



Epilepsy & Seizures
Febrile seizures
Jun. 02, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Focal clonic seizures are a type of neocortical seizure emanating from the contralateral primary motor cortex. They are repetitive, brief and rhythmic, unilateral, and rapidly alternating contractions and relaxations of localized groups of muscles. They may affect the thumb only, the thumb and ipsilateral side of the lips, the hand, the whole arm, or any other contralateral to the focus body part. They may remain focal or progress to a Jacksonian march and hemiclonic or generalized tonic-clonic seizures. Etiology is variable, ranging from genetic, metabolic, or any type of focal structural lesions. Rolandic epilepsy is a common idiopathic syndrome with focal clonic seizures. Brain tumors, infections, malformation of cortical development, and trauma are other common structural causes. In this updated article, the author details the historical aspects, classification, clinical manifestations, pathophysiology, EEG, differential diagnosis, and management of patients with focal clonic seizures.
• Focal clonic seizures are a type of neocortical seizure emanating from the contralateral primary motor cortex. | |
• They are repetitive, brief and rhythmic, unilateral, and rapidly alternating contractions and relaxations of localized groups of muscles. They may affect the thumb only, the thumb and ipsilateral side of the lips, the hand, the whole arm, or any other contralateral to the focus body part. | |
• They may remain focal or progress to a Jacksonian march and hemiclonic or generalized tonic-clonic seizures. | |
• They often occur in conjunction with tonic, atonic, myoclonic, somatosensory, and other ictal manifestations. These may precede, occur concurrently, or present at some stage after the onset of the clonic convulsions. | |
• Postictal paresis (Todd paralysis) is common. | |
• Etiology is variable, ranging from genetic, metabolic, or any type of focal structural lesions. Rolandic epilepsy is a common syndrome with focal clonic seizures. Brain tumors, infections, malformation of cortical development, and trauma are other common causes. | |
• Investigative procedures include brain MRI and EEG. | |
• Prognosis and management depend on the underlying cause. |
The first account of focal clonic seizures can be found as early as 1050 BC in the twenty-fifth Babylonian cuneiform tablet devoted to miqtu (a disease in which the person loses consciousness and foams at the mouth) (75):
In the time of his possession, while he is sitting down, his (left) eye moves to the side, a lip puckers, saliva flows from his mouth, and his hand, leg and trunk on the left side jerk (or, twitch) like a (newly)-slaughtered sheep, it is miqtu. If at the time of the possession his mind is consciously aware, (the demon) can be driven out; if at the time of the possession his mind is not so aware, (the demon) cannot be driven out. |
However, every student of neurology is familiar with the contributions of Louis Francois Bravais (08) and John Hughlings Jackson (31; 32; 33; 29; 30) in the clinical description of motor seizures from the primary motor cortex (ie, Bravais-Jackson seizure, Jacksonian seizure) (42; 15; 54).
The following are exact quotations from Jackson relevant to focal clonic seizures and postictal paralysis:
Convulsions and other paroxysms are owing to (1) sudden, (2) excessive, and (3) temporary nervous discharges.
The quantity of cortical grey matter varies not so much with the size of the muscles of a part of the body, as with the number of movements of that part. Thus the small muscles of the fingers will be represented by much more grey matter in the cortex than will be the voluminous muscles of the upper arm, because the former serve in more numerous, different, and in more specialized movements. Greater differentiation of function implies larger representation in the brain. I mention epileptiform seizures first because their localisation is not doubtful. (They were first described by Bravais in 1824). They are "middle level fits"—that is, they are produced by excessive discharges beginning in parts of the middle level (motor province) of the cerebral system ("motor region"). The three commonest varieties of epileptiform seizures are: (1) Fits starting in the hand (most often in the thumb or index finger or in both). (2) Fits starting in one side of the face (most often near the mouth) or in the tongue, or in both these parts. (3) Fits starting in the foot (nearly always in the great toe). Of necessity the three Varieties depend on the fact that the local discharging lesion is of cells of different parts of the "motor region"—of hand, face, and foot centre respectively. The starting point is almost invariably the same in each patient, but not always… Often enough there is "tingling" or some other crude sensation in the place of onset before convulsion starts. Admitting that there are many ranges, it is allowable to make arbitrary divisions (they must not be taken for real distinctions) into four ranges: (1) Terminal fit—the spasm involves, say the hand, or some part of it only; (2) monospasm—the arm becomes involved; (3) hemispasm; (4) bilateral convulsion—the second side being gained, the fit becomes universal. Observe the use of the word "become." In the range (4) there is a March of spasm from the part first seized all over the body, answering to increased spreading of the central excessive discharge beginning in some particular part of the motor region of the first half, and may be extending to the second half, of the brain. The second side of the body (in 4) is affected later, and commonly less than, the first side. We have not only to note how much of the body is ultimately involved, but also the order in which the several parts involved are affected—the March of the convulsion. There is not a simple, but a compound sequence of spasm; the convulsion does not cease in one part when another is involved. To observe, to give a simple example, how much of the arm has been involved when convulsion appears in the face will, I think, help us to clearer notions of localisation of movements of those two separate parts of the body in the centre discharging (anatomical localisation); or if not, at any rate as to the time relations of different elements of different centres (physiological localisation). From increasing discharge of a motor centre there is a double effect; there is not simply "more convulsion," there is (1) greater amount of convulsion of the part first seized, and there is (2) extension of convulsion to the next part of the same muscular region, or to some other part represented in the centre discharging (or in another centre connected with the one primarily discharging by particular time relations). The term post-epileptiform paralysis will be used to include all paralyses the immediate sequels of epileptiform seizures. Arbitrary divisions may be made, speaking of range only. (1) Terminal paralysis, as of a hand; (2) monoplegia, as of an arm; (3) hemiplegia; and (4) a range, which is not generally admitted—some degree of slight universal paralysis. I hold the hypothesis (essentially that of Todd* and Alexander Robertson) that there is exhaustion of central nervous elements, including fibres of the second segment of the kinetic route, and that this is produced by the sudden and excessive discharge in the prior paroxysms. These nervous elements are exhausted not otherwise injured. |
*This paralysis was long ago described by Todd under the name "epileptic hemiplegia."
The magnitude of Jackson’s contributions should be appreciated considering that his inferences encountered strong opposition during his lifetime, and the contemporary view was the notion that the cerebral convolutions were "for mentation" only (73). Gordon Holmes said of Jackson (28):
Local motor epilepsy was, it is true, recognized and described by Bravais in 1827, but it was Hughlings Jackson who first associated it with disease of the opposite hemisphere of the brain; his observations on it threw the first light on the localization and organization of the cortical motor apparatus. …. The chief features of Jacksonian epilepsy are well known, but its study helps to explain some of the phenomena of the rarer forms of local epilepsy. |
Foerster said of Jackson (20):
Jackson's ideas were revolutionary. In his days the prevalent view as to the cause of epilepsy was that it depended upon disturbances of the medulla or the pons, and nobody believed that the cortex of the brain, the organ of mind, contained motor nervous elements and that an epileptic convulsion was nothing but a sudden, rapid and excessive discharge of these cortical motor cells… Postparoxysmal paralysis was studied carefully by Jackson. He refers to previous observations of Todd and Robertson on epileptic hemiplegia, but Jackson was the first to give the physiological explanation of the paralysis subsequent to epileptic convulsions, an explanation which has been accepted by all succeeding investigators. The paralysis is due to exhaustion of the cortical motor nerve-cells, and on this depends the transient loss of function. |
It was Charcot who coined the name “Jacksonian epilepsy” in 1877, and later “epilepsie Bravais-Jacksonienne” in 1887 (15).
Robert Bentley Todd described "epileptic hemiplegia," which is now known as Todd paralysis (05). Todd held that some patients “who recover from a severe fit, or from frequently repeated fits of epilepsy, are often found to labor under hemiplegia, or other modifications of palsy" (69). He believed that this resulted from "undue exaltation [resulting in] a state of depression or exhaustion. A paralytic state remains sometimes after the epileptic convulsion. This is more particularly the case when the convulsion has affected only one side or one limb: that limb or limbs will remain paralytic for some hours, or even days, after the cessation of the paroxysm, but it will ultimately perfectly recover” (69).
Aleksei Kozhevnikov described epilepsia partialis continua and its association with chronic encephalitis in 1894 (37; 55; 71; 24).
The history of the brain mapping with electrical stimulation can be found in the writings of Penfield and associates, from Fritsch and Hitzig’s research in electrical stimulation (who, by applying galvanic currents through bipolar electrodes to the anterior half of the dog's hemisphere, obtained movements of muscle groups in the opposite half of the body) to the pioneering work of Foerster, Penfield and Jasper, and Penfield’s formulation of the homunculus (59; 60; 61).
Terminology and classification. The 2010 and 2014 ILAE reports define focal clonic seizures as follows (04; 12):
Focal epileptic seizures are conceptualized as originating within networks limited to one hemisphere. They may be discretely localized or more widely distributed. Focal seizures may originate in subcortical structures. For each seizure type, ictal onset is consistent from one seizure to another, with preferential propagation patterns that can involve the contralateral hemisphere. In some cases, however, there is more than one network, and more than one seizure type, but each individual seizure type has a consistent site of onset. Focal seizures do not fall into any recognized set of natural classes based on any current understanding of the mechanisms involved. In the 2014 ILAE epilepsy diagnosis manual, focal motor seizures are classified among those with elementary motor features that involve a stereotyped contraction of a muscle or group of muscles. Such motor features may be predominantly convulsive (rhythmic jerking (clonic activity)) (12). |
In 1981, the ILAE Commission of Classification and Terminology classified focal clonic seizures amongst “partial seizures with motor signs” (10):
A. Partial Seizures 1. With motor signs. Any portion of the body may be involved in focal seizure activity depending on the site of origin of the attack in the motor strip. Focal motor seizures may remain strictly focal or they may spread to contiguous cortical areas producing a sequential involvement of body parts in an epileptic “march.” The seizure is then known as a Jacksonian seizure. Consciousness is usually preserved; however, the discharge may spread to those structures whose participation is likely to result in loss of consciousness and generalized convulsive movements. Other focal motor attacks may be versive with head turning to one side, usually contraversive to the discharge. If speech is involved, this is either in the form of speech arrest or occasionally vocalization. Occasionally a partial dysphasia is seen in the form of epileptic palilalia with involuntary repetition of a syllable or phrase. Following focal seizure activity, there may be a localized paralysis in the previously involved region. This is known as Todd paralysis and may last from minutes to hours. When focal motor seizure activity is continuous it is known as epilepsia partialis continua. Postictal Paralysis (Todd Paralysis): This category refers to the transient paralysis that may occur following some partial epileptic seizures with focal motor components or with somatosensory symptoms. Postictal paralysis has been ascribed to neuronal exhaustion due to the increased metabolic activity of the discharging focus, but it may also be attributable to increased inhibition in the region of the focus, which may account for its appearance in non-motor somatosensory seizures. |
The ILAE core group classified focal clonic seizures as follows (17):
A. Local | ||
(1) Neocortical | ||
(a) Without local spread. Focal clonic seizures are brief focal motor events that are distinguished from focal myoclonic seizures by their rhythmic repetition. Localization to the primary motor cortex is implied. | ||
(b) With local spread. Jacksonian march seizures refer to the clinical manifestations of the slow ephaptic propagation of epileptic discharge along the motor cortex, although similar progression can sometimes be seen in other primary cortical areas as well. | ||
B. With ipsilateral propagation to: | ||
(1) Neocortical areas (includes hemiclonic seizures) | ||
The most recent ILAE positional papers on the operational classification of seizure types classify seizures of focal onset as follows:
• Aware or impaired awareness | |
• Motor onset (automatisms, atonic, clonic, epileptic spasms, hyperkinetic, myoclonic, tonic) | |
• Nonmotor onset (autonomic, behavior arrest, cognitive, emotional, sensory) | |
• Focal to bilateral tonic-clonic |
According to the various ILAE classifications and definitions, clonic seizures are mainly distinguished from myoclonic seizures by their rhythmicity. This has created significant overlap in what to call clonic or myoclonic seizures, and these terms are often used interchangeably. Relevant ILAE terminologies are cited in Tables 1 and 2.
The 2022 ILAE report from The Task Force on Nosology and Definitions uses the terms focal clonic and hemiclonic interchangeably (77).
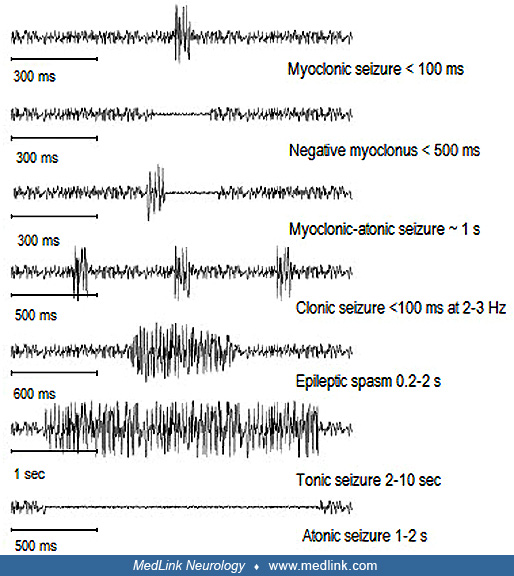
Clonic | Myoclonus that is regularly repetitive and involves the same muscle groups, at a frequency of approximately 2 to 3 c/s, and is prolonged. Synonym: rhythmic myoclonus. |
Focal | A seizure whose initial semiology indicates, or is consistent with, initial activation of only part of one cerebral hemisphere. Synonym: partial. |
Jacksonian march | Traditional term indicating spread of clonic movements through contiguous body parts unilaterally. |
Lateralizing [Todd (or Bravais)] phenomenon | Any unilateral postictal dysfunction relating to motor, language, sensory, and/or integrative functions, including visual, auditory, or somatosensory neglect phenomena. |
Myoclonic (adjective) | Sudden, brief (< 100 ms), involuntary single or multiple contraction(s) of muscles(s) or muscle groups of variable topography (axial, proximal limb, distal) |
| |
Clonic | Jerking, either symmetric or asymmetric, that is regularly repetitive and involves the same muscle groups |
Focal onset | Originating within networks limited to one hemisphere. They may be discretely localized or more widely distributed. Focal seizures may originate in subcortical structures. |
Focal to bilateral tonic-clonic | Replaces the older term “secondarily generalized tonic-clonic” |
Focal aware | Corresponds to the prior term “simple partial seizure” |
Focal impaired awareness | Corresponds to the prior term “complex partial seizure” |
Focal aware or impaired awareness | Optionally may further be characterized by one of the motor-onset or nonmotor-onset symptoms reflecting the first prominent sign or symptom in the seizure. Seizures should be classified by the earliest prominent feature. |
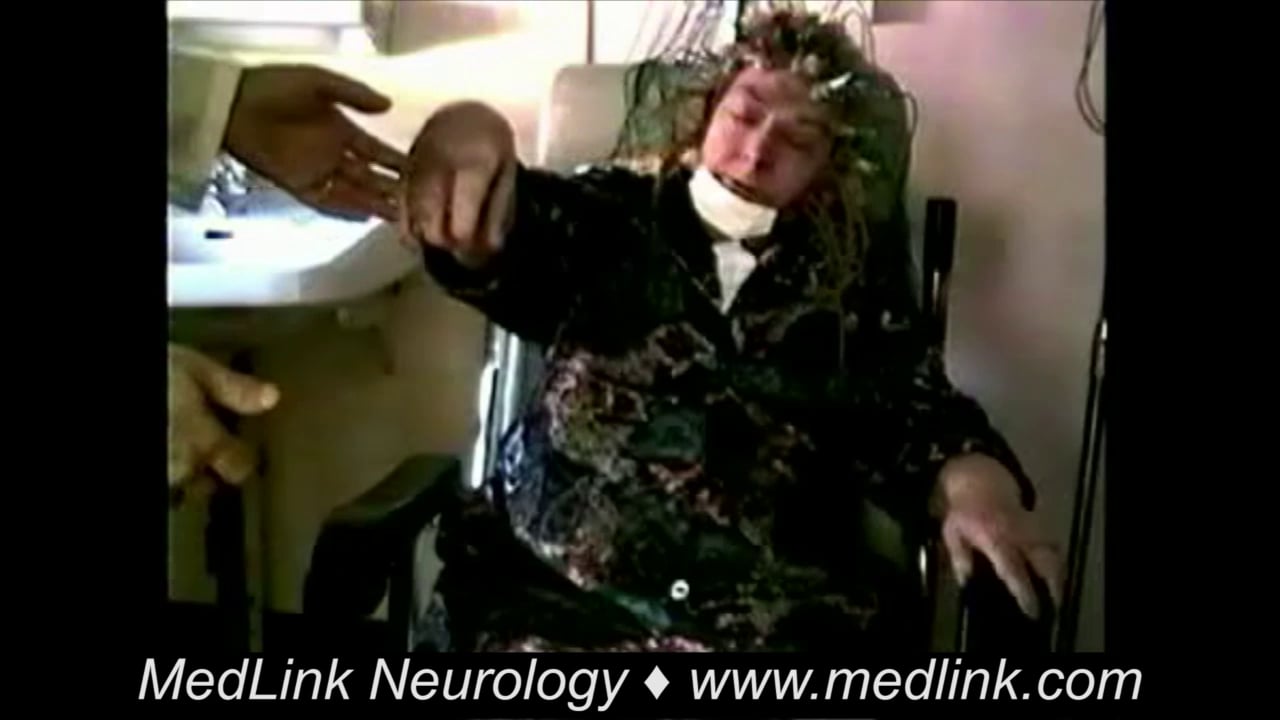
Fencer motor | A focal motor seizure type with extension of one arm and flexion at the contralateral elbow and wrist, giving an imitation of swordplay with a foil; also called a supplementary motor area seizure |
Jacksonian | Traditional term indicating spread of clonic movements through contiguous body parts unilaterally |
Myoclonic | Sudden, brief (< 100 msec) involuntary single or multiple contraction(s) of muscles(s) or muscle groups of variable topography (axial, proximal limb, distal). Myoclonus is less regularly repetitive and less sustained than is clonus. |
| |
Focal clonic seizures manifest with repetitive, brief and rhythmic, unilateral movements of rapidly alternating contractions and relaxations of a localized group of muscles. They are contralateral to the epileptogenic focus in the primary motor cortex. The seizures consist of localized clonic movements that may affect the thumb only, the thumb and ipsilateral side of the lips, the hand, the whole arm, or any other contralateral to the focus body part. Distal segments are more frequently affected than proximal segments. Hand and mainly thumb, and face and mainly lips, are preferentially affected because of their larger cortical representation (homunculus of Penfield) (59). Consciousness is usually intact, and the patient is fully aware of the seizures. By definition of clonic seizures, these should be rhythmic though they are also arrhythmic (and, hence, the confusion with myoclonic seizures).
These ictal motor manifestations may remain highly localized for the whole of the seizure (focal clonic seizures without march) or march in an ordinary anatomical fashion to neighboring motor regions, which constitutes the classical Jacksonian (or Bravais-Jackson) seizure (focal clonic seizures with march) (73; 65; 22; 42; 17; 54). Thus, convulsions might begin in the hand, spread to the arm and face and down the leg to the foot, or they may follow a reverse manner from the foot to the leg, down the arm, and to the face.
Focal clonic seizures rarely manifest by themselves only. They often occur in conjunction with tonic, atonic, myoclonic, somatosensory, and other ictal manifestations. These may precede, occur concurrently, or present at some stage after the onset of the clonic convulsions. This should not be surprising considering that they are all manifestations of the primary motor cortex and its neighboring somatosensory cortex.
Tonic manifestations of the affected muscles, such as initial flexion of the fingers and hand, are frequent, and these may occur prior to the clonic convulsions as documented by intracranial recordings (26). Tonic and clonic contractions may merge. Clonic jerks can be associated with tonic postural signs or turning towards the opposite direction.
It is generally regarded as characteristic of the Jacksonian convulsion that it is clonic, but most observers have recorded the fact that it may be tonic at first, and for a few seconds, then becoming clonic. In general it may be said that the convulsion that remains restricted to a narrow field of musculature and is prolonged tends to be clonic from the outset, but the rapidly spreading convulsion may be tonic for a few moments before the spasm begins to intermit, ie, to become clonic (73). |
Isolated focal tonic manifestations are rare (22).
Sensory-motor seizures of rolandic epilepsy are typical with regards to concurrent sensory and motor manifestations, and epilepsia partialis continua with regard to concurrent clonic and myoclonic jerks.
Sensory concomitants of convulsion: In a considerable number of cases of convulsion beginning unilaterally, there are no subjective or objective sensory concomitants in the convulsed or in other parts, but in not a few cases of the kind subjective sensory phenomena may precede or accompany spasms or loss of power. These may appear in the part weakened or convulsed, or in some other part. Thus Jackson records the development of sensations of numbness in the hand to be followed immediately by clonic spasm in the face. This numbness may accompany weakness of the part affected, or occur without this (73). Other motor concomitants of convulsion: Loss of power. A sudden loss of power in a part is a familiar feature in the range of Jacksonian attacks. It may on occasion be the sole manifestation, it may precede convulsion in a given part, or it may precede convulsion in another, muscle series. In yet other cases, loss of power may characterize some fits, convulsion others (73). |
Furthermore, focal clonic seizures may be less well localized and appear to involve larger areas simultaneously. Thus, an entire extremity, or even half of the body, may begin to convulse almost simultaneously with or without loss of consciousness (hemiclonic seizures) (23; 22; 51).
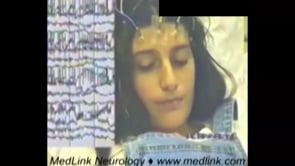
In rolandic epilepsy, hemifacial seizures occur in approximately one-third of patients (57; 54). These may be entirely localized in the lower lip, manifesting with sudden, continuous, or bursts of clonic contractions usually lasting seconds to one minute. Involvement of the ipsilateral eyelids is not unusual. More rarely, ictal symptoms of clonic convulsions may appear nearly simultaneously or spread to the ipsilateral upper extremity. Involvement of the leg is rare. Hemifacial motor seizures may be the only ictal manifestation, but this is often associated with inability to speak, hypersalivation, localized paresthesia, and tonic contraction of the mouth to one side, which may progress or appear synchronously with oropharyngolaryngeal symptoms and other buccal symptoms. Any combination of these symptoms is possible, although focal clonic seizures may occur alone and may also be the only seizure type in a child’s lifetime.
Focal clonic seizures usually last for 30 seconds two to three minutes, but they may continue for long periods constituting focal clonic status epilepticus. Focal clonic status epilepticus is defined as focal clonic seizures occurring repetitively, continuously, or briefly interrupted for more than 30 minutes, and may last for hours or days. Focal clonic convulsions mainly affect the face and hand, wax and wane in intensity and frequency, and may be very subtle. Epilepsia partialis continua, Kozhevnikov-Rasmussen syndrome, and cerebrovascular stroke are the most common causes.
The video shows continuously repetitive lower facial rhythmic and arrhythmic jerks without impairment of consciousness and without discernible ictal EEG changes. Each jerk lasts a few milliseconds and repeated every 1 to 4 seco...
Those fits that remain restricted to the small group of muscles in which they arise, eg, thumb and index muscles, tend to last the longest, and such an attack may last for many minutes or even for a considerable fraction of an hour. The faster and the farther a fit spreads, the shorter usually its duration (73). |
Postictally, patients may quickly return to normality, but some may show transient postictal paresis of the affected muscles.
Focal clonic seizures may be accentuated, induced, or inhibited by active or passive movement of the affected muscles. Some patients may abort focal motor seizures by moving or rubbing the part of the body in which the initial symptoms occur.
The influence of peripheral stimuli on the convulsion beginning unilaterally. One of the most interesting phenomena of the Jacksonian convulsion, and one very early recognized, is the influence of peripheral stimuli applied to the part convulsed. It is less widely recognized that this influence may be in the direction of (a) increasing, as well as of (b) diminishing or abolishing the convulsion (73). |
Prognosis is variable depending on the underlying etiology and syndrome. It is benign in rolandic epilepsy, but very severe in Kozhevnikov-Rasmussen syndrome.
Case 1. Focal clonic seizures and focal clonic status epilepticus due to right-sided cerebrovascular stroke. Onset is with focal-tonic contraction. A 72-year-old man had postvascular epilepsy related to a right ischemic stroke that occurred three years prior and involved the right superficial sylvian artery area. He also had several episodes of focal clonic status epilepticus related to alcohol intake and noncompliance. The patient was admitted to the hospital following serial hemifacial clonic seizures that were not controlled by intravenous diazepam 20 mg. The emergency video-EEG showed left simple focal hemifacial seizures that began with a left tonic hemifacial contraction followed by sustained left hemifacial, high-frequency, clonic jerks. During these seizures, neither consciousness nor language was impaired. Intravenous fosphenytoin 1200 mg was necessary to stop the seizures. The patient was discharged with carbamazepine 1200 mg/day and levetiracetam 1500 mg/day, and he had had no further seizures at follow-up.
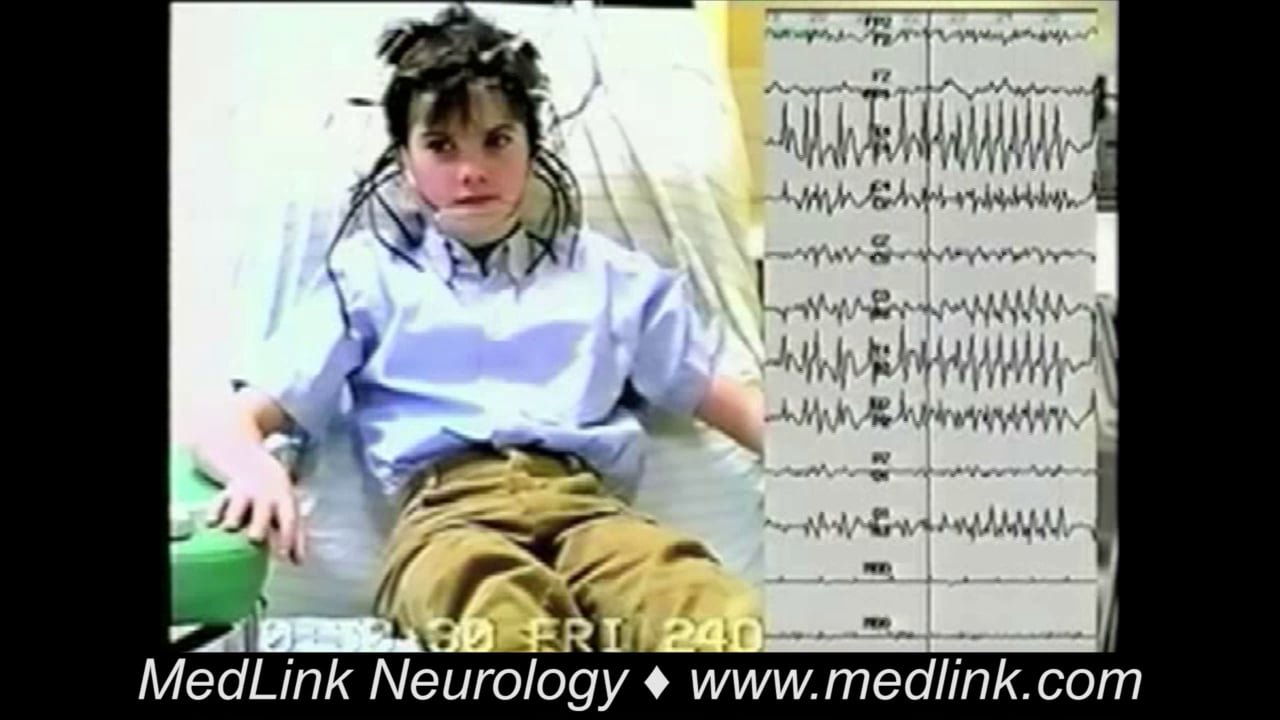
Case 2. Rolandic seizure starting with focal tonic-clonic convulsions and progressing to secondarily generalized tonic-clonic seizures. A 9.5-year-old girl, at the top of her class, had 5 to 10 seizures that started at the age of 8, mainly in successive nights. All of the seizures occurred approximately half an hour after she had gone to sleep. Her parents believed that they could predict when she was about to have an attack because she felt nauseated for a day or so prior to the attack and did not seem herself. She looked rather pale and became tired easily.
The very beginning of the seizures was never witnessed. Her parents became aware of them because they had a baby alarm in her room, and they heard a strange gurgling noise. They found her unresponsive and drooling, with her face twisted or deviated to one side; this was followed by unilateral clonic convulsions. Her first seizure was prolonged for 40 minutes, during which she had repeated ictal vomiting (rolandic seizures combined with symptoms of Panayiotopoulos syndrome). She had two routine awake EEGs. The first was normal and the second was reported as showing runs of occipital intermittent rhythmic delta waves, interpreted elsewhere as suggestive of localized or systemic pathology. Brain MRI was normal. When followed at 18 years of age, she was entirely normal and unmedicated.
Case 3. Epilepsia partialis continua of unknown cause. An intelligent 12-year-old girl had epilepsia partialis continua, which started at the age of four and continued to date with increasing frequency and duration. Fast (3 to 10 Hz) twitching of the left eyelid occurred simultaneously with the left rectus abdominis (she pointed close to the midline by the umbilicus) and a muscle in the armpit (likely the latissimus dorsi). This lasted from hours to two to three days and was continuous, day and night. This was interspersed with left-sided, focal tonic-clonic motor seizures mainly affecting the face and upper limb. Additionally, there was postictal, and probably ictal, left hemiparesis, mainly of the upper limb. She did not lose consciousness during these attacks and communicated well. She was also able to understand, but could not speak, during the focal motor seizures. She had never had a full-blown generalized tonic-clonic seizure.
Initially, the seizures occurred once or twice per year, but increased to every two weeks. Neurologic and mental status was normal. High-resolution brain MRI was normal. All appropriate tests for metabolic or other diseases associated with epilepsia partialis continua were normal. Drug treatments had failed; only rectal diazepam provided temporary relief during the attacks.
Epilepsia partialis continua is a focal motor status epilepticus that may occur in a number of diverse conditions and has a number of different causes, eg, Kozhevnikov-Rasmussen syndrome in a child (54). However, the patient’s good clinical status over the years, as well as her normal MRI and the lack of background EEG abnormalities, is rather against this diagnosis, though some exceptional cases of Kozhevnikov-Rasmussen syndrome in which deterioration occurs 10 years after onset of epilepsia partialis continua have been described.
Case 4. Hemifacial clonic status epilepticus in rolandic epilepsy (52). A 30-year-old engineer had his first seizure at the age of 11. He had just fallen asleep when he was awakened by numbness in his mouth; his tongue was tightened, and he was unable to speak for one minute, after which he went back to sleep. The next year he had two brief, similar nocturnal seizures. Postictally and briefly in one of these episodes, he could not see his mother’s head and a glass in front of him. Treatment with phenobarbital 50 mg nocte was initiated. Six months later, he had a diurnal seizure after some partial sleep deprivation. While skating, he felt that the left side of his tongue was numb and that he could not see well. Within seconds, this was followed by repetitive and continuous left-sided clonic hemifacial spasms involving the mouth and eye that ended 40 minutes later with left hemiconvulsions. There was postictal Todd paralysis. In addition, he had frequent right-sided headaches. Brain scan was normal, but EEG showed right midtemporal spikes that appeared in only two EEGs, taken at the ages of 12 and 13. His first EEG, taken eight days postictally, had shown mainly right-sided slow waves. He was treated with carbamazepine for four years. No further seizures and no additional serious headaches occurred in the next 18 years. He does not suffer from migraines.
An interesting fact, not generally recognised, is that convulsive attacks are occasionally replaced by other manifestations, the most common of which is a sudden but transient paresis of that portion of the limb which in other attacks is involved in the clonic spasms. Usually the primary paresis appears in the part in which the spasms commence, but it may affect another region the cortical representation of which is adjacent to the site of discharge; in a case recorded by Hughlings Jackson, for instance, clonic movements of the face were immediately followed by paralysis of the hand of the same side. The following case is a good example of one in which at times defect or negative symptoms only occurred. CASE 1. The patient received a gunshot wound on his head in the region of the posterior end of the second left frontal convolution. Some months after being wounded he began to have epileptiform seizures of the Jacksonian type. All began with a clonic flexion of the right, thumb and fingers, particularly of the index and medius. Within a few seconds all the fingers were involved in clonic flexion spasms, which then spread to the wrist, and later to the elbow. Many attacks ended at this stage, but in others the arm was abducted and protracted at the shoulder, the clonic twitching extended to the right side of his face, and jerked his head and eyes to the right. Each attack was followed by weakness of the right fingers and wrist which according to his own story, varied with the severity and duration of the clonic spasms. No numbness, tingling, or other sensory disturbance accompanied the clonic movements. Sometimes, however, attacks occurred in which there were neither tonic nor clonic spasms, or movements of any kind; he simply lost power suddenly and without any warning in the hand. If reading, his book might fall unexpectedly, or his cane might drop from his hand as he walked. The loss of power was complete for a time, but after a few minutes voluntary movement returned and gradually reached the normal. |
Focal clonic seizures emanate from the contralateral precentral gyrus of the primary motor cortex. The face, hand, and body manifestations correspond to the homunculus of Penfield (59). However, there is great variability in the clinical and EEG manifestations of focal clonic seizures due to the complex and varied patterns of spread and propagation within the frontal lobe from and to extrafrontal areas and, particularly, the primary sensory cortex (65; 03; 07; 26; 27; 67; 16).
Seizures from the precentral area of the primary motor cortex include the following:
• Simple focal motor clonic or tonic-clonic seizures with or without Jacksonian march | |
• Myoclonic seizures that may be unilateral or bilateral and are predominantly distal or facial, eg, epilepsia partialis continua | |
• Tonic postural motor seizures associated with clonic movements that are unilateral or bilateral and asymmetric | |
• Focal negative motor responses (atonia and/or paresis) |
Etiology may be genetic, metabolic, structural, or unknown (see underlying disorders).
The pathophysiology is similar to that of other neocortical focal seizures (21; 68). The Jacksonian march is due to the slow ephaptic propagation of epileptic discharge along the motor cortex (17; 46).
A thorough investigation of the electrophysiology of focal clonic seizures in humans was published by Hamer and associates (26). In this study, coregistration of subdural EEG of the motor strip, recordings from subthalamic nucleus electrodes, and surface EMG were performed in an attempt to clarify the pathophysiology of focal clonic seizures in humans. The findings suggested that focal clonic seizures are indeed focal tonic-clonic seizures and originate from the motor strip. The epileptic clonus consisted of simultaneous contractions of agonistic and antagonistic muscles at regular intervals and was generated by localized polyspike-wave activity in the cortical primary motor area. Activation of the subthalamic nucleus was not essential in the generation of clonic seizures.
Focal clonic seizures happen in 30% of patients with rolandic epilepsy, which occurs in approximately one-fourth of all epileptic seizures in children between 5 and 14 years of age (57; 56). In a population of 8938 patients more than three years of age with epilepsy, somatosensory (1.4%) and Jacksonian clonic seizures (2.23%) were found to be the less frequent types of focal seizures (47). Of 154 cases of focal epilepsy with seizure onset in the first three years of life, 57 patients had hemiclonic seizures, and eight patients had clonic seizures (51). In a study of 252 adult patients with focal epilepsy, 352 seizure types were identified, and these comprised 14 clinical groups. Somatosensory seizures (including focal clonic seizures) occurred in 26 patients, and Jacksonian clonic seizures occurred in 12 patients. A focal somatosensory onset was reported in 26 seizure types (7.4%) in 26 patients, 12 of whom had a typical Jacksonian progression (44). According to one report, “generalized” onset clonic seizures were the most common ictal event in patients with Panayiotopoulos syndrome and convulsive status epilepticus, and one third required admission to intensive care units (72). However, it is doubtful that these are of generalized onset clonic seizures in this syndrome, which is a focal epilepsy (54).
Avoidance of head trauma and prevention of brain infections is important.
The typical focal clonic seizure with or without Jacksonian march is unlikely to impose any diagnostic difficulties.
Nonepileptic facial nerve hemifacial spasm and facial tics may sometimes cause problems in the differential diagnosis (01; 76; 43). This peripheral type of hemifacial spasm is defined as unilateral, involuntary, irregular clonic or tonic movement of muscles innervated by the seventh cranial nerve. It manifests with unilateral, painless, irregular, and continuous clonic twitching of the facial muscles. It mainly affects women, aged 50 to 60 years, without known antecedent causes other than Bell palsy in a few cases. The spasms usually begin in the orbicularis oculi and gradually spread to other facial muscles and the platysma of the face. Most frequently attributed to vascular loop compression at the root exit zone of the facial nerve, the spasm-related electromyogram activity is probably generated by ephaptic transmission due to local demyelination at the entry zone of the facial nerve root. However, there are many other etiologies of unilateral facial movements that must be considered in the differential diagnosis. In all of these cases, EEG is normal during the hemifacial spasms. Spread and variable synkinesis on blink reflex testing and high-frequency discharges on EMG with appropriate clinical findings are diagnostic. Stimulation of one branch of the facial nerve may spread and elicit a response in a muscle supplied by a different branch. Synkinesis is not present in essential blepharospasm, dystonia, or seizures. Needle EMG shows irregular, brief, high-frequency bursts (150 to 400 Hz) of motor unit potentials that correlate with clinically observed facial movements.
In one case, a 50-year-old woman had nearly continuous twitching in the left side of her face for two months (53). The diagnosis of hemifacial spasms was made by eminent neurologists. However, MRI and subsequent surgery revealed a large glioblastoma in the right side of the brain.
Limb-shaking transient ischemic attacks associated with severe stenosis of the internal carotid artery may also imitate focal clonic seizures (62). Limb shaking is precipitated by rising or exercise, and the arm is more frequently involved than the leg. The attacks usually last less than five minutes and are often accompanied by paresis of the involved limb.
Extremely sustained startle-induced clonus is a nonepileptic motor attack mimicking clonic seizures in children with encephalopathy (45). Clonus is a pathological motor pattern characterized by involuntary, rhythmic, and brisk muscular contractions in response to peripheral stimuli producing muscle stretching. It indicates pathological involvement of the corticospinal tract and can be considered as a functional spastic movement disorder of variable clinical presentation and duration. Severe and prolonged episodes of startle-induced clonic attacks associated with severe apnea may rarely occur in infants with severe encephalopathy. The clinical characteristics of such episodes are very similar to those of clonic epileptic seizures (45).
Focal clonic seizures may occur in the following epilepsies:
• Structural with lesions of any type localized in the primary motor cortex (tumors, malformations of cortical development, granulomas, injuries) (16) | |
• Probably genetic (rolandic epilepsy and Panayiotopoulos syndrome) (54) | |
• Unknown causes, but probably chronic encephalitis (Kozhevnikov-Rasmussen syndrome) and other etiologies of epilepsia partialis continua (see relevant article) | |
• Hemiconvulsion-hemiplegia-epilepsy syndrome | |
• Focal clonic seizures may also be induced by drugs such as mirtazapine. |
Diagnosis is based on a careful clinical history. Diagnostic procedures depend on suspected etiology. Brain imaging and particularly MRI is mandatory, except in straightforward cases of rolandic epilepsy. In developing countries where infections involving the nervous system are common, brain CT scan should be carried out in all children with focal motor seizures (58). SPECT and PET scans are needed for neurosurgical purposes only if the epileptogenic focus cannot be determined otherwise.
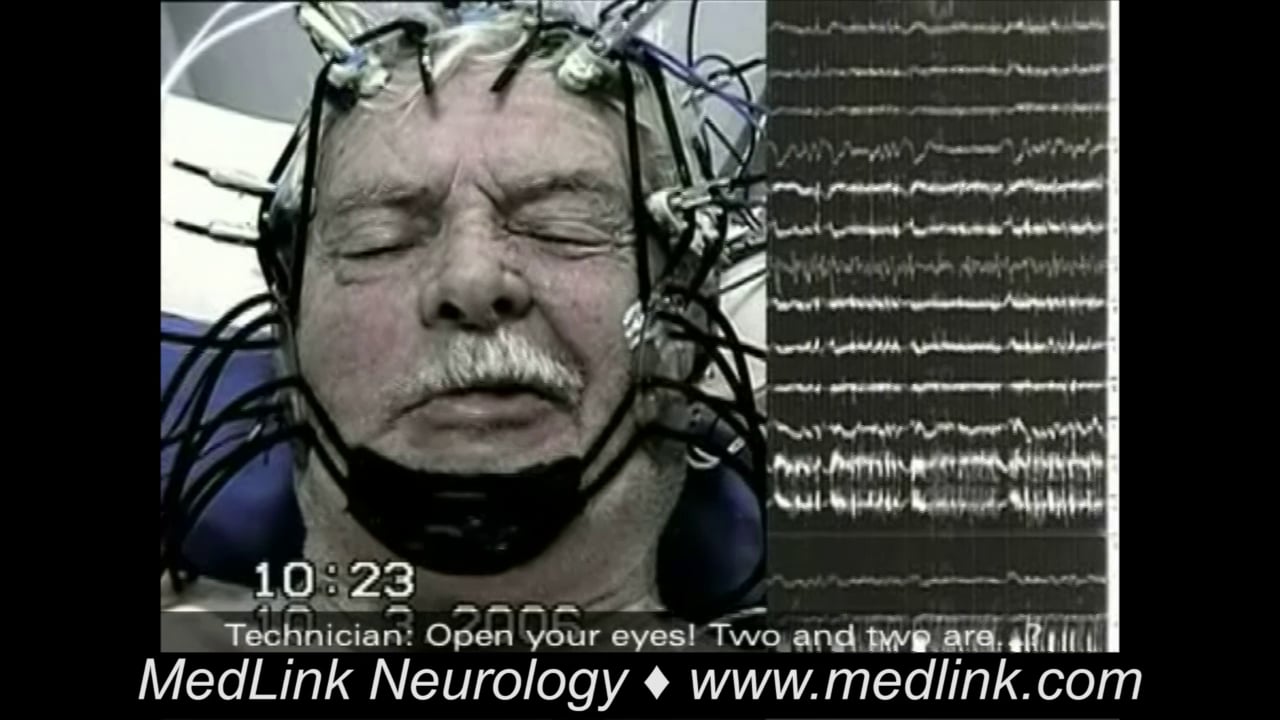
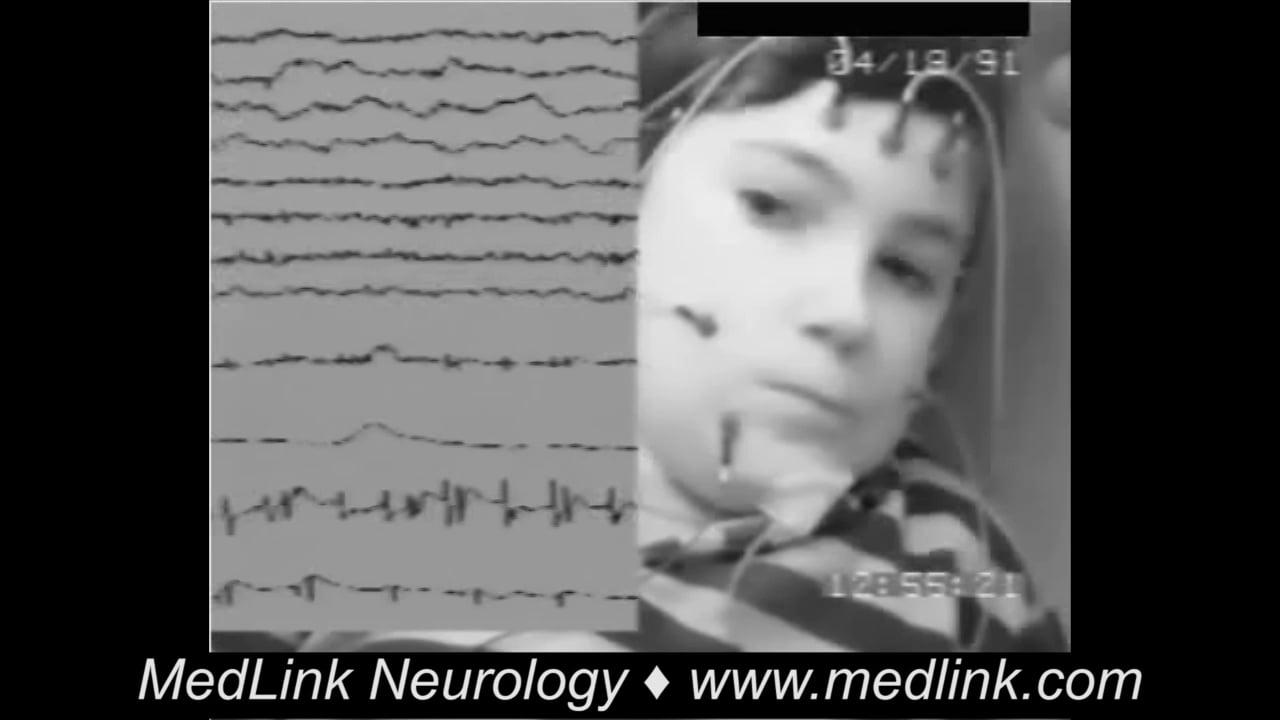
Interictal EEG may show no abnormality; background asymmetry; or focal spikes, sharp waves, or slow waves over the rolandic electrodes (11; 67; 35; 36). Typical are the centrotemporal spikes in rolandic epilepsy, which are accentuated in sleep EEG.
Ictal EEG is normal in 75% of focal clonic seizures, although clonic muscle artifact may be present (67; 35; 36). If abnormal, EEG shows ictal fast activity in the parasagittal electrodes. Ictal EEG of focal sensory-motor seizures in rolandic epilepsy show an initial paucity of spontaneous centrotemporal spikes before the onset of the ictal discharge, which appears in the rolandic regions contralateral to the clinical manifestations. The most frequent ictal EEG pattern is characterized by low-voltage activity of fast rhythmic spikes, increasing in amplitude and decreasing in frequency (09). Another ictal pattern consists of slow waves intermixed with spikes (56).
Ictal intracranial EEG shows fast activity that is initially entirely focal, but may then spread to adjacent electrode contacts. In one excellent report, focal clonic seizures were always associated with a polyspike-wave pattern in the EEG of the primary motor area (frequency range 1.6±3.4 Hz), whereas neighboring electrodes not overlying the precentral gyrus showed different EEG patterns (26). The seizures consisted of an initial tonic, with a gradual transition to clonic, phase over several seconds. The tonic phase was usually mild (often clinically inconspicuous, but detected with EMG) and of short duration (median 19.5 s; range 8 to 28 seconds). The focal clonic seizures had a median duration of 30.5 seconds (range 14 to 202 seconds) and consisted of bursts of compound muscle action potentials that occurred synchronously in agonistic and antagonistic muscles and were separated by periods of complete muscle relaxation.
At seizure onset, ictal EEG derived from the precentral gyrus subdural electrodes consisted of low-voltage fast activity followed by repetitive spiking for 8±28 seconds (median 19.5 seconds), accompanied by a continuous increase in muscle tone. During the tonic muscle contraction, EMG showed a complete interference pattern in which single compound muscle action potentials were not recognizable (26). This evolved to a pattern of polyspike-wave complexes that were associated with clinical clonus and lasted for 14±202 seconds (median 30.5 seconds). The waveform of each polyspike-wave complex consisted of two to six polyspikes recurring with a frequency of 12 to 45 Hz, followed by a negative slow wave. The duration and amplitude of the polyspikes and EEG slow waves varied within and between subjects. The frequency of the polyspike-wave complexes was in the range of 1.6 to 3.4 Hz (median 2.25 Hz) and slowed down towards the end of the focal clonic seizures. However, when the focal clonic seizure evolved to a generalized tonic-clonic seizure, the frequency tended to increase before the occurrence of the secondarily generalized seizure. The polyspike-wave complexes could also be recorded by scalp electrodes. Each series of compound muscle action potentials followed the polyspikes in the EEG with a latency of 17±50 ms. The periods of muscle relaxation occurred during the EEG slow waves.
The main antiepileptic drugs for monotherapy in focal clonic seizures, in alphabetical order, are carbamazepine, lamotrigine, levetiracetam, oxcarbazepine, phenytoin, and topiramate. Clobazam, brivaracetam, lacosamide, perampanel, pregabalin, and zonisamide may be used in polytherapy (54; 02; 49; 40).
Ketogenic diet is indicated in severe infantile epileptic encephalopathies with clonic seizures including migrating partial seizures in infancy (50).
There is a growing interest in the use of cannabidiol for the treatment of drug resistant epilepsies like Dravet and Lennox-Gastaut syndrome (13; 14; 39; 48; 66). In a double-blind, placebo-controlled trial in a 14-week treatment period of patients with Dravet syndrome, cannabidiol was significantly beneficial, particularly in convulsive-seizures, with 5% of patients becoming seizure free, though associated with higher rates of adverse events (13; 14). In June 2018, Epidiolex (cannabidiol oral formulation) from GW Pharmaceuticals obtained marketing authorization by the FDA for the treatment of Dravet and Lennox-Gastaut syndrome in the US market (see Dravat syndrome pipeline review 2018. See also package insert for epidiolex at Epidiolex (cannabidiol) oral solution.
Many children with rolandic epilepsy do not need prophylactic antiepileptic drug treatment.
See MedLink Neurology articles: Hemiclonic seizures; Dravet syndrome; Rasmussen syndrome; Benign childhood epilepsy with centrotemporal spikes.
Intramuscular botulinum toxin was found effective in three patients with refractory focal motor seizures (41). Neurosurgery may be useful in structural focal epilepsy. The face area can usually be safely resected, and the hand area may be amenable to subpial transection, but the foot area should be avoided (67; 38).
Lesion-focused stereotactic radio-frequency thermo-coagulation is a new approach for intractable focal clonic seizures in patients with epileptogenic lesions, such as of focal cortical dysplasia (74). Seizure outcome following primary motor cortex-sparing resective surgery for perirolandic focal cortical dysplasia is associated with an excellent early seizure-freedom rate and no permanent neurologic deficits (25).
Based on the effect of subthalamic nucleus stimulation on penicillin-induced focal motor seizures in primates, Prabhu and colleagues suggested that high frequency stimulation of subthalamic nucleus could be used as an experimental therapy when other therapeutic strategies are not possible or have failed in humans suffering from motor epilepsy (64).
Non-EEG devices are commercially available for long-term home monitoring of focal convulsive epileptic seizures (70). A real-time automated detection of clonic seizures in newborns has been described based on a low complexity image processing approach extracting the differential average luminance from videotaped body movements (63). This algorithm reliably detects neonatal clonic seizures and differentiates them from noise, random movements, or other seizure types.
Prognosis is variable depending on the underlying etiology and syndrome. It is benign in rolandic epilepsy, but very severe in Kozhevnikov-Rasmussen syndrome.
See MedLink Neurology article, Pregnancy and epilepsy.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Henry Hasson MD
Dr. Hasson of Albert Einstein College of Medicine has no relevant financial relationships to disclose.
See Profile
Solomon L Moshé MD
Dr. Moshé of Albert Einstein College of Medicine has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Epilepsy & Seizures
Jun. 02, 2026
Neuropharmacology & Neurotherapeutics
May. 11, 2026
Epilepsy & Seizures
May. 08, 2026
Epilepsy & Seizures
May. 01, 2026
Epilepsy & Seizures
Apr. 30, 2026
Epilepsy & Seizures
Apr. 17, 2026
Epilepsy & Seizures
Apr. 13, 2026
Epilepsy & Seizures
Mar. 24, 2026