Epilepsy & Seizures
Febrile seizures
Jun. 02, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Epilepsy with myoclonic-atonic seizures is a genetic generalized epilepsy that begins between the ages of 7 months and 6 years (peak age 2 to 4 years) in previously normal children. It starts with frequent and usually lengthy febrile and afebrile generalized tonic-clonic seizures. Myoclonic, atonic, myoclonic-atonic, and absence seizures usually follow a few weeks later. Seizures are frequent and may cause traumatic falls. Nonconvulsive status epilepticus, sometimes lasting for many hours or days, is common. Interictal EEG shows brief, generalized 2 to 4 Hz spike/polyspike-wave discharges. Ictal EEG depends on the seizure type. Myoclonic-atonic seizures manifest with discharges of irregular spike-wave or polyspike-wave complexes at a frequency of 2.5 to 3 Hz, or faster. Atonia is usually concurrent with the slow wave of a single- or polyspike-wave complex. Prognosis is uncertain because of various diagnostic criteria and treatment strategies. In general, seizures remit 2 to 4 years after onset, but some cognitive impairment may occur. Treatment is usually with a combination of valproate and small doses of lamotrigine; other antiepileptic drugs such as ethosuximide and clonazepam may be used. Carbamazepine, phenytoin, and vigabatrin are amongst the contraindicated drugs. Ketogenic diet or modified Atkins diet may be therapeutic in drug-resistant cases. In this article, the author details the clinical manifestations, etiology, EEG, differential diagnosis, prognosis, and optimal management of patients with epilepsy with myoclonic-atonic seizures, also known as Doose syndrome.
|
• Epilepsy with myoclonic-atonic seizures (also known as Doose syndrome) is a genetically determined generalized (idiopathic/genetic) epilepsy with onset in infancy and early childhood. | |
|
• It manifests with frequent and multiple types of seizure (generalized tonic-clonic, myoclonic-atonic, atonic, and absence seizures). Myoclonic-atonic seizures with falls are the defining symptoms. | |
|
• EEG shows frequent generalized discharges of spike/polyspike-slow wave at varying frequencies of 2.5 to 3 Hz or higher. | |
|
• Differential diagnosis is often demanding and requires exclusion of structural epilepsies and epileptic encephalopathies that may imitate epilepsy with myoclonic-atonic seizures. | |
|
• The prognosis of seizures is usually good, but cognition may be affected. | |
|
• Early onset of treatment, mainly with a combination of valproate and small doses of lamotrigine, may prevent the development of cognitive disturbances. Ketogenic diet or modified Atkins diet should be considered for drug-resistant cases, and it may be therapeutic. Other antiepileptic medications have also been used to treat specific seizure types as part of myoclonic-atonic epilepsy, including topiramate, ethosuximide, rufinamide, and cannabidiol. | |
|
• Vagal nerve stimulation may offer some benefit in seizure reduction. Responsive neurostimulation treatment has not been reported in this population. |
Doose and colleagues introduced the concept of a specific clinical entity with myoclonic-atonic seizures being the core of the disorder, which he called “centrencephalic myoclonic astatic petit mal” (15; 18). This disorder has been accepted as an epileptic syndrome by the ILAE, initially under the name “myoclonic-astatic epilepsy” (11) and “epilepsy with myoclonic-atonic seizures” (03; 10). It is also referred to as “Doose syndrome” (31), particularly the pure form of genetic nonstructural epilepsy with myoclonic-atonic seizures (54).
The ILAE Task Force considered “epilepsy with myoclonic-astatic seizures” to be an idiopathic generalized epilepsy (21), a view similar to that of Doose. Epilepsy with myoclonic-astatic seizures belongs to the epilepsies with primarily generalized seizures, including absence epilepsies, juvenile myoclonic epilepsy, and infantile and juvenile idiopathic epilepsy with generalized tonic-clonic seizures. Like these types of epilepsy, epilepsy with myoclonic-astatic seizures is genetically determined, with little nongenetic variability. Doose proposed the following features: genetic predisposition (high incidence of seizures and/or genetic EEG patterns in relatives); mostly normal development and no neurologic deficits before onset; primarily generalized myoclonic, astatic, or myoclonic-astatic seizures, short absences, and mostly generalized tonic-clonic seizures; generalized EEG patterns (spikes and waves, photosensitivity, 4 to 7 Hz rhythms); and no multifocal EEG abnormalities (but often pseudofoci) (16).
Nowadays, the problem of defining epilepsy with myoclonic-atonic seizures may reflect a lack of specific diagnostic criteria and undefined boundaries of certain epileptic syndromes. Several epilepsy syndromes manifest with myoclonic-atonic seizures, such as Dravet syndrome, Lennox-Gastaut syndrome, and atypical benign partial epilepsy of childhood. Cases of benign and severe myoclonic epilepsy in infants may have been included in epilepsy with myoclonic-atonic seizures (16; 62). However, it is generally accepted that children with myoclonic-atonic epilepsy are otherwise normal, with no discernible causes other than a strong genetic epileptic background, and these cases probably represent the genuine, genetic (idiopathic) syndrome of epilepsy with myoclonic-atonic seizures or “Doose syndrome.” This distinguishes “Doose syndrome” from structural epilepsies and epileptic encephalopathies with myoclonic and atonic seizures.
The ILAE “epilepsy diagnosis” manual considers epilepsy with myoclonic-atonic seizures as a childhood epilepsy syndrome of an epileptic encephalopathy and is described as follows:
|
Epilepsy with myoclonic-atonic seizures (previously known as epilepsy with myoclonic astatic seizures, or Doose syndrome) is a syndrome characterized by the presence of myoclonic-atonic seizures in an otherwise normal child who may have a history of febrile and/or afebrile seizures. There is often a family history of seizures. Clinical context: This syndrome is characterized by seizures that have onset between 6 months and 6 years of age (peak 2 to 4 years). In two thirds of children, febrile seizures and generalized convulsive seizures precede the onset of myoclonic-atonic and atonic seizures. Both sexes are affected, with a male predominance (ratio 2:1). Antecedent and birth history are unremarkable. Neurologic examination and head size are normal. Development and cognition are typically normal; however, impairments may develop at or after seizure onset. Caution: Glucose transporter disorders should be excluded. Note: Epilepsy with myoclonic-atonic seizures is considered an 'epileptic encephalopathy'. This term denotes the concept that the epileptic activity itself might directly contribute additional cognitive and behavioral impairments over those expected from the underlying etiology alone, and that suppression of epileptic activity might minimize this additional impairment. The ILAE has published a classification and diagnosis position paper on epilepsy syndromes with onset in childhood and refers to myoclonic atonic epilepsy as epilepsy with myoclonic atonic seizures (61). Seizures (61) Mandatory seizures. Myoclonic-atonic seizures are mandatory seizure types of this syndrome. Atonic and myoclonic seizures are frequent. Myoclonic-atonic status epilepticus is common. May have: • Febrile seizures • Absence seizures—seen in half the patients, typically with myoclonic jerks, facial myoclonia, and atonia (not just loss of awareness). • Atypical absence seizures • Tonic seizures are rare; the presence of tonic seizures is linked to a higher frequency of cognitive impairment. • Generalized convulsive seizures • Nonconvulsive status epilepticus is common, lasting for hours to days and manifests as cognitive impairment with repetitive myoclonic (affecting face, eyelids, and limbs) and atonic seizures. Exclusionary: • Epileptic spasms or infantile epileptic spasm syndrome Clinical: • Presence of developmental delay and cognitive impairment prior to seizure onset should alert the clinician to evaluate for other diagnoses. • Focal neurologic signs on exam should also prompt further evaluation for alternate diagnoses. Genetics. Pattern of inheritance. Complex (polygenic) inheritance with variable penetrance. Known genes. A minority of cases may be explained by variants in SCN1A and SLC2A1. Family history of seizures/epilepsy. There is frequently a family history of febrile seizures (in 50% of cases); other seizures/epilepsy syndromes may also occur in families. Epilepsy with myoclonic-atonic seizures is seen in families with other individuals with genetic epilepsy with febrile seizures plus, suggesting common genetic etiological factors. EEG (61) Background. The background may be normal or show generalized slowing. Background biparietal theta is usually seen. Caution: Focal slowing consistently over one area is not seen; consider a structural brain abnormality. Interictal. Generalized 2 to 6 Hz spike-and-wave and polyspike-and-wave occur. Caution: Generalized paroxysmal fast activity in sleep, generalized slow spike-and-wave complexes less than 2 Hz, photoparoxysmal response at low flash frequencies. Exclusion. Persistent focal discharges consider structural brain abnormality. Activation. Intermittent photic stimulation may trigger generalized spike-and-wave, polyspike-and-wave, and myoclonic-atonic seizures. EEG abnormality is enhanced by sleep deprivation and by sleep. Generalized spike-and-wave often becomes fragmented with sleep deprivation or during sleep. Fragmented generalized spike-and-wave can appear focal or multifocal but usually is not consistently seen in one area. The morphology of the focal spike-and-wave typically appears similar to the generalized spike-and-wave. Ictal. The myoclonic component is associated with a generalized spike or polyspike. The atonic component is associated with the following high voltage slow wave. Imaging. Neuroimaging is normal. Differential diagnoses. • Atypical childhood epilepsy with centrotemporal spikes |
A survey from a large group of experts identified preferred diagnostic tests, electroclinical features, and investigations for myoclonic-atonic epilepsy. Clinically, there should be a history suggestive of myoclonic-atonic seizures, an EEG with generalized discharges, normal neuroimaging, and normal development before seizure onset. Recommended work up includes EEG, MRI, amino acids, organic acids, fatty acid/acylcarnitine profile, microarray, epilepsy genetic panel, lactate/pyruvate, and CSF glucose and lactate studies (44).
Epilepsy with myoclonic-atonic seizures is characterized by myoclonic-atonic seizures that often occur together with atonic, myoclonic, and absence seizures; myoclonic-atonic status epilepticus is common (18; 17; 16; 49; 42; 31; 65; 30; 44).
Myoclonic-atonic epilepsy begins between 7 months and 6 years of age (peak age 2 to 4 years). Before the onset of seizures, 85% of affected children are of normal development; the remainder show moderate psychomotor deficits, mainly affecting speech. Onset before 6 months or after 6 years is most unlikely (44).
In two-thirds of patients, febrile and afebrile generalized tonic-clonic seizures appear first, prior to the onset of myoclonic-atonic seizures. They are usually prolonged, recurring frequently and, mainly, during the daytime. After a period of repeated generalized tonic-clonic seizures lasting a few weeks or months, myoclonic absences, atonic seizures, and myoclonic-atonic seizures appear. These occur several times a day. This period of frequent seizures lasts 1 to 4 years.
Myoclonic-atonic epilepsy may also encompass a spectrum of phenotypic variability ranging from epileptic encephalopathy to genetic generalized epilepsy (63). In a large study, 20% of patients had developmental or speech delay before seizure onset, indicating an epileptic encephalopathy symptomatology. This variability in signs and symptoms of myoclonic-atonic epilepsy is further supported by the observation that more than 50% of patients with the suspected diagnosis go through multiple diagnosis switching through the natural course (63).
Itoh and colleagues studied video-EEGs of epileptic drop attacks in children with symptomatic epilepsy of early childhood in comparison with children with myoclonic-atonic epilepsy (28). In the former group, most of the drop attacks were caused by epileptic spasms corresponding to generalized biphasic slow discharges, sharp-and-slow wave complexes, or the flattening of ongoing background activity. Atonic seizures were associated with runs of generalized spike-and-wave complexes. The drop attacks in epileptic spasms usually occur in periodic clustering. Interictal EEG revealed generalized irregular multiple spikes-and-waves with focal or multifocal accentuations. Conversely, in myoclonic-atonic epilepsy, drop attacks were caused by myoclonic-atonic seizures corresponding to generalized high-amplitude spikes or single polyspike-and-wave complexes. The authors concluded that epileptic drop attacks, often seen in young children with symptomatic epilepsy, were most frequently caused by flexor type epileptic spasms and rarely by myoclonic-atonic seizures, a hallmark seizure type of myoclonic-atonic epilepsy. In a clinical setting, the occurrence of periodic clusters and independent focal or multifocal accentuations of generalized spike-and-wave complexes in interictal EEG may indicate epileptic drop attacks caused by epileptic spasms.
Myoclonic-atonic seizures. Myoclonic-atonic seizures are the defining symptoms in 100% of cases (16; 44). These manifest with symmetrical myoclonic jerks and are immediately followed by loss of muscle tone (post-myoclonic atonia).
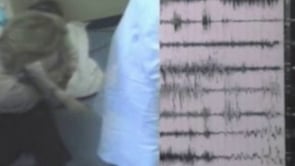
A typical myoclonic-atonic seizure consists of a myoclonic jerk of the upper limbs or the trunk and is immediately followed by an atonic fall straight down, landing on the buttocks (48; 49; 45; 65). There is no propulsive or retropulsive falling. It is like the collapse of a marionette when all its strings are simultaneously cut. The whole episode lasts approximately 1 second or less, and the patient quickly rises from the floor with no impairment of consciousness. In falls from the standing position, the patient suddenly flexes at the waist and knees, followed by further knee flexion, and then drops straight down and lands on the buttocks. When sitting, the patient falls forward or backward depending on the position of the center of gravity. The seizure is abrupt with no warning, which is also the reason for frequent traumas.
The myoclonic jerks preceding atonia, which are sometimes difficult to detect without video-EEG recordings, vary greatly in intensity and extent. The jerks may be limited to the facial or neck muscles. Similarly, atonic manifestations may be slumping of the head (head drops, head nodding) and of the upper trunk, without falls. The same patient may have myoclonic-atonic seizures with all variations of intensity, from mild to severe.
In addition to myoclonic-atonic seizures, atonic and absence seizures occur frequently, sometimes many times per day, in the active period of the disease.
Atonic seizures. Atonic seizures are sudden and brief and involve severe loss of postural tone involving the whole body or only the head. Attacks are brief, 1 to 4 seconds, and frequent. Generalized loss of postural tone causes a lightning-like fall. The patient collapses on the floor irresistibly. In brief and milder attacks, there is only head nodding or bending of the knees. Consciousness usually remains clear during pure atonic seizures, and the child can resume the original posture immediately. Rarely, pure atonic seizures occur as the only manifestation of the disorder (18).
Myoclonic jerks. Myoclonic jerks precede or, less often, intersperse with the atonic manifestations.
They usually involve the arms and shoulders symmetrically and are accompanied by head nodding. The myoclonic jerks are brief and vary in intensity: some may be so violent that the arms are flung upward, and some so mild that they are palpable rather than visible. Irregular twitching of facial muscles, especially of the perioral and periocular musculatures, may also be seen. A brief yell, probably a result of contraction of the diaphragm, can also accompany the myoclonic jerks.
Absence seizures. Absence seizures alone and without clinical symptoms, other than impairment of consciousness, are exceptional. However, more than half of the cases have brief absence seizures that occur together with myoclonic jerks, facial myoclonias, and atonic manifestations.
Tonic seizures. Tonic seizures may occur later in the course of the illness and are associated with a poorer outcome (61).
Myoclonic-atonic status epilepticus. Myoclonic-atonic status epilepticus lasting hours, or even days, is common and affects one third of patients. This manifests with varying degrees of usually severe cognitive impairment or cloudiness of consciousness, interspersed with repetitive myoclonic and atonic fits. Facial myoclonus of eyelids and mouth may be continuous, together with irregular jerks of the limbs and atonic seizures consisting of head nodding or falls. There may be associated drooling, unsteadiness, and speech impairment (61). Myoclonic-atonic status epilepticus may occur several times during a period of 1 or 2 years. During each episode, features of atypical absences, myoclonus, and atonia are present in various degrees, with persisting consequences on cognitive abilities. The child appears apathetic, hypokinetic, and stuporous. Barely discernible myoclonic contractions and irregular twitching of facial muscles and of the hands can be detected (18; 19).
Drop attacks. Drop attacks may result from pure atonic, myoclonic-atonic, or atypical absence seizures (46). Oguni and colleagues studied the nature of drop attacks with video and slow-motion analysis in five patients. Recovery to the preictal position was observed in 0.3 to 1 second. The drop attacks were characterized by sudden forward flexion of the head and trunk, as well as both arms, which caused the patient to fall. In myoclonic-atonic seizures, patients showed brief myoclonic flexor spasms, immediately followed by atonic falling. Atonic seizures showed abrupt atonic falling, with and without transient preceding facial expression change and/or twitching of extremities.
There is a period of frequent seizures and episodes of myoclonic-atonic status epilepticus, which lasts for approximately 2 to 4 years. Then the general condition improves, and the patient may become seizure-free, but some patients may show cognitive impairment (see Prognosis and complications).
The prognosis is unclear, probably because of different inclusion and exclusion criteria, methodology, and treatments (44). In general, it appears that there is a significant difference in the prognosis between seizure remission and cognitive and behavioral outcome (65). Speech or developmental delay may be apparent before seizure onset in about one third of patients. After seizure onset, one third of patients go through a period of increased seizure frequency, multiple seizure types, and a higher risk of nonconvulsive status epilepticus, which are often resistant to seizure medications. During this period of increased seizure frequency, developmental delays, regression, behavioral, and sleep disorders commonly occur. These symptoms may also improve or resolve once seizures are controlled (24). Fifty to 89% of patients have seizure remission within 3 to 4 years of onset. Half of patients achieve a seizure-free state and normal or near-normal development (32; 65). In a large, retrospective study of 166 patients, seizure freedom was seen in 57% and normal development in 47%, and 12% had a delay in one developmental domain (43). Conversely, most patients show cognitive (usually mild) and behavioral abnormalities (usually a tendency of withdrawn/depressed and aggressive behavior) (65). If untreated, spontaneous remission with normal development has been observed in a few untreated cases, but these may belong to benign myoclonic epilepsy in infancy. The others that continue with refractory seizures, severe impairment of cognitive functions, and behavioral abnormalities probably belong to structural cases or other severe epileptic encephalopathies (16). Ataxia, poor motor function, dysarthria, and poor language development may emerge in such cases, but these may also represent epileptic encephalopathy syndromes.
Unfavorable outcome is related to frequent generalized tonic-clonic seizures, particularly if these occur during sleep; tonic seizures; recurrent nonconvulsive status epilepticus; and EEG findings of frequent generalized discharges, slow spike-and-wave discharges, or generalized paroxysmal fast activity (61).
Doose and colleagues reported that only 26% of individuals had normal cognition in their original paper (18), but this is much higher in subsequent reports: 59% normal development and 20% mild developmental delay (52); 43% normal development and 52% mild developmental delay (32); 66.7% normal development after a mean period of 4.4 years, with the remaining showing mild and less frequent moderate cognitive impairment (65).
In addition, Tang and colleagues looked at neurodevelopmental comorbidities in a large cohort of myoclonic-atonic epilepsy patients and found that in a cohort of 97 patients, 62.8% had intellectual disability, 24.1% had autism spectrum disorder, and 37.8% had ADHD symptoms (63). These are higher incidences of neurodevelopmental comorbidities than previously reported.
Inoue and associates reported that seizure onset at a younger age and the presence of focal spike discharges on EEG are indicators of poor prognosis. Onset in patients with refractory seizures was earlier than that in those with a favorable prognosis, and all cases with a poor prognosis had focal spike discharges (27). It should also be noted that focal discharges are not typical of this syndrome, and other differential diagnoses should be evaluated as well (47).
In a large multicenter study by Nickels and colleagues, persistent global developmental delay/regression after onset of epilepsy, seizures on subsequent EEGs, and failure to respond to dietary therapy were associated with failure to achieve seizure freedom (43). In this same study, poor developmental outcome was associated with global developmental delay/regression after onset of seizures; seizure onset prior to age 24 months; EEGs showing slowing, epileptiform discharges, or seizures (after the initial EEG); and having refractory seizures.
Rarely, patients with childhood epilepsy with myoclonic-atonic seizures who achieved complete remission during childhood may develop pharmaco-responsive absence seizures during early adolescence (05).
A 2-year-old boy with normal neurocognitive development had a febrile generalized tonic-clonic seizure. Two months later, he started having frequent febrile and non-febrile generalized tonic-clonic seizures, which were often prolonged. There was a family history of idiopathic generalized epilepsies. Interictal EEG had a normal background, but there were frequent brief, generalized discharges of 2.5 to 3 Hz spike/polyspike and slow wave, mainly in sleep. Treatment with valproate had an initial beneficial effect on generalized tonic-clonic seizures, but 3 months later, the boy started having myoclonic jerks, absences, and falls—some of which were traumatic. Despite adding lamotrigine, and later clonazepam, multiple seizures continued on a daily basis, and the boy also had lengthy episodes of myoclonic-atonic status epilepticus. Video-EEGs documented myoclonic, absence, and myoclonic-atonic seizures. Epileptic seizures improved in frequency and severity after the age of 4 years and finally remitted at the age of 5 years. The boy has mild cognitive impairment. He attends mainstream school, but his performance is poor.
Epilepsy with myoclonic-atonic seizures may be genetically determined in a multi-factorial polygenic fashion with variable penetrance and involving different families of genes (18; 17; 16; 64). One third of patients have familial seizure disorders and mainly idiopathic generalized epilepsies. The prevalence is higher in siblings (16%) than in parents (6%). The prevalence of abnormal EEG patterns (photosensitivity, 4 to 7 Hz rhythms, spike and wave) without clinical seizures among relatives is even higher. EEG abnormalities can be detected in 46% of siblings. The type of seizure in affected relatives is variable; febrile or afebrile generalized tonic-clonic seizure predominates, followed by absence, myoclonic, or myoclonic-atonic seizures. Cases associated with generalized tonic-clonic seizures have an even higher prevalence (36%) of seizures among their parents or siblings than those without the generalized tonic-clonic seizures (12%) (17). However, some age-related modifying factors also seem to contribute, facilitating an unfavorable outcome by producing a pattern that resembles a symptomatic etiology (ie, Lennox-Gastaut).
Epilepsy with myoclonic-atonic seizures shows underlying genetic heterogeneity with only a few cases linked to mutations in genes reported in developmental and epileptic encephalopathies (57). Of significant interest are the clinical and molecular studies of epilepsy with febrile seizures plus, where myoclonic-atonic seizures are common (59). Epilepsy with febrile seizures plus has strong genetic links to Dravet syndrome. However, mutations in the SCN1A gene are rarely found in patients with epilepsy with myoclonic-atonic seizures (29; 59). In a study of 22 sporadic patients with myoclonic astatic epilepsy, no mutation was found in the three major genes of epilepsy with febrile seizures plus (SCN1A, SCN1B, and GABRG2) (41). In particular, SCNA1 mutation could be excluded, confirming that the condition is distinct from Dravet syndrome (41). This was confirmed by further studies (20).
An important finding is that four of 84 probands (5%) with epilepsy with myoclonic-atonic seizures had a mutation of SLC2A1 on sequencing, suggesting that a minority of patients with epilepsy with myoclonic-atonic seizures should be tested for glucose transporter 1 (GLUT1) deficiency (40). Diagnosis of GLUT1 deficiency is a strong indication for early use of the ketogenic diet, which may substantially improve the outcome of this severe disorder (71). In a study, Larsen and associates examined the role of SLC2A1 mutations in myoclonic-atonic epilepsy and absence epilepsy, and the estimated frequency of GLUT1 deficiency syndrome in the Danish population (35). None of the 120 patients with myoclonic-atonic epilepsy harbored SLC2A1 mutations. The estimated frequency of SLC2A1 mutations in the Danish population was approximately 1:83,000. Thus, this study failed to support the notion that SLC2A1 aberrations are a cause of epilepsy with myoclonic-atonic seizures without associated features such as movement disorders (35).
In another study, targeted resequencing of 644 individuals with epileptic encephalopathies led to the identification of 6 SLC6A1 mutations in seven individuals, all of whom have myoclonic-atonic epilepsy. There were two truncations and four missense alterations, all of which most likely lead to loss of function of GAT-1 and, thus, reduced GABA reuptake from the synapse. These individuals share many of the electrophysiological properties of Gat1-deficient mice, including spontaneous spike-wave discharges. Overall, pathogenic mutations occurred in six of 160 individuals with myoclonic-atonic epilepsy, accounting for approximately 4% of unsolved cases of epilepsy with myoclonic-atonic seizures (09).
There are several reports of single gene mutations identified in myoclonic-atonic epilepsy, including SCN1B (found in an individual in a family with GEFS+), SCN2A, GABRG2, CHD2, and the more recently described STX1B, SLC6A1, and GABRB3 genes (39).
A study done in Japan looked to identify genetic causes of myoclonic-atonic epilepsy and analyzed genomic DNA from 29 patients and their parents (26). Of the 29 patients, four had pathogenic variants, with two of them exhibiting possible candidate genes: HNRNPU and STS. Haploinsufficiency is thought to cause the pathologic phenotype of the HNRNPU mutation and is known to cause intellectual disability with or without dysmorphic features and infantile epileptic encephalopathy. Variants in this gene mutation can have variable epileptic seizure semiology, such as generalized tonic, atonic, or absence, and generally present by age 5 years. However, in this study of myoclonic-atonic epilepsy, the proband patient did not have intellectual disability or dysmorphic features. X-linked ichthyosis is caused by an STS mutation, which encodes steroid sulfatase. X-linked ichthyosis is associated with intellectual disability and neurobehavioral signs such as ADHD and autism, but is not associated with epilepsy. The patient in this study had dry skin and epilepsy, suggesting that the mutation may be causative of myoclonic-atonic epilepsy.
Tang and colleagues studied 101 patients with myoclonic-atonic epilepsy, performing exome sequencing and analysis on 85 patients. Pathogenic mutations were found in 12 patients (14.1%) in genes KCNA2, K1AA2022, SCN2A, SLC6A1, STX1B, MECP2, KCNB1, SYNGAP1, ASH1L, CHD4, and SMARCA2, proposing the last three as candidate genes.
Mutations in the SYNGAP1 gene can cause a phenotypic spectrum of myoclonic-atonic epilepsy. In a study of 57 patients with mutations in SYNGAP1, the varying phenotypes were evaluated (69). Eleven of these patients had a phenotype consistent with myoclonic-atonic epilepsy, though with atypical features. Children had atonic seizures, myoclonic-atonic seizures, and absence seizures; however, most had developmental delay prior to seizure onset and were also found to have subtle facial dysmorphisms.
Moeller and associates aimed to identify neuronal networks underlying generalized spike-and-wave discharges in epilepsy with myoclonic-atonic seizures (38). Simultaneous EEG-fMRI recordings were performed in 13 children with this epileptic syndrome. Individual generalized spike-wave discharge–associated blood oxygenation level-dependent (BOLD) signal changes were analyzed in every patient. A group analysis was performed to determine common syndrome-specific hemodynamic changes across all patients. Generalized spike-wave discharges were recorded in 11 patients, all showing generalized spike-wave–associated BOLD signal changes. Activation was detected in the thalamus (all patients), premotor cortex (six patients), and putamen (six patients). Deactivation was found in the default mode areas (seven patients). The group analysis confirmed activations in the thalamus, premotor cortex, putamen, and cerebellum and deactivations in the default mode network. The authors concluded that in addition to the thalamocortical network, which is commonly found in idiopathic generalized epilepsies, generalized spike-wave discharges in patients with myoclonic-atonic epilepsy are characterized by BOLD signal changes in brain structures associated with motor function. The results are in line with animal studies demonstrating that the somatosensory cortex, putamen, and cerebellum are involved in the generation of myoclonic seizures. The involvement of these structures might predispose to the typical seizure semiology of myoclonic jerks observed in epilepsy with myoclonic-atonic seizures.
Case reports. In a case report, a child with epilepsy with myoclonic-atonic seizures and a de novo SLC6A1 mutation had an excellent clinical response to the ketogenic diet (53). In another case report of a child with a balanced translocation of the SLC6A1 gene, the seizures responded well to valproic acid (39).
A chromodomain helicase DNA-binding protein 2 (CHD2) gene frameshift mutation was found in one patient (c.4256del19) with epilepsy with myoclonic-atonic seizures (66). This was a 17-year-old boy with no familial history of epilepsy and normal development before epilepsy onset. Epilepsy onset was at 3 years and 5 months: he presented with myoclonic-atonic seizures, head drops, myoclonic jerks, and absences. Interictal EEGs revealed slow background activity associated with generalized epileptiform abnormalities and photoparoxysmal response. His seizures were highly responsive to valproic acid, and an attempt to withdraw it led to seizure recurrence. Neuropsychological evaluation revealed moderate intellectual disability. However, the authors emphasized that chromodomain helicase DNA-binding protein 2 is not the major gene associated with epilepsy with myoclonic-atonic seizures.
Haploinsufficiency of the STX1B gene is also associated with this syndrome as documented in a case report of an 18-year-old male patient with myoclonic-atonic epilepsy, moderate to severe intellectual disability, behavioral problems, several dysmorphisms, and a 1.2-Mb de novo deletion on chromosome 16p11.2 (68). This deletion results in haploinsufficiency of STX1B and other genes.
The incidence of epilepsy with myoclonic-atonic seizures is 16.4 per 100,000 children, based on a population-based study looking at children with developmental and/or epileptic encephalopathies and intellectual disability with epilepsy (55). The accurate prevalence of the syndrome is not known. The condition is estimated to occur in 1% to 2% of all childhood epilepsies (17) and 5.5% of children between 1 and 9 years old with a diagnosis of generalized epilepsy (65). Two-thirds are boys (30).
To date, no prevention is possible. However, the most important thing is to diagnose the patient early and initiate treatment quickly, which may help improve cognitive and seizure outcomes (44).
The diagnosis of myoclonic-atonic epilepsy can be considered if myoclonic-atonic seizures start in a previously normal child with preexisting generalized tonic-clonic seizures and familial seizure disorders. In general, children with myoclonic-atonic epilepsy are normal prior to the development of seizures, have a strong family history of idiopathic generalized epilepsy, and have normal background EEG and brain imaging.
The following conditions should be considered in the differential diagnosis.
Lennox-Gastaut syndrome. Differential diagnosis problems with Lennox-Gastaut syndrome probably reflect ill-defined inclusion and exclusion criteria. Although drop attacks are common to both Lennox-Gastaut syndrome and myoclonic-atonic epilepsy, the predominant seizure type in Lennox-Gastaut syndrome is tonic. Tonic seizures are exceptional in myoclonic-atonic epilepsy and, if they exist, they mainly occur during sleep (29). Atypical absence may occur independently in Lennox-Gastaut syndrome, whereas the absence seizures in myoclonic-atonic epilepsy usually occur with myoclonic and atonic episodes. The EEG of Lennox-Gastaut syndrome shows a pattern of diffuse slow (2 Hz) spike waves and polyspike waves superimposed on slow background activity, whereas myoclonic-atonic epilepsy shows more rapid (2 to 3 Hz) spike-wave discharges.
Progressive myoclonic epilepsies. Progressive myoclonic epilepsies, such as myoclonic epilepsy with ragged red fibers, Lafora disease, neuronal ceroid-lipofuscinoses type 2, and late-infantile GM2-gangliosidosis, may initially imitate epilepsy with myoclonic-atonic seizures (45). However, their associated relevant neurologic abnormalities and, often, their relentless progression and deterioration will establish the diagnosis.
Developmental epileptic encephalopathy with “spike-wave activation in sleep” and epileptic encephalopathy with “spike-wave activation in sleep” (DEE-SWAS and EE-SWAS). Children with DEE-SWAS or EE-SWAS may also have drop attacks due to atypical absences or negative myoclonus, which are characterized by lapses of muscle tone that appear when the patient stretches his arm out in front, but do not show when the patient is lying down (23).
Infantile epileptic spasms syndrome. Epileptic spasms are brief (less than 3 seconds) tonic contractions of axial muscles that tend to occur in clusters. Onset is between 1 and 24 months old, with peak incidence between 3 and 12 months of age (73). Myoclonic-atonic epilepsy, on the other hand, has a more variable seizure pattern (myoclonic, atonic, myoclonic-atonic, absence, generalized tonic-clonic, clonic, and tonic seizures). The seizures are more prolonged, and the EEG shows regular and irregular bilaterally synchronous 2 to 3 Hz spike waves and polyspike patterns, with a 4 to 7 Hz background.
Malformations of cortical development. Malformations of cortical development include another group of conditions that potentially cause structural epilepsy with myoclonic-atonic seizures (45). They display generalized spike-and-wave or multiple spike-and-wave discharges similar to the EEG pattern of myoclonic-atonic epilepsy and may have localized frontal lobe EEG foci. Although patients with diffuse cortical malformations have significant learning difficulties, this is not so in cases of focal cortical dysplasia, which can imitate the clinical and EEG features of myoclonic-atonic epilepsy.
Non-epileptic myoclonus may, rarely, raise a diagnostic problem with epilepsy with myoclonic-atonic seizures.
By definition, imaging and EEG background are normal. Brain imaging may show mild brain subcortical atrophy in some cases (33; 34). Testing for glucose transporter 1 (GLUT1) deficiency is probably mandatory (40).
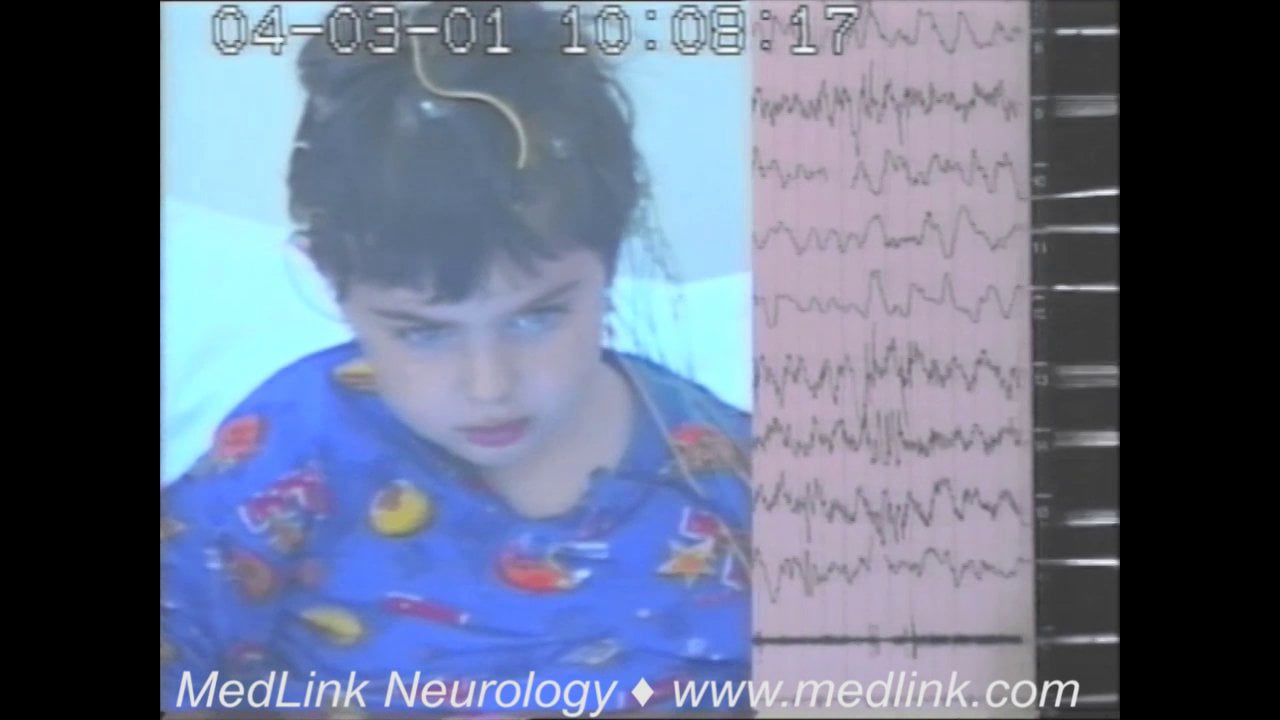
Although the interictal EEG may be normal, rhythmic theta activity in the parasagittal regions may be present. When myoclonic-atonic seizures occur, they are associated with clusters of 2 to 6 Hz generalized spike/polyspike-wave discharges, interrupted by high-amplitude slow waves in cases with predominant atonic or myoclonic-atonic seizures (61). Sleep facilitates the appearance of spike-wave or polyspike-wave discharges.
Ictal EEG of myoclonic and atonic seizures manifests with discharges of irregular spike-wave or polyspike-wave complexes at a frequency of 2.5 to 3 Hz or more.
Atonia is usually concurrent with the slow wave of a single- or polyspike-wave complex, and the intensity of the atonia is proportional to the amplitude of the slow wave. Drop attacks are associated with diffuse electromyography (EMG) paucity, indicating their true atonic nature (48; 49). The intensity of the myoclonic seizure may be correlated with the depth of the positive sharp wave component of the spike-polyspike wave complex (51; 47). In myoclonic-atonic status epilepticus, the EEG shows continuous or discontinuous and repetitive 2 to 4 Hz or higher spike/polyspike-wave discharges.
Genetic testing should be considered in patients with epilepsy with myoclonic atonic seizures. In addition to SLC2A1, other genes that have reported association with myoclonic-atonic epilepsy may need to be considered and tested: SCN1B, SCN1A, SCN2A, SCN8A, GABRB3, CACNA1H, CHD2, HNRNPU, IQSEC2, NEXMIF, POL3B, SYNGAP1, KCNA2, STX1B, SLC6A1, SLC2A1, TBC1D24, KIAA2022, KCNT1, STXBP1, SYNGAP1, and MECP2 (01; 63; 24). Whole-exome sequencing is the most comprehensive approach for identifying genetic causes of epilepsy with myoclonic atonic seizures, analyzing rarer genes, and enabling future reanalysis of gene variants (24).
Treatment of myoclonic-atonic epilepsy is empirical, and it is dictated by the type of seizures. The Pediatric Epilepsy Research Consortium survey identified valproate as a first-line recommended therapy (24). The consensus is that valproate, which is effective in myoclonic jerks, atonic seizures, and absences, is the most efficacious of the antiepileptic drugs (42; 45; 32; 70; 14; 44). Small, add-on doses of lamotrigine prior to starting valproate decrease the occurrence of rash from the introduction of lamotrigine while the patient is on valproate. However, valproate may rarely induce tonic status epilepticus (22). Valproate and clobazam were recommended as first-line treatment by the international Delphi consensus (24).
Topiramate reduces the frequency of atonic seizures. Ethosuximide (for absences) and clonazepam (for myoclonic seizures) are also beneficial. Rufinamide was found to be effective in a small retrospective European multicenter study of eight patients with myoclonic-atonic epilepsy (70). In small retrospective series, felbamate showed more than a 50% decrease in seizures in patients with epilepsy with myoclonic-atonic epilepsy (74). However, because there is a concern for significant side effects (aplastic anemia, hepatic failure), no recommendations can be given for felbamate. Levetiracetam has also been used, but occasionally there is a dose-related worsening in the frequency of the myoclonic and atonic seizures (36).
Cannabidiol-enriched cannabis has been assessed as very beneficial by parents of four children with epilepsy with myoclonic-atonic seizures (56; 12). However, safety and tolerability data for cannabidiol-enriched cannabis use among children are not available. An open-label noncontrolled trial of purified cannabidiol was studied in refractory epilepsy due to four etiologies: CDKL5 deficiency disorder, Aicardi syndrome, Dup15q syndrome, and Doose syndrome. In the Doose syndrome subgroup, there was a median decrease of seizures of 58.6% at 12 weeks and 28.8% at 48 weeks of treatment. Responder rate (> 50% decrease in seizure frequency from baseline) in the Doose group was 43% at 12 weeks and 57% at 48 weeks (13). In a retrospective study of patients with epilepsy with myoclonic astatic seizures or Sturge-Weber syndrome with myoclonic-atonic seizures, responder rate to cannabidiol (CBD) was 57.7%, with decreased frequency of myoclonic/atonic, tonic, and tonic-clonic seizures (08).
In myoclonic-atonic status epilepticus, intravenous benzodiazepines are often efficacious, but may, rarely, precipitate tonic status epilepticus. In resistant cases, ketogenic diet is often therapeutic (07; 32; 04; 37). Ketogenic diet should be started as soon as the signs of epileptic encephalopathy with cognitive deterioration appear. In non-responders to the diet and in those who do not tolerate it, the indication of adrenocorticotropic hormone or steroids should be considered. The treatment needs to be prolonged in order to prevent relapse during the period of risk, which lasts 1 to 2 years. Ketogenic diet is the treatment of choice in those 5% of patients with epilepsy with myoclonic-atonic seizures found to have glucose transporter 1 (GLUT1) deficiency (40; 53; 71) (see details in Etiology and pathogenesis section).
In the multicenter study by Nickels and colleagues, 5% of patients were seizure-free on the first antiseizure medication (43). Most of the patients (81%) were started either on levetiracetam or sodium valproate; the rest were started on a variety of other antiseizure medications. A greater than 50% reduction of seizures in response to the first three antiseizure medications was seen in only 26% of patients. Of the 166 patients in the study, 57% of the children were put on the ketogenic or modified Atkins diet after failure of greater than two antiseizure medications. Of these patients, 79% of the patients had a greater than 50% reduction in seizures, demonstrating the superiority of dietary therapy over the first three antiseizure medications (43). In practice, the best therapy is to begin valproate from the first seizure in which the EEG shows signs consistent with this diagnosis in a child aged 2 to 5 years (generalized spike waves and slowing of the background). At this point, it may be indicated to start on a combination of valproate with lamotrigine because no case will be controlled by valproate monotherapy, and the introduction of lamotrigine requires 6 weeks, which is long regarding the usually acute expression of the disease. Another approach is to begin with a low dose of lamotrigine and then add valproate, to avoid the complication of a rash, as when lamotrigine is started after valproate.
In a retrospective study of nine patients with myoclonic-atonic epilepsy, both the modified Atkins diet and ketogenic diet were found to be efficacious in complete seizure control and allowed other medications to be stopped in seven patients (60). Two patients had greater than 90% seizure control without medications: one was on the ketogenic diet and the other on the modified Atkins diet. Seizure freedom has ranged from 13 to 36 months, and during this time, four patients have been fully weaned off of diet management. One patient was found to have a mutation in SLC2A1. The authors concluded that strictly defined patients with myoclonic-atonic epilepsy respond to the modified Atkins diet with prolonged seizure control. Some patients may require the ketogenic diet for seizure freedom, suggesting a common pathway of increased requirement for fats. Once controlled, those fully responsive to the diet(s) could be weaned off traditional seizure medications and, in many, subsequently off the modified Atkins diet and ketogenic diet.
In a report, the modified Atkins diet was applied in 30 patients with epilepsy with myoclonic-atonic seizures. By the end of the observation period, 83% experienced a seizure reduction by 50% or more, and 47% were seizure-free (72).
Caution: Long-term ketogenic diet treatment stimulates liver parenchymal injury, hepatic steatosis, and gallstone formation. Therefore, patients should be monitored by screening liver enzymes and abdominal ultrasonography in order to detect these side effects (02).
Vagus nerve stimulation may be an option for drug-resistant epilepsy in these patients. In a meta-analysis of children with genetic epilepsy that was drug resistant, patients with epilepsy with myoclonic atonic seizures (Doose Syndrome) showed benefits in seizure reduction, duration of seizure, duration of postictal state, and cognitive status. However, there was a limited number of patients, and broader conclusions could not be drawn (25).
Responsive neurostimulation in patients with epilepsy with myoclonic atonic seizures has not been specifically studied. There is blood-oxygen-level-dependent (BOLD) signal activation in the thalamus during generalized spike and wave discharges, which may support the use of thalamic responsive neurostimulation, though this has not been specifically reported (58).
Early evaluation of executive functioning using both questionnaires and standardized tools is necessary for early detection of executive functioning deficit and initiating tailored rehabilitation (06).
Family support and psychotherapeutic approaches should be considered (67).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Aparna Polavarapu MD
Dr. Polavarapu of Albert Einstein College of Medicine and Montefiore Medical Center has no relevant financial relationships to disclose.
See Profile
Solomon L Moshé MD
Dr. Moshé of Albert Einstein College of Medicine has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Epilepsy & Seizures
Jun. 02, 2026
Neuropharmacology & Neurotherapeutics
May. 11, 2026
Epilepsy & Seizures
May. 08, 2026
Epilepsy & Seizures
May. 01, 2026
Epilepsy & Seizures
Apr. 30, 2026
Epilepsy & Seizures
Apr. 17, 2026
Epilepsy & Seizures
Apr. 13, 2026
Epilepsy & Seizures
Mar. 24, 2026