Epilepsy & Seizures
Febrile seizures
Jun. 02, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Generalized onset tonic seizures are the predominant epileptic seizures in Lennox-Gastaut syndrome and also occur in neonates and infants with severe epilepsy, like Ohtahara syndrome. Clinically, they manifest with symmetrical sustained increase in muscle contraction, usually lasting a few seconds to 1 minute. Severity varies from inconspicuous to marked clinical manifestations with falls depending on the extent and groups of muscles involved and violence of the attack. Ictal autonomic alterations are prominent. Consciousness is usually lost or impaired (cloudiness). They predominantly occur in sleep. Patients with generalized onset tonic seizures also suffer from concurrent atypical absences, tonic-clonic, atonic, and other types of epileptic seizures according to the primary epileptic syndrome. Interictal and ictal EEG are usually abnormal, but their features depend on the age of the patient and the syndrome with which these occur. Management is usually difficult, and prognosis is that of the underlying disorder. In this article, the author details the clinical manifestations, pathophysiology, EEG, neuroimaging, and optimal management of patients with generalized onset tonic seizures and related epileptic syndromes.
• Generalized onset tonic seizures are epileptic seizures of mainly severe epilepsies of neonates, infants, and children with learning difficulties who also suffer from frequent seizures of other types. | |
• Lennox-Gastaut syndrome is the prototype disorder of generalized onset tonic seizures. | |
• Generalized onset tonic seizures manifest with abrupt onset and termination of sustained increase in muscle contraction, usually lasting a few seconds to 1 minute. | |
• Severity varies from inconspicuous to marked clinical manifestations with falls depending on the extent and group of muscles involved and violence of the attack. Ictal autonomic alterations are prominent. Consciousness is usually lost or impaired (cloudiness). The seizures predominantly occur in sleep. | |
• Interictal and ictal EEG are usually grossly abnormal. | |
• Prognosis (usually poor) and management (usually difficult) is that of the underlying disease or syndrome. |
ILAE classification and nomenclature. The ILAE Commission of 1981 classified tonic seizures as generalized seizures and cited the description of Gowers for their clinical manifestations:
…a rigid, violent muscular contraction, fixing the limbs in some strained position. There is usually deviation of the eyes and of the head toward one side, and this may amount to rotation involving the whole body, (sometimes actually causing the patient to turn around, even 2 or 3 times). The features are distorted; the color of the face, unchanged at first, rapidly becomes pale and then flushed and ultimately livid as the fixation of the chest by the spasms stops the movements of respiration. The eyes are open or closed; the conjunctiva is insensitive; the pupils dilate widely as cyanosis comes on. As the spasm continues, it commonly changes in its relative intensity in different parts, causing slight alterations in the position of the limbs (41; 18). |
According to this ILAE classification:
• Tonic axial seizures with extension of head, neck, and trunk may also occur. | |
• Ictal EEG consists of “low voltage, fast activity or a fast rhythm of 9 to 10 c/sec or more, decreasing in frequency and increasing in amplitude” | |
• The EEG interictal expression is of “more or less rhythmic discharges of sharp and slow waves, sometimes asymmetrical. Background is often abnormal for age” (18). |
Tonic seizures are defined as follows in the ILAE glossary: “a sustained increase in muscle contraction lasting a few seconds to minutes” (12).
The ILAE core group on classification classifies tonic seizures as generalized and explains, “The mechanism of tonic seizures is probably not the same as that of the tonic phase of generalized tonic-clonic seizures. Generalized tonic seizures typically occur in Lennox-Gastaut syndrome and occasionally in epilepsy with myoclonic astatic seizures” (30). The more recent proposals of the ILAE Commission maintain that tonic seizures should be classified amongst generalized epileptic seizures (08; 17).
The ILAE Commission diagnostic manual of the epilepsies describes generalized tonic seizures as follows (17):
A generalized tonic seizure involves bilaterally increased tone of the limbs typically lasting seconds to a minute. They often occur out of sleep and in runs of varying intensity of tonic stiffening. The individual is unaware during these events. At the beginning of tonic seizures with more intense stiffening, individuals may make an expiratory sound. More severe and prolonged tonic seizures may have a vibratory component which may be confused with clonic jerking. Tonic seizures often occur in individuals with intellectual impairment. Caution: Although asymmetry can occur in a generalized tonic seizure, if consistent focal features are seen from seizure to seizure, then consider focal seizure involving the frontal lobe. NOTE: Tonic seizures are one type of seizure that can result in a ‘drop attack’ (also known as astatic seizure), other causes of drop attacks include myoclonic (especially in younger children), atonic and myoclonic-atonic seizures. EEG Background/Interictal/Activation: Please refer to specific syndromes and etiologies in which this seizure type occurs. EEG Ictal: Tonic seizures show diffuse or generalized accelerating low amplitude paroxysmal fast activity, which is often bilateral and predominates in the anterior and vertex regions. Caution: Consistent focality of spikes or maximal amplitude of ictal rhythm, then consider focal seizure. Differential diagnosis: Epileptic spasms: the motor contraction is often shorter in duration (< 2 seconds), epileptic spasms often occur in a series; Focal seizure - supplementary sensorimotor cortex of frontal lobe; Syncope; Non epileptic seizures. Related syndromes: Lennox-Gastaut syndrome; Epilepsy with myoclonic-atonic seizures (17) |
The latest ILAE position paper of the operational classification of seizure types recognizes that tonic seizures can be of focal or generalized onset. The generalized tonic seizures are classified as motor generalized seizures together with tonic-clonic, clonic, myoclonic, myoclonic-tonic-clonic, myoclonic-atonic, atonic, and epileptic spasms (32; 33).
Historical aspects. Gowers described tonic seizures as cited above whereas tonic axial seizures were first described by Hughlings Jackson who called them "trunk fits" or "respiratory fits" (41; 45; 46) and considered them as "lowest level fits" proposing that a primary discharge of the pontobulbar centers induced discharges in the spinal motor centers and secondarily involved higher centers via ascending pathways. He distinguished tonic seizures from epileptic seizures arising in the cerebral hemisphere. However, these were also frequently referred to as "tetanoid attacks" or "tonic postural attacks" and confused with nonepileptic tonic attacks in subjects suffering from intracranial hypertension often related to a tumor in the posterior fossa, the "cerebellar fit" ("cerebellar attack," posterior fossa seizure or decerebrate seizure) (37). They often have also been grouped with other nonepileptic attacks, sometimes referred to as "extrapyramidal seizures" (which are usually localized, or at least asymmetrical, and are frequently related to known cerebral pathology), as well as with the syndrome of "paroxysmal choreoathetosis" (37).
Our current knowledge of tonic seizures is mainly based on the meticulous investigations by Henri Gastaut and his school in Marseilles who studied 30 cases in which tonic seizures were observed and recorded cinematographically, electroencephalographically, electromyographically, electrocardiographically, and pneumographically (38; 37). They found and pointed out:
• Tonic seizures occur almost uniquely in children in association with or precedence of infantile myoclonic encephalopathy with hypsarhythmia (one quarter of cases) and diffuse subcortical pathology. | |
• Clinically, they manifest as (a) tonic axial seizures, (b) tonic axo-rhizomelic seizures, (c) global tonic seizures, (d) asymmetrical and unilateral tonic seizures, and (e) tonic seizures terminating with a brief clonic phase. Autonomic manifestations are common and consist of extreme tachycardia, irregular tachypnea mixed with apneic periods, systolic hypertension, a positive psychogalvanic reflex, mydriasis, vasomotor phenomena, increased intravesicular pressure, glandular hypersecretion, and occasional piloerection. There is loss of consciousness and occasional postictal confusion. Generally, reflexes are diminished during and increased after the seizure. Their duration is between several seconds to 1 minute (usually 10 to 20 sec). Nocturnal predominance regularly occurs. | |
• Status epilepticus is to be especially feared as with passing seizures the striking motor manifestations diminish whereas impairment of consciousness and autonomic phenomena (especially bronchial secretions) are enhanced. | |
• An association with other seizure types is common, especially the spasms of hypsarhythmia or tonic-clonic epilepsy. | |
• Ictal EEG always shows bilateral, symmetrical, and synchronous discharges. Rapid desynchronization with or without subsequent rapid synchronization, pure hypersynchronization at 10 Hz (the epileptic recruiting rhythm), termination by added slow wave activity appearing as spike-waves, or other patterns may be seen. In one patient similar seizures may be associated with different EEG elements, or similar tracings with different intensities of seizures. | |
• On EMG there is a tendency for brief seizures to start acutely and long seizures to start progressively. | |
• It is important to differentiate them from other tonic seizures of epileptic and nonepileptic origin, with particular emphasis on generalized tonic-clonic seizures, the infantile spasms with hypsarhythmia, partial adversive seizures, other epileptic seizures, convulsive syncopes, cerebellar fits, tetanus, rabies, strychnine poisoning, and hysteria. | |
• Possible pathophysiological mechanisms: a type of generalized seizures outside of the classical dichotomy of generalized tonic clonic and absence seizures. | |
• Intractability to treatments and the significance of urgent management of status epilepticus. |
Tonic seizures have been mainly studied in Lennox-Gastaut syndrome (38; 16) and neonatal epileptic encephalopathies such as Ohtahara syndrome (24; 63).
Clinically generalized onset tonic seizures manifest as follows:
• Abrupt start and end of symmetrical and sustained tonic contraction of the muscles, which is usually brief for approximately 10 seconds. | |
• Severity varies from inconspicuous to marked clinical manifestations, depending on the extent and group of muscles involved and violence of the attack. | |
• Autonomic alterations are prominent. | |
• Consciousness is usually lost or impaired (cloudiness). |
The seizures occur mainly during sleep where they are abundant. They occur together with other types of seizure such as clonic, tonic-clonic, atonic, absence, or focal seizures.
Generalized onset tonic seizures have been best described in Lennox-Gastaut syndrome where they are the most common (probably in 100% of patients) and most characteristic type of seizures (38; 37).
Generalized onset tonic seizures consist of sustained tonic contraction of the muscles involved. They are commonly brief, lasting for usually 10 seconds. They may be briefer (1 to 5 seconds) or longer (up to 1 minute). Longer tonic seizures may progress to a vibratory stage of the whole body with extremely rapid and small amplitude clonic convulsions (clonias).
The extent of motor involvement in tonic seizures varies. Mild tonic seizures may be limited to a minimal upward deviation (sursum vergems) or opening of the eyes with a brief respiratory disturbance. Severity varies from inconspicuous to marked clinical manifestations depending on the extent and groups of muscles involved and violence of the attack. Their frequency in each patient is probably grossly underestimated because tonic seizures may be brief, more frequent during sleep, and their clinical manifestations are often difficult to recognize without video-EEG.
Tonic seizures are usually symmetrical, but even predominantly unilateral tonic seizures may infrequently happen. Series of tonic seizures, reminiscent of infantile spasms but of longer duration, may occur especially when Lennox-Gastaut develops from West syndrome. Depending on the extent and groups of muscles involved, they may appear as axial, axo-rhizomelic, and global (38; 37).
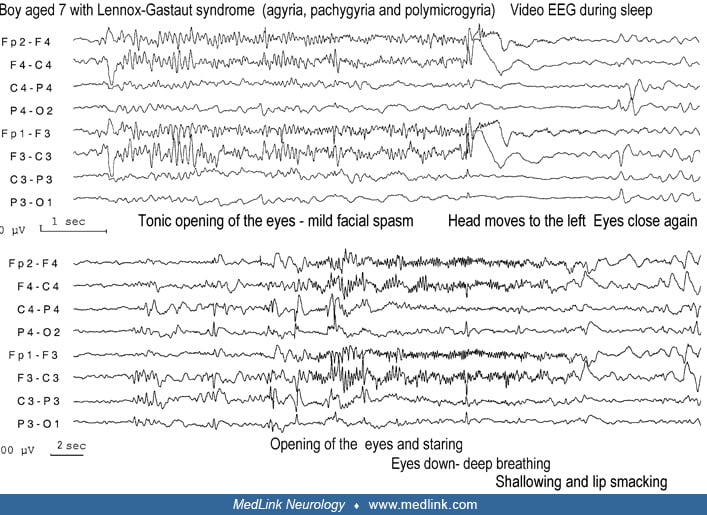
Axial tonic seizures. Axial tonic seizures affect facial, nuchal, trunk, paraspinal, respiratory, and abdominal muscles alone or in combination. Tonic neck spasms are usually flexor, raising the head from the pillow. Facial tonic spasms may cause sudden elevation of the eyebrows, opening of the eyes, upward deviation of the eyeballs, opening of the mouth, or stretching of the lips to a fixed smile.
There is generally eyelid retraction, staring, mydriasis, and, frequently, upward deviation of gaze. Respiratory disturbance and apnea are common associated symptoms. Breathing may stop or may become shallow or deep, fast or slow. An “epileptic cry” is common at onset of the attacks. This brief yell is probably due to the contraction of the diaphragm, forcing air out through the closed glottis. Minimal tonic seizures restricted to eye opening, with a brief change in respiration, are common during sleep and are often mistaken as normal movements.
Axo-rhizomelic tonic seizures. Axo-rhizomelic tonic seizures are axial seizures also involving proximal (rhizomelic) muscles of upper and, less often, lower limbs. Elevation, abduction, or adduction of upper limbs and shoulders occurs together with the other symptoms of axial tonic seizures. Elevation and abduction of the legs occurs less often.
Global tonic seizures. Global tonic seizures also involve the distal part of the limbs. The arms are forced upwards, abducted, and semiflexed with clenched fists "like that of a child defending himself from a facial blow.” The lower limbs are forced in triple flexion at hip, knee, and ankle or in extension. Global tonic seizures often cause forceful sudden falls and injuries.
Other associated ictal symptoms in generalized onset tonic seizures include impairment of consciousness and autonomic disturbances.
Consciousness. Consciousness is generally severely impaired though occasionally awareness of the event may exist. The patient may be aroused and fall asleep again.
Tonic seizures are always accompanied by loss or at least some clouding of consciousness. Return to consciousness after the seizure is typically very rapid. However, in some cases the tonic seizure is followed by a state of confusion lasting from several seconds to a few minutes during which simple automatisms, particularly movements of gesticulation (for example tapping), may be observed. On the other hand, after exceedingly long and intense seizures causing the patient to fall, a brief period of coma associated with important vegetative phenomena may persist” (38). |
Autonomic manifestations. Autonomic manifestations are common and sometimes severe. All types of tonic seizures are associated with autonomic phenomena such as tachycardia, apnea, flushing of the face, cyanosis, salivation, mydriasis, incontinence of urine, and lacrimation. Autonomic phenomena may occasionally be the prominent symptom of the attacks.
These are probably as characteristic of tonic seizures as the convulsive manifestations. However, as they are less evident, they are more often neglected. The respiratory manifestations are never missed if one takes the precaution of recording them with a pneumogram. One sees important respiratory embarrassment even where a marked polypnea, an apnea or a cry would not have gained the attention of the observer. The most common findings are an increase of frequency and a diminution of amplitude of the respiratory movements during and occasionally after the motor components. Cardiovascular phenomena are very frequent. A tachycardia almost always occurs during which the heart rate may double. We have been able to record the arterial pressure only in one case but then during several seizures. Each was accompanied by a considerable increase in systolic pressure. A positive psychogalvanic reflex is always noted starting immediately after the first EEG change. Mydriasis is an equally frequent phenomenon. It progresses continuously throughout the contraction to reach its maximum towards the end of the seizure. It is often accompanied by a slow upward deviation of the ocular globes occasionally associated with convergence. It is not rare for the observer to see at the end of the seizure several nystagmoid movements or even a hippus. Vasomotor phenomena are equally frequent. The face reddens during the seizure. Occasionally the lips become cyanotic. From time to time one may even observe salivary, lacrymal and sudoral hypersecretion with or without pilo-erection. The intravesicular pressure has been registered only in one patient but during several seizures. It showed each time a considerable increase. This explains, no doubt, the emission of urine sometimes seen during tonic seizures” (38). |
Automatisms, gestural or ambulatory, may follow the tonic stage in 7%, particularly of mainly late onset Lennox-Gastaut syndrome.
Circadian distribution and precipitation of tonic seizures. Tonic seizures occur far more frequently during sleep than wake states; some patients may have hundreds of them during sleep. They do not occur during REM sleep. In my experience with all-night video-EEG monitoring of patients with Lennox-Gastaut syndrome, it was not unusual to record over 70 to 100 tonic seizures per night whereas only a few of them (5 to 10) were reported by the nursing staff in charge of the patient.
Prognosis of conditions associated with generalized onset tonic seizures is usually poor (see relevant epileptic syndromes in which these occur). In only one report of 10 children with early onset of generalized seizures who had normal development and normal EEG background, the response to treatment was good and freedom from seizures was achieved (53; 54).
Ohtahara syndrome. A boy, born after normal pregnancy and delivery, started having frequent tonic seizures at 4 days of age. Seizures consisted of a forward tonic flexion of his body and limbs lasting 1 to 10 seconds. They occurred while he was awake and when he was asleep, singular or clusters, 100 times every 24 hours. An EEG showed a suppression-burst pattern that continued while asleep and awake. Metabolic and other relevant tests were normal, but MRI showed extensive microgyria in the right hemisphere. There was no benefit from appropriate medication. Severe psychomotor delay soon became evident with marked quadriplegia. At 4 months, the EEG became hypsarrhythmic, and the child had typical salaam spasms. There was some temporary seizure improvement with treatment with ACTH and vigabatrin. The child died 2 months later.
Generalized onset tonic seizures are most common in structural or metabolic epilepsies of neonates, infants, and children, mainly with neurocognitive impairments, irrespective of underlying cause (structural or metabolic). Therefore, their etiology is age-related. It should be noted that having a generalized onset tonic seizure excludes a diagnosis of idiopathic generalized epilepsy (42).
Generalized onset seizures are conceptualized as originating at some point within, and rapidly engaging, bilaterally distributed networks. Such bilateral networks can include cortical and subcortical structures, but do not necessarily include the entire cortex. Although individual seizure onsets can appear localized, the location and lateralization are not consistent from one seizure to another. Generalized seizures can be asymmetric (08). |
For generalized onset tonic seizures, the consensus is that there is a significant brainstem involvement (13; 78; 24; 36; 31; 69). Because the brainstem also projects widely to the cortex, a central role for the brainstem would also explain how generalized onset tonic seizures occur even in patients with focal pathology.
According to one hypothesis, the presence versus absence of tonic seizures in neonatal encephalopathies indicates the severity of the brainstem pathology or dysfunction at the time of presentation of the syndrome. It is hypothesized that brainstem alterations are already present at the onset of Ohtahara syndrome whereas they appear with time in early myoclonic encephalopathy, either because they are initially less severe than those observed in Ohtahara syndrome or because they emerge as a result of a kindling process or a release of the brainstem from cortical control as the disease progresses (24). Secondary bilateral synchrony from cortical, mainly frontal, foci has been implicated in “generalized” epileptic seizures in epileptic encephalopathies (39; 10; 11; 50; 65). Secondary bilateral synchrony refers to bilateral and synchronous EEG discharges generated by a unilateral cortical focus. Secondary bilateral synchrony has been considered the main pathophysiological mechanism in a third of cases of typical Lennox-Gastaut syndrome (61).
The ILAE commission considered that “the mechanism of tonic seizures is probably not the same as that of the tonic phase of generalized onset tonic-clonic seizures” (30). In favor of this stance is the fact that tonic seizures have a different pattern of EMG activation in relation to the tonic phase of generalized tonic-clonic seizures (19). There is a shift toward higher frequencies during the tonic seizures, which may be due to excessive activation of the extrapyramidal system. Conversely, the increase in amplitude characteristics of the EMG signal during the tonic phase of the tonic-clonic seizures could be attributed to an excessive activation of the corticospinal pathways, as opposed to the possible extrapyramidal dominance during the tonic seizures (19).
Intusoma and associates sought to reveal the networks involved in generalized onset tonic seizures by comparing ictal and interictal single-photon emission computed tomography (SPECT) of seven patients with Lennox-Gastaut syndrome (median age 11 years; range 1 to 38) (43). They performed a voxel-wise comparison of ictal and interictal SPECT studies across the group. The evolution of blood flow changes was explored by examining early and late injection groups. They found that the median duration of tonic seizures was 10 s (range 6 to 29 s), and injection latency from seizure offset was -8 to 48 s. In the early injection group (< 10 s; three studies), there was hyperperfusion over pons and cerebellar hemispheres (p < 0.05 cluster corrected family wise error), and hypoperfusion bilaterally over the pericentral region, with a trend toward hyperperfusion over bilateral superior and middle frontal gyri, and lateral parietal cortex. In the late injection group, there was hyperperfusion over midline and lateral cerebellar regions, with hypoperfusion widely over bilateral frontal regions. The authors concluded that these findings suggest that the tonic seizures of Lennox-Gastaut syndrome result from activity in a network, containing bilateral frontal and parietal association areas and the pons. They also postulated that tonic seizures recruit the corticoreticular system, which connects frontal attentional areas to the pontine reticular formation and is normally responsible for postural tone and orienting behavior (43). The same group of authors conceptualized Lennox-Gastaut syndrome as a “secondary network epilepsy,” where the epileptic activity is expressed through large-scale brain networks, particularly the attention and default-mode networks. Cortical lesions, when present, appear to chronically interact with these networks to produce network instability rather than triggering each individual epileptic discharge. The epileptic manifestations of the disorder reflect the networks being driven, rather than the specific initiating process. (01).
Generalized onset tonic seizures are common in Lennox-Gastaut syndrome and also occur in neonates and infants with severe epilepsy, like Ohtahara syndrome.
There is no major differential diagnosis of generalized onset tonic seizures when these happen together with other epileptic seizures as part of the manifestations of Lennox-Gastaut or Ohtahara syndrome. The primary problem in diagnosis is their differential diagnosis from other types of seizures and mainly epileptic spasms and focal onset tonic seizures. Also, it is possible to not recognize them because their manifestations may be inconspicuous and mainly occur in sleep. Video-EEG is often needed in order to identify generalized onset tonic seizures.
Epileptic spasms. Epileptic spasms (a term that includes infantile spasms) are sudden and brief bilateral tonic contractions in flexion or extension of the axial and proximal limb muscles with abrupt onset and termination. They usually last for approximately 1 second (range 0.2 to 2 seconds) and, thus, they are of longer duration than myoclonic jerks (shorter than 0.1 second) but of shorter duration than tonic seizures (usually 2 to 10 seconds); their characteristic muscular contraction reaches a peak more slowly than a myoclonic jerk but more rapidly than a tonic seizure. Epileptic spasms mainly occur in neonates and infants with severe epileptic encephalopathies such as West syndrome, but they may also continue past or even occur de novo after infancy (15; 40). The updated ILAE report classifies them as seizures of unknown origin because of inadequate knowledge to make a firm decision of whether they are focal, generalized, or both (08). They are distinguished from generalized onset tonic seizures because of their brief duration and the associated interictal and ictal EEG changes (17). However, “epileptic spasms” are often reported as lasting longer than 2 seconds as, for example, in Ohtahara syndrome where their duration may be 10 seconds. There is the so-called “epileptic spasm-tonic seizure” sequence whereby an epileptic spasm progresses in continuity to a tonic seizure (21; 35). Recordings of this sequence reveal that during the spasm the EEG shows a multiphasic slow-wave transient pattern with some sharp components, and EMG shows a diamond-shaped pattern. During the tonic phase of the seizure, the EEG shows an attenuation with superimposed fast activity and the EMG a sustained increase in the muscle activity (21).
Focal onset tonic seizures. Focal tonic seizures are seizures with tonic posturing of the extremities in extension or flexion and are classified as of neocortical origin (30). They mainly originate from the supplementary motor area (60% or more) though they also occur in patients with parietal, temporal, and occipital focal epilepsy (84; 47; 14). Tonic posturing can be unilateral or bilateral and is predominantly asymmetrical. The onset of the seizures is often explosive; upper and lower extremities are involved, and often there is associated head version and eye deviation. The patient may assume a typical fencing posture with head version to an asymmetrical extended arm. Focal tonic seizures of frontal lobe origin may start with a somatosensory aura; visual and epigastric auras are commonly experienced in those of extrafrontal origin. Consciousness is frequently intact. Seizures are usually brief (lasting less than 1 minute), frequent, and mainly nocturnal. The interictal EEG is usually normal. The ictal EEG may be normal or show focal discharges.
Seizures with tonic posturing originating from the supplementary sensorimotor area (SSMA) show some differences from those of extra-SSMA origin. The SSMA patients have a higher incidence of preservation of consciousness and there is a propensity for having unilateral or bilateral asymmetrical tonic limb posturing. In contrast, extra-SSMA patients have a statistically significantly higher incidence of bilateral symmetrical tonic limb posturing (71; 14).
Faciobrachial dystonic seizures are focal onset tonic stereotyped contractions of face and arm, occurring up to 200 times in a subgroup of patients with leucine-rich glioma inactivated 1 (LGI1) autoantibodies. Their recognition is important because they usually precede other symptoms of typical limbic encephalitis, which is responsive to immunotherapy, including corticosteroids, but not anticonvulsant medication (72; 44; 77).
Considering their clinical and EEG characteristics, focal tonic seizures should not be difficult to differentiate from generalized onset tonic seizures, bearing in mind that they usually occur in neurocognitively normal patients who do not have other types of seizure (myoclonic, atonic, or absence) that are so common in patients with generalized onset tonic seizures. However, the problem may be when the latter are from focal epileptogenic foci with secondary bilateral synchrony.
Nonepileptic tonic attacks in infants and children. There are numerous nonepileptic tonic attacks in infants and children that should be differentiated from generalized onset tonic epileptic seizures. These include gastroesophageal reflux and Sandifer syndrome, self-stimulatory behavior, hyperekplexia, cataplectic attacks, benign paroxysmal tonic up-gaze deviation, benign nonepileptic myoclonus of early infancy (Fejerman syndrome), and nonepileptic tonic reflex seizure of early infancy (81; 80).
Also, in children with other medical illnesses, a different list of causes is usually considered (60). Sudden drops in blood pressure, reduction of cardiac output, or increases in intracranial pressure can all cause diffuse tonic postures. Distress or pain of any sort can cause diffuse tonic posturing in the immature patient, and a careful evaluation to uncover the source in all circumstances is needed.
The diagnosis in all these cases should not be difficult with a complete clinical history based on accurate description of the events, provocative maneuvers, circadian distribution, associated symptoms and signs, other concurrent paroxysmal attacks, and neurocognitive state of patients.
Lennox-Gastaut syndrome. Lennox-Gastaut syndrome is the prototypic disorder of generalized onset tonic seizures that occur in all patients (see historical aspects and clinical description).
Generalized onset tonic seizures also occur in:
• Neonates with severe epilepsy and mainly with hypoxic-ischemic encephalopathy (02), Ohtahara syndrome, and, less often, early myoclonic epilepsy (63; 64). | |
• Dravet syndrome (59) | |
• Patients with malformations of cortical development (04), chromosomal abnormalities such as ring chromosome 14 syndrome (85), and 1p36 deletion syndrome (05) or mutations in the X-linked CDKL5 gene (06) | |
• Epilepsy with myoclonic atonic seizures (though there are conflicting reports) (74). | |
• Otherwise normal children (53; 54) (though this may be exceptional). | |
• Startle epilepsy (36). |
Neonates. Generalized onset tonic seizures are reported as 5% of all seizures in neonates (83), and they are a predominant type of neonatal seizures of the first week of life (29%), although most of them (71%) occur concomitantly with other types of seizure (02).
Ohtahara syndrome and early myoclonic epilepsy. Generalized onset tonic seizures are frequently reported in Ohtahara syndrome and less often in early myoclonic encephalopathy (3 to 4 months after the onset of the myoclonic seizures) (09; 24; 36; 67; 07). However, most of them may be tonic epileptic spasms or tonic seizures occurring in continuity with a preceding epileptic spasm (so-called epileptic spasm-tonic seizure sequence; see differential diagnosis section). Genuine generalized onset tonic seizures occur at later stages when Lennox-Gastaut syndrome is developed.
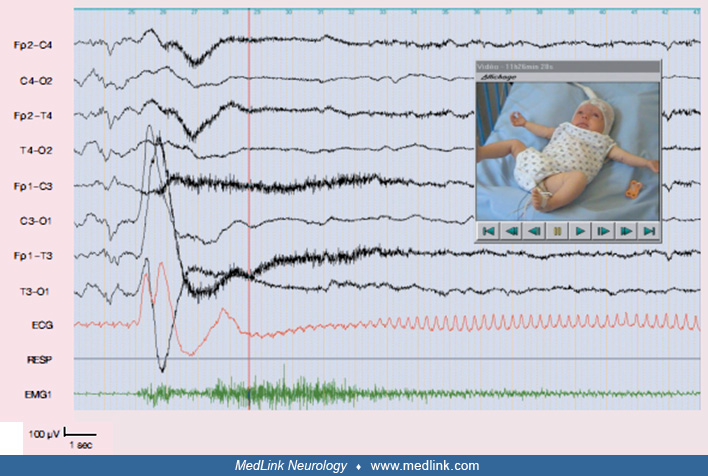
In Ohtahara syndrome, onset of seizures is mainly around the first 10 days of life, sometimes within the uterus or up to 3 months after birth (62; 63; 64). The syndrome manifests with clinico-EEG features of mainly tonic epileptic spasms and suppression-burst EEG that remains unchanged during both wakefulness and sleep. Tonic epileptic spasms usually cause a forward flexion of the body and head, last for 1 to 10 seconds (often longer than the classical epileptic spasms of West syndrome), and may be singular or occur in clusters. Each cluster consists of 10 to 40 spasms with interictal periods of 9 to 15 seconds. Seizures are frequent, sometimes hundreds within 24 hours, both while awake and asleep. These seizures may be bilateral or show an alternating or consistent side emphasis. Approximately one third of patients with Ohtahara syndrome will also develop other seizures. In addition to tonic epileptic spasms, partial motor seizures, erratic focal motor seizures, hemiconvulsions, and generalized onset tonic or tonic-clonic seizures occur in one third of the cases (63). Ohtahara syndrome results mostly from structural malformations.
Early myoclonic encephalopathy usually starts in the first days of life, sometimes immediately after birth. It is associated mostly with metabolic abnormalities. Erratic myoclonus appears first, followed by simple focal seizures and later (2 to 4 months from onset) by tonic epileptic spasms. They manifest with truncal tonic contraction, which usually also involves the limbs. They occur during wakefulness and sleep.
Early onset epileptic encephalopathies caused by KCNQ2 mutation. KCNQ2 mutations have been found in patients with benign familial neonatal seizures, myokymia, or early onset epileptic encephalopathy. Kato and associates delineated the clinical spectrum of early onset epileptic encephalopathies associated with KCNQ2 mutation (49). A total of 239 patients with early onset epileptic encephalopathies, including 51 cases with Ohtahara syndrome and 104 cases with West syndrome, were analyzed by high-resolution melting (HRM) analysis or whole-exome sequencing. Detailed clinical information including EEG and brain MRI were collected from patients with KCNQ2 mutation. A total of nine de novo and one inherited mutations were identified (two mutations occurred recurrently). The initial seizures, which were mainly tonic seizures, occurred in the early neonatal period in all 12 patients. A suppression-burst pattern on EEG was found in most. Only three patients showed hypsarrhythmia on EEG; eight patients became seizure free when treated with carbamazepine, zonisamide, phenytoin, topiramate, or valproic acid. Although the seizures were relatively well controlled, moderate-to-profound intellectual disability was found in all except one patient who died at 3 months. The authors concluded that de novo KCNQ2 mutations are involved in early onset epileptic encephalopathies, and most of the cases are diagnosed as Ohtahara syndrome (49). These cases showed distinct features with early neonatal onset, tonic seizures, a suppression-burst EEG pattern, infrequent evolution to West syndrome, and good response to sodium channel blockers, but poor developmental prognosis. Genetic testing for KCNQ2 should be considered for patients with early onset epileptic encephalopathies.
Mutations in the X-linked cyclin-dependent kinase-like 5 (CDKL5) gene. Mutations in the X-linked cyclin-dependent kinase-like 5 (CDKL5) gene are responsible for a severe encephalopathy with early epilepsy (06). The epilepsy course reveals three successive stages: (stage I) early epilepsy (onset 1 to 10 weeks of age) with normal interictal EEG despite frequent convulsive seizures; (stage II) epileptic encephalopathy with infantile spasms and hypsarrhythmia. At the age of evaluation, patients may become seizure-free or develop refractory epilepsy; (stage III) with tonic seizures and myoclonia. Patients carrying CDKL5 mutations causing a truncation of the catalytic domain tend to develop refractory epilepsy more frequently than patients with mutations located downstream (06).
Dravet syndrome. In Dravet syndrome, tonic seizures were not initially reported (26), and they are considered an unusual finding in later reports (nine of 105 patients) (27; 28). However, tonic seizures were found in 12 of 82 patients with Dravet syndrome in one study (59). They were frequently seen in adolescent patients with a particular EEG pattern (three of five patients) combining frontal slow bi-tri spikes followed or not by slow waves when awake and activated by sleep with 5- to 10-second discharges of 8 to 9 Hz spikes (59). According to the authors, this EEG pattern should not be considered as a change into Lennox-Gastaut syndrome but as a previously overlooked unusual pattern in the adolescent course of Dravet syndrome (59).
Developmental and epileptic encephalopathy due to SCN8A gene variants. This disorder is characterized by drug-resistant early-onset epilepsy associated with severe intellectual disability. Different seizure types have been reported as well as a sequence of autonomic manifestations such as brady-/tachycardia, irregular breathing, and cyanosis. In a video-EEG report, generalized tonic seizures were identified as the major seizure type at epilepsy onset (73). Seizure severity could vary from subtle to marked clinical manifestations, depending on the extent and groups of muscles involved and association with autonomic modifications. Autonomic signs were found in 80% of cases, often with a stereotyped sequence of ictal events for most of the seizures. Autonomic signs occurred in rapid sequence: flushing of the face, sometimes associated with sialorrhea, bradycardia, and hypopnea appeared within the first 1 to 2 seconds. Tachycardia, polypnea, perioral cyanosis, and pallor occurred later in the course of the seizure. Generalized tonic seizures are rarely described in other genetic epileptic conditions of early infancy because of ion channel mutations, such as in developmental and epileptic encephalopathy due to KCNQ2 or SCN2A gene mutations whereby seizures are most frequently reported as focal to bilateral tonic. The authors concluded that generalized symmetric tonic seizures with autonomic signs can be considered a clinical hallmark for diagnosis of SCN8A-related developmental and epileptic encephalopathy and relevant for therapeutic implications (73).
Epilepsy with myoclonic-atonic seizures. Epilepsy with myoclonic-atonic seizures (also known as Doose syndrome) is a genetic generalized epilepsy that begins between 7 months and 6 years of age (peak 2 to 4 years) in previously normal children. It manifests with frequent and multiple types of seizure though myoclonic atonic seizures with falls are the defining symptoms. It starts with frequent and usually lengthy febrile and afebrile generalized tonic-clonic seizures. Myoclonic, atonic, myoclonic-atonic, and absence seizures follow usually after a few weeks from onset. Seizures are frequent and may cause traumatic falls. Nonconvulsive status epilepticus, sometimes lasting for many hours or days, is common. Tonic seizures were not considered as part of the syndrome, “no tonic seizures or tonic drop attacks during daytime (except for some rare cases with a most unfavorable course)” (25), but these are now well described in more recent reports (36; 74; 17).
Generalized onset tonic seizures in otherwise normal children. In a retrospective analysis of patients with seizure onset prior to age 16 years, 25 patients with generalized onset tonic (n = 10) or tonic-clonic (n = 15) seizures were identified (54). These patients constituted 5.7% of patients with epilepsy aged 1 month to 16 years. Both seizure types were characterized by early onset of generalized seizures that appear in normally developed children with a normal electroencephalographic background. The children usually responded quickly to antiepileptic drugs, and 95% of them were seizure-free at the end of a long follow-up period. There were no significant differences between the two groups in regard to age of onset, family history, and seizures at follow-up. The authors concluded that the natural history of patients with generalized onset tonic seizures is similar to the benign course of those with generalized onset tonic-clonic seizures (54).
Startle epilepsy. Startle epilepsy is characterized by seizures triggered by unexpected sudden sensory stimuli, usually sound or touch. Most patients have intractable seizures, evident static encephalopathy, and neurologic deficits (infantile hemiplegia is common) with corresponding abnormal imaging. The startle response consists of axial, usually asymmetric, tonic posturing, frequently causing falls, which can often be traumatic. Concurrent symptoms such as marked autonomic manifestations, automatisms, laughter, and jerks may occur. Less commonly, startle-induced seizures may be atonic or myoclonic, particularly in patients with cerebral anoxia. Seizures are frequent, occurring many times a day, and sometimes progress to status epilepticus. Tonic seizures in startle epilepsy may not be of generalized onset; they are usually attributed to epileptogenesis from the primary motor cortex.
The diagnostic workup is relevant to the epileptic syndrome with which the generalized onset tonic seizures occur (see Lennox-Gastaut and Ohtahara syndrome). A thorough clinical neurodevelopmental assessment as well as ophthalmological and ultraviolet skin examination may reveal the underlying cause. The cause may already be known in those who develop Lennox-Gastaut from Ohtahara and West syndrome. Biochemical, hematological, metabolic, and other relevant screenings are rarely abnormal depending on the cause. Brain imaging with high-resolution MRI is abnormal in nearly all patients with the exception of epilepsy with myoclonic atonic seizures.
Interictal and ictal EEG are usually abnormal, but their features depend on the age of the patient and the syndrome with which these seizures occur (51; 52).
The following description mainly refers to patients with Lennox-Gastaut syndrome.
Interictal EEG. The interictal EEG is nearly always abnormal and reflects the underlying pathology as well as the specific epileptic encephalopathy of the patient. The background activity is usually slow for the average age of the subject; diffuse and asymmetric slow activity predominates in the posterior regions and does not respond to opening and closing of the eyes. High amplitude delta and theta waves are superimposed on this background, and this is usually diffuse with some focal predominance. In addition, there may be frequent spikes in various locations and, more frequently, episodic epileptiform discharges of slow spike-wave, polyspikes, and fast rhythms. The latter are usually of brief duration and asymmetrical with no apparent clinical manifestations. However, in addition to these asymptomatic discharges, there are also ictal discharges associated with a variety of seizures (tonic, atonic, myoclonic, and atypical absences).
Ictal EEG. The ictal EEG shows accelerating fast paroxysmal activity, which is bilateral and often predominates in the anterior regions and the vertex (38; 37).
This may be of two types:
(1) Very rapid (20 ± 5 Hz) and initially of low amplitude, progressively increasing to 50 to 100 µV and
(2) A more ample and less rapid rhythmic discharge at 10 Hz, identical to that of the tonic phase of the generalized tonic-clonic seizures (epileptic recruiting rhythm), except that it may be of high amplitude from the onset.
Flattening of all EEG activity alone or in combination with fast paroxysms is also common. Fast ictal paroxysms may be preceded by slow generalized spike-wave discharges or EEG suppression.
Sleep significantly increases any type of paroxysmal discharges and tonic seizures.
Ictal EMG activation. Quantitative analysis of the surface EMG during generalized onset tonic seizures is characterized by a significant increase in the frequency of the EMG activity. This is unlike the tonic phase of generalized tonic-clonic seizures, which is characterized by increased amplitude of the EMG signal (19). In other words, tonic seizures are produced by a significant shift toward the higher frequency bands, whereas the tonic phase of tonic-clonic seizures is produced by an increase in the amplitude characteristic. These differences between the tonic seizures and tonic phase of the tonic-clonic seizures are not merely a function of time, as there is no significant difference in duration between the two seizure types, and the quantitative EMG parameters that are differentiated between them do not show a correlation with the duration of the tonic muscle activation.
Seizures in syndromes with generalized onset tonic seizures are usually intractable. Antiepileptic drugs may reduce them but do not control them, and it is doubtful if they affect the outcome. Rational polytherapy is often required though this is rarely successful. Avoiding drugs that may cause drowsiness or worsen seizures, cognition, and behavior is an important aspect of management.
Lennox-Gastaut syndrome, Ohtahara syndrome, Dravet syndrome, and other epileptic severe disorders with generalized onset tonic seizures require a multidisciplinary approach to management that is demanding and often frustrating for the family and the treating healthcare professionals and can only be supportive and palliative (67; 58; 03; 70; 20; 75; 79). A management strategy should include the following elements:
• Treatment of epileptic seizures with appropriate medications and nonpharmacological methods. |
Antiepileptic drugs effective in tonic seizures are valproate, lamotrigine, phenytoin, clobazam, topiramate, rufinamide, perampanel, and zonisamide. Combinations of these antiepileptic drugs are often needed (67; 58; 03; 70; 20; 79). Vigabatrin should be considered for tonic seizures associated with tuberous sclerosis complex (76). Caution should be exercised with antiepileptic drugs that cause drowsiness that precipitates seizures.
The ketogenic diet may have significant beneficial effects in epileptic encephalopathies, alone or combined with corticosteroids (56; 29; 82).
Neurosurgical interventions are now possible and effective in selected cases (48). Callosotomy has a beneficial effect on tonic or atonic drop attacks (57).
Adjunctive cannabidiol treatment has attracted significant media attention for the treatment of intractable epilepsies in children (34). In Dravet syndrome a double-blind, placebo-controlled trial in a 14-week treatment period showed that cannabidiol was significantly beneficial, particularly in convulsive seizures, with 5% of patients becoming seizure-free though associated with higher rates of adverse events (22; 23). Also, cannabidiol has been found to be beneficial in patients with Lennox-Gastaut syndrome presenting with intractable seizures uncontrolled by concomitant antiepileptic drugs (55). In June 2018, Epidiolex® (cannabidiol oral formulation) from GW Pharmaceuticals obtained marketing authorization by the U.S. Food and Drug Administration, becoming the first drug to be approved for the treatment of Dravet syndrome in the United States market.
Seizures in syndromes with generalized onset tonic seizures are usually intractable.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Henry Hasson MD
Dr. Hasson of Albert Einstein College of Medicine has no relevant financial relationships to disclose.
See Profile
Solomon L Moshé MD
Dr. Moshé of Albert Einstein College of Medicine has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Epilepsy & Seizures
Jun. 02, 2026
Neuropharmacology & Neurotherapeutics
May. 11, 2026
Epilepsy & Seizures
May. 08, 2026
Epilepsy & Seizures
May. 01, 2026
Epilepsy & Seizures
Apr. 30, 2026
Epilepsy & Seizures
Apr. 17, 2026
Epilepsy & Seizures
Apr. 13, 2026
Epilepsy & Seizures
Mar. 24, 2026