable cortical zones as treatment targets via surgery, ablation, or stimulation. Functional MRI and MEG studies have identified potential spatial targets for neurostimulation (159). Vagus nerve stimulation has also been used for treatment-resistant childhood absence epilepsy (134). A study of nine patients found a mean reduction in seizure frequency of 53.5±60.3% (p=0.04) with vagus nerve stimulation (07). Animal models of absence epilepsy have shown reduction in spike-wave discharges in response to invasive and noninvasive stimulation devices (94; 23); this is an emerging area of study.
Recognizing GLUT1 deficiency syndrome is important, because a ketogenic diet is effective in improving epileptic seizures and may also reduce some neurologic symptoms. Most antiepileptic drugs are contraindicated either because they are ineffective or because they exaggerate seizures (85; 90). Phenobarbital, but also chloral hydrate and diazepam, may further impair GLUT1 and should not be used in these patients. Ketogenic diets have also shown promise in treating absence seizures in the absence of GLUT1 deficiency. A review of prior studies found that 69% of patients with absence seizures had over 50% reduction in seizures after starting the ketogenic diet; however, it was not documented whether these patients included some with GLUT1 deficiency (65).
Withdrawing antiepileptic medication. Withdrawal of the medication is dependent on the syndrome (120). In the pure form of childhood absence epilepsy--with no aberrant electroclinical features (the syndrome), drug therapy can be gradually withdrawn (over 3 to 6 months) after 2 to 3 seizure-free years (38; 83). In others, such as juvenile absence epilepsy, juvenile myoclonic epilepsy, or eyelid myoclonia and absences, treatment may be lifelong (118; 119; 120; 40).
In general, early correct diagnosis and management lead to better outcomes. The evolution of epilepsies with typical absence seizures in their phenotype is variable from excellent to less favorable, depending on the underlying epilepsy syndrome. Childhood absence epilepsy typically has very favorable outcomes, with around 60% of patients achieving long-term remission (06). In a video-EEG recording analysis of 213 typical absence seizures from 61 patients with idiopathic generalized epilepsy, the presence of polyspikes had a high positive predictive value for unfavorable therapeutic outcome. In contrast, none of the semiological features, alone or in combination, were predictors of therapeutic outcome (170). EEG may help predict the likelihood of seizure recurrence after treatment discontinuation (83).
See epilepsy and pregnancy.
There are no special considerations for anesthesia in absence seizures. Optimal seizure control should be targeted prior to receiving anesthesia and potentially epileptogenic anesthetic agents should be avoided (39).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Emily Grew MD
Dr. Grew of NYU Langone Health has no relevant financial relationships to disclose.
See Profile
Parmpreet Dhillon MD
Dr. Dhillon of NYU Langone Health has no relevant financial relationships to disclose.
See Profile
Solomon L Moshé MD
Dr. Moshé of Albert Einstein College of Medicine has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Sleep Disorders
Jul. 05, 2026
General Child Neurology
Jun. 24, 2026
General Child Neurology
Jun. 10, 2026
Epilepsy & Seizures
Jun. 02, 2026
General Neurology
May. 13, 2026
General Child Neurology
May. 12, 2026
Neuropharmacology & Neurotherapeutics
May. 11, 2026
Epilepsy & Seizures
May. 08, 2026