
















Peripheral Neuropathies
Clinical evaluation of peripheral neuropathies
Jul. 16, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
There are numerous syndromes of multiple cranial nerve involvement. This article describes a segmental classification that can help organize and differentiate the various cranial polyneuropathies.
|
• Several diseases can cause multiple cranial nerve palsies, including cancer, benign tumors, infections, traumatic brain injury, and vascular lesions. |
Galen (131 AD to 201 AD) was the first to classify cranial nerves, but he counted only seven pairs of nerves (53). This classification was employed until the 17th century. Galen demonstrated the existence of 11 of the 12 cranial nerves (he regarded the olfactory nerve as an extension of the brain) but combined several and arrived at a total of seven. Although Flemish anatomist Andreas Vesalius (1514-1564) followed the classification of Galen, his excellent dissections of the brain showed nine pairs of cranial nerves (100). English physician and anatomist Thomas Willis (1621-1675) described the arteries at the base of the brain and showed 10 pairs of cranial nerves (104). This was the first reclassification of cranial nerves since the time of Galen, and it provided the correct numbering up to cranial nerve X. He described the accessory nerve but did not give it the number it has now: XI. Complete classification of all 12 cranial nerves did not take place until a century later by German physician and anatomist Samuel Thomas von Sömmerring (1755-1830) (92).
This image was prepared from an original photograph for a special museum exhibit. (From: Lanska DJ. Dissection of the Brain in Woodcut: A Visual Exploration of Renaissance Anatomy from Gersdorff to Vesalius. 23rd Annual Meeting...
This image was prepared from an original photograph for a special museum exhibit. (From: Lanska DJ. Dissection of the Brain in Woodcut: A Visual Exploration of Renaissance Anatomy from Gersdorff to Vesalius. 23rd Annual Meeting...
This image was prepared from an original photograph for a special museum exhibit. (From: Lanska DJ. Dissection of the Brain in Woodcut: A Visual Exploration of Renaissance Anatomy from Gersdorff to Vesalius. 23rd Annual Meeting...
From Sömmerring's De Basi Encephali Et Originibus Nervorum Cranio Egredientium Libri Quinque: Cum IV. Tabulis Aeneis, 1778. The olfactory bulb and tract, the optic chiasm, and the third nerve (exiting the brainstem from the int...
The best summary of the 19th-century understanding of cranial nerve disorders was by British neurologist Sir William Gowers (1845-1915), who devoted 150 pages to cranial nerve disorders in his 1886 Manual of Diseases of the Nervous System (32).
Engraving from Gowers’ A Manual of Diseases of the Nervous System, 1888. The cranial nerves are indicated by Roman numerals. Abbreviations: Th, thalamus; t c, tuber cinereum; h, pituitary body ("hypophysis cerebri"); P, peduncl...
Several syndromes involving particular combinations of multiple cranial nerve palsies were described toward the end of the 19th century and at the beginning of the 20th century.
Terms used for multiple cranial nerve palsies include “polyneuritis cranialis,” "cranial polyneuropathy," "multiple cranial neuropathies," and "multiple cranial nerve palsies," although the latter term would strictly apply only to cranial neuropathies involving the motor cranial nerves (III, IV, VI, VII, IX, X, XI, and XII).
|
• Among inpatients with simultaneous or serial involvement of two or more different cranial nerves, cranial nerves VI, VII, V, and III are most often affected. | ||
|
• Among inpatients with simultaneous or serial involvement of two or more different cranial nerves, the most common combinations of involved cranial nerves were (in descending order of frequency): III and VI; V and VI; V and VII; VII and VIII; III, IV, and VI; V, VI, and VII; II, III, and VI; X and XII; V, VII, and VIII; and VI and XII. | ||
|
• Among inpatients with simultaneous or serial involvement of two or more different cranial nerves, the cavernous sinus (26%), brainstem (22%), and individual nerves (19%) were the most frequent sites. | ||
|
• Among inpatients with cranial polyneuropathy, frequent anatomical patterns were cavernous sinus syndrome, orbital apex syndrome, and jugular foramen syndrome. | ||
|
• Cranial neuropathies can be classified segmentally: intracranial (intramedullary and extramedullary), cranial (skull base, foramina, cavernous sinus), or extracranial (retropharyngeal and retroparotid space). | ||
|
• Supranuclear or internuclear disorders, disorders of the neuromuscular junction, and disorders of cranial nerve-innervated muscles can present as pseudo-cranial nerve disorders and so fall within the differential diagnosis of cranial polyneuropathies and must certainly be distinguished from disorders of the cranial nerves themselves. | ||
|
• Consideration of cranial neuropathies depends first on identification (by history and examination) of the abnormal cranial nerves or constellations of disrupted functions served by cranial nerves, recognizing that dysfunction may involve only some nerve functions or may be incomplete for any given function. | ||
|
• The characteristic patterns of involvement of cranial-nerve-innervated muscles due to supranuclear or internuclear pathology (eg, Foix-Chavany-Marie syndrome, unilateral and bilateral internuclear ophthalmoparesis, gaze palsy, Fisher's one-and-a-half syndrome, and the vertical one-and-a-half syndrome—pseudo-cranial neuropathy type I) should be distinguished from partial or combined cranial neuropathies involving the third, fourth, and sixth cranial nerves. | ||
Scope. For this article, the following will not be considered among the cranial polyneuropathies discussed: bilateral olfactory, optic, auditory, or vestibular dysfunction due to end-organ pathology. Although these could conceivably be considered with cranial polyneuropathies, these traditionally have been considered separately, and their inclusion here would greatly complicate the explication of classical cranial polyneuropathies. Also not considered are one-off case reports, unless they provide significant neuroanatomical insights.
Frequency of cranial nerve involvement in cranial polyneuropathy. In a consecutive series of 979 unselected inpatients with simultaneous or serial involvement of two or more different cranial nerves, cranial nerves VI, VII, V, and III were most often affected (46). The most common combinations of involved cranial nerves were (in descending order of frequency): III and VI; V and VI; V and VII; VII and VIII; III, IV, and VI; V, VI, and VII; II, III, and VI; X and XII; V, VII, and VIII; and VI and XII (46). Although the locations were diverse, the cavernous sinus (26%), brainstem (22%), and individual nerves (19%) were the most frequent sites (46).
In a hospital-based prospective observational study from 2015 to 2017 of 54 consecutive patients with multiple cranial palsies, the most frequently involved cranial nerve was the abducens (76%), and the most frequent anatomical patterns were cavernous sinus syndrome (37%), orbital apex syndrome (22%), and jugular foramen syndrome (11%) (55).
In a less useful and conceptually problematic retrospective case series of 142 adult cases in Colombia of purportedly multiple cranial neuropathy, the most commonly affected cranial nerves were III and VII, with the most prevalent combinations being III-IV, III-VI, and V-VII (59). The most frequently identified etiologies were autoimmune, vascular, and neoplastic. The most common locations included the extra-axial cranial nerves (misrepresented in the paper as "peripheral nerves"), neuromuscular junction (sic, also not cranial nerves per se), cavernous sinus, and lateral medulla. Unfortunately, it is not possible, from the data presented, to remove the 22 cases of myasthenia gravis and recalculate the results.
Cranial neuropathies can be classified segmentally: specifically, cranial nerve pathology may involve intracranial (intramedullary and extramedullary), cranial (skull base and foramina), or extracranial (retropharyngeal and retroparotid space) structures (Table 1) (69; 11). Others approach cranial polyneuropathies as constellations of different syndromes, an approach that is also useful for focusing the differential and guiding management plans but that can be overwhelming with the large number of syndromes and eponyms (77). Several particular syndromes are important to recognize: orbital apex syndrome, cavernous sinus syndrome, cerebellopontine angle syndrome, and jugular foramen syndrome.
Even if supranuclear or internuclear disorders, disorders of the neuromuscular junction, and disorders of cranial nerve-innervated muscles are not considered cranial neuropathies per se, these pseudo-cranial nerve disorders fall within the differential diagnosis and must certainly be distinguished from disorders of the cranial nerves themselves, so they are worth considering within the framework of the classic cranial polyneuropathies.
|
Intracranial segments | |||
|
• Intramedullary (brainstem) | |||
|
- Nuclear | |||
|
• Extramedullary | |||
|
- Subarachnoid space | |||
|
Cranial (skull base) segment | |||
|
Extracranial (retropharyngeal and retro-parotid space) segment | |||
|
Multiple (eg, contiguous segments or diffuse involvement) | |||
|
• Congenital | |||
|
• Acquired (eg, toxic or metabolic, neoplastic or paraneoplastic, vascular) | |||
|
Pseudo-cranial polyneuropathies | |||
|
• Type I (central) | |||
|
- Supranuclear | |||
|
• Type II (peripheral) | |||
|
- Neuromuscular junction | |||
|
Botulism | |||
|
- Disorders of cranial-nerve-innervated muscles (including restrictive extraocular myopathies) | |||
|
Chronic progressive external ophthalmoplegia? (some) | |||
|
• Type III (psychogenic or malingered) | |||
|
Unclear | |||
(1) Identify abnormal cranial nerves, constellations of disrupted functions served by cranial nerves, and associated neurologic findings. Consideration of cranial neuropathies depends first on identification (by history and examination) of the abnormal cranial nerves or constellations of disrupted functions served by cranial nerves, recognizing that dysfunction may involve only some nerve functions or may be incomplete for any given function. With cranial polyneuropathies, or with a cranial mononeuropathy in conjunction with other neurologic manifestations, the collective pattern of neurologic involvement may directly suggest a specific pathologic locus to parsimoniously explain the collective abnormalities.
Paired cranial neuropathies represent a special case of cranial polyneuropathies. Paired cranial neuropathies result from lesions in specific loci where a single lesion can affect both cranial nerves, or special susceptibilities of individual pairs of cranial nerves to a pathogenic agent (eg, bilateral olfactory nerve shear injuries with head trauma, bilateral optic neuropathy with methanol poisoning or Leber optic neuropathy, bilateral oculomotor nerve palsy with pituitary apoplexy, bilateral traumatic trochlear nerve palsy, bilateral trigeminal neuropathy with mixed connective tissue disease, bilateral abducens palsy with Wernicke encephalopathy of increased intracranial pressure, bilateral peripheral facial neuropathy with Lyme disease or Melkersson-Rosenthal syndrome, bilateral acoustic neuropathy with neurofibromatosis type 1 or clival epidural hematomas, and bilateral hypoglossal nerve palsies with radiotherapy or bilateral carotid endarterectomies).
(2) Consider pseudo-cranial neuropathies (types I and II). Second, consider (and substantiate if possible) whether the nerves themselves are dysfunctional or whether their functions are disrupted by either (1) central nervous system pathology (ie, supranuclear or internuclear disorders for motor functions of the cranial nerves or thalamocortical radiations of sensory functions) or (2) peripheral pathology (eg, neuromuscular junction, cranial nerve-innervated muscles, or special sensory end organs [ie, eyes, ears, nose, mouth]). These I will label as pseudo-cranial neuropathy Types I and II, respectively.
One could, of course, dismiss dysfunction of cranial nerves resulting from supranuclear lesions as not strictly cranial neuropathies and, therefore, not of direct relevance. I think this narrow view is not ideal, however. By employing an extended idea of a cranial "neuropathy" (as is routinely done for central and peripheral facial paresis), or at least routinely considering supranuclear or internuclear CNS dysfunction, and peripheral dysfunction (eg, neuromuscular junction and cranial-nerve-innervated muscles), as well as psychogenic and malingered dysfunction (however, one wishes to classify these), one is forced to be more systematic and is consequently much better positioned to correctly localize lesions.
Pseudo-cranial neuropathy type I (supranuclear or internuclear cranial nerve dysfunction). Unilateral intracranial intramedullary lesions of the cerebral hemisphere (eg, stroke, demyelination, mass lesions) produce a pattern of contralateral supranuclear cranial nerve involvement that may include, depending on size and location, homonymous hemianopsia, gaze palsy, facial hemihypesthesia, central facial paresis, dysarthria, and dysphagia (105).
Bilateral anterior opercular lesions may produce Foix-Chavany-Marie syndrome, a cortical-subcortical pseudobulbar palsy characterized by volitional paralysis of masticatory, facial, pharyngeal, and lingual muscles innervated by cranial nerves V, VII, IX, X, and XII, with preserved autonomic and emotional innervation of these muscles. Thus, although volitional movements are absent, emotional smiling, laughter, crying, and automatic yawning are preserved--an automatic-voluntary dissociation of movements served by multiple motor cranial nerves.
The characteristic patterns of involvement of cranial-nerve-innervated muscles due to supranuclear or internuclear pathology (eg, Foix-Chavany-Marie syndrome, unilateral and bilateral internuclear ophthalmoparesis, gaze palsy, Fisher's one-and-a-half syndrome, and the vertical one-and-a-half syndrome--pseudo-cranial neuropathy type I) should be distinguished from partial or combined cranial neuropathies involving the third, four, and sixth cranial nerves.
Pseudo-abducens palsy (pseudo-sixth cranial nerve palsy; pseudoabduction defect; thalamic esotropia), which may be bilateral (103; 43; 65), is a neurologic limitation in ocular abduction with an intact abducens nerve as demonstrated, for example, by full abduction with vestibular stimulation (eg, the vestibulo-ocular reflex/caloric stimulation); the intact vestibulo-ocular reflex indicates the integrity of the infranuclear abducens nerve and establishes that the observed abduction impairment is caused by supranuclear pathology (27; 71; 103; 96; 74; 29; 43; 23; 99; 65; 76). This was recognized initially by Canadian neurologist C Miller Fisher (1913-2012) in 1959 (27). Lesions near the meso-diencephalic junction are usually implicated in pseudo-abducens palsy (type I). For example, isolated pseudo-abducens palsy may occur with thalamic hemorrhage or infarction. Strategic unilateral midbrain lesions may result in a contralateral pseudoabducens palsy due to damage of supranuclear projections within the rostral mesencephalon (ie, interrupting supranuclear corticofugal fibers from the frontal eye field and colliculofugal fibers from the superior colliculus) (02). In addition, tonic activation of the medial rectus (falsely suggesting abducens paresis) may occur with interruption of descending inhibitory convergence pathways that traverse the paramedian thalamus and decussate in the subthalamic region to innervate the contralateral oculomotor nucleus (29).
Paramedian thalamopeduncular infarction, damaging descending supranuclear pathways, may cause isolated inferior rectus palsy (95), and isolated medial longitudinal fasciculus infarction in the midbrain may cause medial rectus weakness (106).
Pseudo-cranial neuropathy type II (neuromuscular junction or muscle). Occasionally, supranuclear or internuclear disorders are mimicked by neuromuscular junction disorders (08; 54); indeed, myasthenia gravis can mimic any pupil-sparing eye movement disorder (56) or lower motor cranial nerve disorder, including cranial polyneuropathies involving individual combinations of oculomotor-, trochlear-, and abducens-innervated muscles). Myasthenia can be distinguished by abnormal muscle fatiguability, the ice-pack test, antibody tests, and electrophysiological methods (the tensilon or edrophonium test has not been available in the United States since 2017, based on a determination by the U.S. Food and Drug Administration). Botulism can also produce a range of cranial nerve dysfunction, affecting particularly the extraocular muscles (16; 94; 36; 68). Similarly, cranial nerve involvement may be seen in up to 70% of patients with Lambert-Eaton myasthenic syndrome; ptosis and diplopia are the most common cranial nerve manifestations, but dysphagia and dysarthria also occur (41).
Restrictive ophthalmopathies (eg, orbital blowout fracture [fracture of the medial orbital wall], trapdoor fracture [orbital floor fracture], and thyroid eye disease) can be established with a forced duction test and orbital imaging.
Pseudo-cranial neuropathy type III (psychogenic or malingered). Cases of psychogenic and malingered forms of cranial nerve dysfunction include some cases with psychogenic bilateral ptosis (32), functional blindness or deafness, hysterical hemianopia (“missing half” field defect) (44), midline-splitting hemifacial sensory loss, monocular diplopia (that does not resolve with a pinhole test), spasm of the near reflex (18; 33; 98; 64; 13; 79; 82; 30; 72; 07; 83; 17; 25; 39; 50; 89; 75; 10; 66; 80; 38; 101), globus hystericus, and “wrong way” tongue deviation (45).
Spasm of the near reflex consists of the triad of intermittent convergent strabismus, accommodative spasm, and pupillary miosis, as first reported by Harvard neuro-ophthalmologist David Cogan (1908-1993) and his fellow Carl Freese Jr. in 1955 (18). Spasm of the near reflex has been confused with abducens palsy or myasthenia gravis (33; 79; 101). Although the vast majority of cases are psychogenic, spasm of the near reflex may uncommonly be present with organic disease (20; 81; 73; 47; 91; 40); although an overlying functional disorder explains some of these cases, it is important not to overlook organic disease in the presence of functional disease.
(3) Localize affected cranial nerves to the involved segments. Considering the various segments that cranial nerves traverse helps avoid diagnostic errors due to so-called false-localizing signs, a concept introduced by English neurologist James Stansfield Collier (1870-1935) in 1904 (19), when the pathophysiology of cerebral dysfunction was less well understood and long before the availability of neuroimaging. A modern rendition considers signs to be falsely localizing “if they reflect dysfunction distant or remote from the expected anatomical locus of pathology, hence, challenging the traditional clinico-anatomical correlation paradigm on which neurological examination is based.” Such false localizing signs typically result from raised intracranial pressure and occur with spinal cord lesions as well. Cranial nerve palsies (especially sixth nerve palsy), hemiparesis, sensory abnormalities (eg, truncal sensory levels), and muscle atrophy may all occur as false localizing signs by this definition (51). However, much of what has been deemed either “localizing” or falsely localizing depends on rather narrow (and somewhat archaic) concepts of how dysfunction is localized. For example, recognizing a third-nerve palsy in its intracranial extramedullary course as part of the uncal herniation syndrome almost always signifies ipsilateral supratentorial pathology with secondary involvement of the ipsilateral third cranial nerve (as it traverses the edge of the tentorium on its way to the superior orbital fissure), rather than primary pathology in the posterior fossa. Recognizing the nature of this third-nerve palsy accurately localizes the responsible lesion as well as its consequences. With this more nuanced idea of localization, such signs are not falsely localizing but instead are truly localizing and provide a much deeper understanding of the responsible pathology.
Recognition of which segments are involved in a particular cranial nerve typically requires consideration of accompanying symptoms and signs. For example, consider different segments of the abducens nerve. Although the abducens nerve innervates just one muscle, the lateral rectus, an intramedullary lesion involving the sixth-nerve nucleus, will produce an abduction defect of the ipsilateral eye and an adduction impairment of the contralateral eye—a gaze palsy. This occurs because the abducens nucleus contains (1) motor neurons that supply the ipsilateral lateral rectus muscle and (2) internuclear neurons that project via the medial longitudinal fasciculus to the contralateral medial rectus subdivision of the contralateral oculomotor nucleus.
(1) Fibers to the ipsilateral lateral rectus in the abducens nerve. (2) Fibers to the contralateral medial rectus through the medial longitudinal fasciculus to the contralateral third nerve nucleus and then through the oculomot...
It is very difficult (practically impossible) to damage the abducens nucleus without damaging the facial colliculus, formed by facial motor neurons looping over the abducens nucleus. Thus, a lesion of the abducens nucleus should be expected to occur with an ipsilateral gaze palsy and an ipsilateral peripheral facial paresis. This combination unequivocally localizes the lesion to the dorsal caudal pontine tegmentum.
When multiple cranial nerves (or cranial nerve functions) are involved, combine the information on involved nerve segments to localize the responsible pathology. Then integrate that with information on age, onset, and course to suggest likely etiologies.
It sometimes makes sense to subdivide some cranial nerve segments. In particular, it is common to divide the course of the facial nerve within the skull into multiple segments rather than a global “cranial segment” or “foraminal segment.” The fallopian canal (facial nerve canal) is the bony canal through which the facial nerve traverses the petrous temporal bone, from the internal acoustic meatus to the stylomastoid foramen. There are four (or by some accounts three) segments of the facial nerve within the canal: (1) meatal, (2) labyrinthine, (3) tympanic, and (4) mastoid. Then, the nerve finally emerges from the stylomastoid foramen to continue in the extracranial segment, which runs through the parotid gland and gives rise to various branches that supply the muscles of facial expression.
It is also worth pointing out that all cranial nerves have an extracranial segment except cranial nerve VIII.
Intramedullary lesions. Intramedullary lesions are distinguished by the co-occurrence of segmental and long tract signs (eg, crossed hemiplegia along with oculomotor nerve deficits indicates midbrain pathology and, specifically, Weber syndrome) or other evidence of brainstem involvement (eg, pontine gaze palsy, internuclear ophthalmoparesis, etc.).
Extramedullary intracranial lesions. Extramedullary intracranial lesions are recognized by specific patterns of cranial nerve involvement without long tract signs or other evidence of brainstem involvement.
The so-called cavernous sinus syndrome is a grab bag that encompasses a variety of disorders with differing pathologies and variably overlapping clinical manifestations--essentially any disease process involving the cavernous sinus. Cavernous sinus syndrome may present with multiple cranial neuropathies (involving cranial nerves III, IV, VI, V1, and V2). Although the optic nerve does not pass through the cavernous sinus, vision (cranial nerve II) may be impaired from the secondary effects on venous drainage of the orbit. Cavernous sinus syndrome is often used as a working diagnosis for signs and symptoms related to the four cranial nerves passing through the cavernous sinus (III, IV, V1, and VI).
The cerebellopontine angle is a triangular space in the posterior cranial fossa where the pons, cerebellum, and petrous part of the temporal bone meet. This area is clinically significant because it contains cranial nerves V, VI, VII, and VIII, as well as the anterior inferior cerebellar artery.
If extramedullary intracranial involvement of multiple cranial nerves occurs with long tract signs, this implies that the process that initially involved a collection of extramedullary intracranial nerves has expanded sufficiently to involve the brainstem, either because of significant mass effect or infiltration of the brainstem. So, for example, an acoustic neuroma may grow sufficiently large that it pushes on the adjacent brainstem causing long tract signs, but in either case, brainstem involvement is a late-stage finding. A careful history of the progression of symptoms and signs generally provides clarification of what was primary and what was a secondary late-stage manifestation.
Bilateral bulbar palsy. Consider now a relatively frequent occurrence: relatively isolated paralysis involving cranial nerves V, VII, IX, X, and XII bilaterally, sparing the eye muscles and without evident long tract signs. What are the possibilities? Or perhaps first, what can be excluded to narrow the range of possibilities?
1. It is extremely difficult to contrive a way that this could occur acutely with brainstem involvement sparing long tract signs given the large expanse of the pons and medulla involved (we will consider chronic, progressive possibilities in a moment).
2. Similarly, it is extremely unlikely to result from skull base lesions spanning the middle and posterior fossas. Although Paget disease can involve these nerves (including the lower cranial nerves through basilar invagination), abnormalities are not restricted to the motor components of mixed cranial nerves, and by far the most common cranial neuropathy with Paget disease involves the eighth nerve manifesting as hearing loss, tinnitus, and sometimes vestibular symptoms (58; 85; 24; 78; 21; 37; 28; 05; 15; 26; 14; 57; 70; 86; 52; 22; 12; 87; 35). Other cranial neuropathies in Paget disease can include optic atrophy, trigeminal neuralgia, and hemifacial spasm.
3. It is also impossible for this constellation of cranial nerves to all be involved with their extracranial segments. If it occurs much more slowly and progressively, and there is evidence of lower motor neuron abnormalities (eg, tongue wasting and fasciculation) on examination, progressive bulbar palsy (a form of motor neuron disease) is a possibility. Even without long tract signs, emotional lability or other manifestations of "pseudobulbar palsy" provide evidence of dysfunction within the parenchyma of the cerebrum or brainstem.
4. Multiple (eg, contiguous segments or diffuse involvement) could not explain this constellation either.
5. Pseudo-cranial polyneuropathy types I (subtype internuclear), II, and III are not viable either. For type II (peripheral), a neuromuscular junction abnormality is unlikely in the absence of ptosis or the extraocular muscles, and no disease of the bulbar muscles alone is known. For type III, there is no precedent for a convincing case with this pattern of pseudo-cranial polyneuropathies on a psychogenic or malingered basis.
What is left then?
1. A chronic, progressive intramedullary process, such as progressive bulbar palsy (thought to be a form of motor neuron disease), if the time course and clinical findings are consistent with this.
2. Extramedullary intracranial (eg, Guillain-Barre syndrome variants) is especially progressive over a subacute time course (03; 31; 34; 60).
3. Pseudo-cranial polyneuropathy type I, subcategory supranuclear, especially if acute. A likely possibility would be Foix-Chavany-Marie syndrome, a cortical form of supranuclear (pseudobulbar) palsy caused by bilateral anterior opercular lesions. In this syndrome, there is an “automatic-voluntary dissociation” of motor function of lower cranial nerves. Manifestations include volitional paralysis of masticatory, facial, pharyngeal, and lingual muscles innervated by cranial nerves V, VII, IX, X, and XII, with preserved autonomic and emotional innervation of these muscles (eg, emotional smiling, laughter, and crying as well as automatic yawning). Pseudobulbar paralysis in Foix-Chavany-Marie syndrome is clinically distinguished from bulbar paralysis and from disorders of the cranial nerves and neuromuscular junction (eg, botulism and myasthenia gravis) by normal eye movements, preserved or hyperactive brainstem reflexes (eg, jaw jerk), the dissociation of automatic and volitional movements of the bulbar muscles with preservation of automatic movements, and the absence of atrophy and fasciculations of the lower motor neuron–innervated muscles.
Idiopathic cranial polyneuropathy. The diagnosis of idiopathic cranial polyneuropathy is usually made after exclusion of other causes. This term is also used for an unusual syndrome of pain in the face and head followed by the onset of multiple cranial nerve palsies occurring days to weeks later (42). Cranial nerves III to VII are involved in various degrees. Involvement of the lower cranial nerves (VIII to XII) is less common. Fifty percent of these patients have mild cerebrospinal fluid lymphocytosis or protein elevation.
Prognosis depends on the pathology of the primary lesions.
|
• Multiple cranial neuropathies occur most commonly with malignancies involving the base of the skull, with infectious and malignant meningeal diseases, and with granulomatous diseases. | |
|
• Multiple cranial neuropathies may also occur with sequelae of radiotherapy for cancer, paraneoplastic diseases, complications of surgical procedures, vascular diseases, intracranial hypertension, metabolic disorders, and idiopathic conditions. | |
|
• Among inpatients with simultaneous or serial involvement of two or more different cranial nerves, tumors, vascular disease, trauma, infection, and the Guillain-Barré and Fisher syndromes are the most frequent causes. Tumors account for about one third of cases. |
Multiple cranial neuropathies occur most commonly with malignancies involving the base of the skull, with infectious and malignant meningeal diseases, and with granulomatous diseases. Leptomeningeal leukemic, lymphomatous, or carcinomatous infiltration of basal meninges often presents with multiple cranial nerve palsies (49). Multiple cranial neuropathies may also occur with sequelae of radiotherapy for cancer, paraneoplastic diseases, complications of surgical procedures, vascular diseases, intracranial hypertension, metabolic disorders, and idiopathic conditions.
In a consecutive series of 979 unselected inpatients with simultaneous or serial involvement of two or more different cranial nerves, tumor (31%), vascular disease (13%), trauma (13%), infection (10%), and the Guillain-Barré and Fisher syndromes (9% combined) were the most frequent causes (46). A functional (psychogenic) cause was identified in 3% of the cases. Some cases had more than one cause. The most common tumors causing multiple cranial neuropathy were (in descending order of frequency) schwannoma, metastases, meningioma, lymphoma, and nasopharyngeal carcinoma (46). Recurrent cranial neuropathy was uncommon (4%), seen usually with diabetes mellitus, self-limited unknown causes, or idiopathic cavernous sinusitis (46).
Although multiple intracranial nerve palsies are not rare, no population-based estimates of incidence or prevalence are available, partly due to the multiplicity of disease processes involved.
The differential diagnosis of multiple cranial nerve palsies involves several important steps:
(1) Localization of nervous system involvement, based on the specific cranial nerves involved, other accompanying neurologic manifestations, and CNS imaging studies.
(2) Exclusion of pseudo-cranial neuropathies (eg, supranuclear and internuclear disorders, Graves' orbitopathy and restrictive myopathy, myasthenia gravis).
(3) Determination of the pathology of the lesion based on laboratory, radiologic, and pathologic studies.
|
• Diagnostic procedures vary according to clinical examination findings. | |
|
• MRI and CT are complementary modalities in evaluating multiple cranial neuropathies. | |
|
• Optimal assessment of skull base lesions requires both MRI and CT with the thin-section bone algorithm. MRI shows the extent of soft tissue lesions, whereas CT allows precise evaluation of the surrounding bone changes. | |
|
• MRI has greater utility for the detection and characterization of cerebellopontine angle processes. | |
|
• The differential diagnosis of cerebellopontine angle lesions can be helpfully categorized into mass lesions, vascular lesions (eg, aneurysm, arteriovenous malformation), and leptomeningeal disease. | |
|
• Neurosarcoidosis, subarachnoid hemorrhage, basal meningitis, viral neuritis, and chronic inflammatory demyelinating polyneuropathy may involve multiple cranial nerves and be demonstrated on MRI as nerve thickening and contrast enhancement. |
The following diagnostic procedures are recommended in a patient who presents with multiple cranial nerve palsies:
|
• Neurologic examination with special emphasis on testing the cranial nerves. | |
|
• Ear, nose, and throat examination to identify or exclude nasopharyngeal carcinoma. | |
|
• Blood count, routine chemistry tests, and erythrocyte sedimentation rate. | |
|
• CT and MRI of the base of the skull. | |
|
• High-resolution MRI is preferred for evaluating most lesions involving the orbital apex. | |
|
• Lumbar puncture with cerebrospinal fluid examination and culture for infectious agents (including syphilis, tuberculosis, and cryptococcus) as well as large-volume cytology for malignant cells. For suspected syphilitic meningitis, include CSF FTA-ABS. | |
|
• Diagnostic procedures for establishing or excluding a cancer diagnosis, according to the neurologic and general physical examinations. | |
|
• Cerebral angiography in select cases for detecting arterial aneurysms and venous sinus thrombosis. | |
|
• A meningeal biopsy may be necessary in some cases. |
Conceptualizing the general segmental architecture of the cranial nerves aids in the evaluation of patients with pathologic conditions affecting or adjoining their course (69; 11).
Blitz and colleagues use a segmental approach for neuroimaging cranial neuropathies that divides the course of the cranial nerves into the (1) nuclear segment, (2) parenchymal fascicular segment, (3) cisternal segment (subarachnoid space), (4) dural cave segment (the region of transition from the cisternal segment centrally to the interdural segment distally; eg, Meckel's cave), (5) interdural segment (eg, cavernous sinus), (6) foraminal segment (extending from the internal orifice of the foramen through the foramen itself to the external orifice), and (7) extraforaminal segment (cranial nerve VIII is the only cranial nerve that does not have an extraforaminal component) (11). This approach is useful for precise neuroradiological diagnosis and anatomic localization, but clinically, I have lumped the cisternal segment with the dural cave because, in both cases, the cranial nerves are surrounded by cerebrospinal fluid. The interdural segment can be considered separately from the foraminal segment where that applies, but it is also reasonable to lump these segments as "skull base" because both have passed out of the subarachnoid space and through a layer of dura and have not yet emerged from the skull. The outer periosteal layer of the dura provides a tubular covering for the cranial nerves as they pass through the skull foramina, but when the cranial nerves exit the foramen, the periosteal layer fuses with the epineurium of the nerves.
Policeni and colleagues similarly use a segmental approach for neuroimaging lower cranial neuropathies that divides the course of the cranial nerves into the (1) brainstem or nuclear segment (combining nuclear and fascicular segments), (2) cisternal segment, (3) skull base segment, (4) suprahyoid segment, and (5) infrahyoid or mediastinum segment (69). Because they consider only the lower cranial nerves, they do not address the dural cave and interdural segments considered by Blitz and colleagues (69; 11).
MRI and CT are complementary modalities in the evaluation of multiple cranial neuropathies. Optimal assessment of skull base lesions (eg, jugular foramen disorders) requires both MRI and CT with the thin-section bone algorithm (63). MRI shows the extent of soft tissue lesions, whereas CT allows precise evaluation of the surrounding bone changes.
MRI has greater utility for the detection and characterization of cerebellopontine angle processes (48). Heavily T2-weighted three-dimensional sequences, such as fast imaging using steady-state acquisition or constructive interference in the steady state, provide high-resolution details of cranial nerves and the blood vessels associated with them as well as ear structures (48). Pre- and post-gadolinium 2-mm T1-weighted sequences should be obtained in the axial and coronal planes. CT is useful as a supplemental imaging modality in select cases for defining any bone changes associated with cerebellopontine angle masses, such as meningiomas.
In evaluating cranial nerves and the skull base, diffusion tensor tractography can be used to delineate cranial nerves not visible on conventional sequences, such as those displaced by tumors (97). In addition, magnetic resonance neurography techniques have been developed that employ vascular and fat suppression (eg, diffusion weighting, motion-sensitized driven equilibrium, and selective water excitation) to facilitate visualization of the extracranial portions of the cranial nerves (97). Diffusion-weighted imaging and dynamic contrast-enhanced MRI may be useful in the differential diagnosis, prognostication, and posttreatment follow-up of skull base lesions (97).
Some MRI findings are very suggestive of pathology and may point to a particular diagnosis or a group of possible diagnoses: for example, (1) enhancement of the intracanalicular or the labyrinthine portion of the facial nerve is always abnormal (93); (2) an intracanalicular-cisternal mass suggests a vestibulocochlear nerve schwannoma, whereas a strictly cisternal mass suggests a meningioma (93); (3) a jugular foramen lesion is most commonly a paraganglioma, but less common possibilities include neural sheath tumors (eg, schwannomas and neurofibromas), meningiomas, and metastases (102; 93); (4) in hypoglossal neuropathy, the most important MRI feature is unilateral signal intensity denervation changes of the tongue musculature (93).
The differential diagnosis of cerebellopontine angle lesions can be helpfully categorized into mass lesions, vascular lesions (eg, aneurysm, arteriovenous malformation), and leptomeningeal disease (48). Enhancing mass lesions include schwannomas, meningiomas, aneurysms, and metastases, whereas nonenhancing mass lesions include epidermoid tumors and arachnoid cysts, and enhancing leptomeningeal disease occurs with meningeal carcinomatosis or lymphomatosis, sarcoidosis, and infection, whereas gradient echo signal loss indicates superficial siderosis.
Neurosarcoidosis, subarachnoid hemorrhage, basal meningitis, carcinomatous meningitis, neurolymphomatosis (a rare lymphoma infiltrating cranial nerves), viral neuritis, and chronic inflammatory demyelinating polyneuropathy may involve multiple cranial nerves and be demonstrated on MRI as nerve thickening and contrast enhancement; lower cranial nerves are more commonly affected (04; 93; 90; 88; 06; 09; 62).
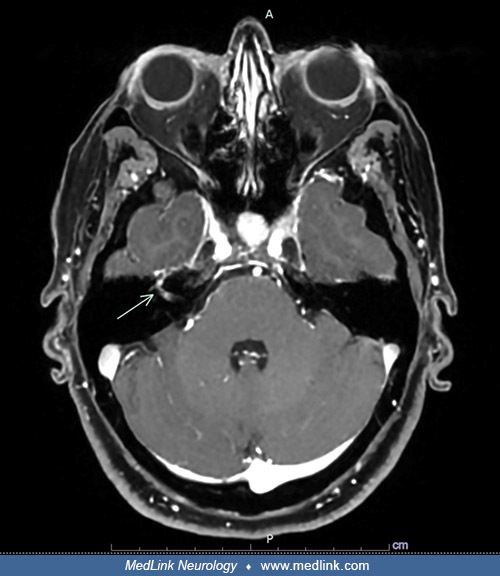
MRI in a 51-year-old woman with neurosarcoidosis who presented with acute cranial nerve polyneuropathy involving the facial (VII), oculomotor (III), and trigeminal (V) nerves, accompanied by systemic constitutional symptoms, in...
|
• Management depends on the underlying etiology. | |
|
• Cancer-related multiple cranial neuropathies that are detected early may be amenable to surgery and radiotherapy. | |
|
• The mainstay of treating rhinocerebral mucormycosis is surgical debridement plus antifungal therapy (amphotericin B). | |
|
• In idiopathic multiple cranial nerve palsy cases where no cause is discovered despite extensive investigations, corticosteroids may be helpful. |
Management depends on the underlying etiology. Because the range of disorders causing multiple cranial neuropathies is very wide, it is not feasible to summarize the treatment for all possible causes of multiple cranial neuropathies.
Cancer-related multiple cranial neuropathies that are detected early may be amenable to surgery and radiotherapy.
Corticosteroids have been used with benefit in inflammatory lesions.
Some infectious granulomas are amenable to appropriate antibiotic therapy.
Management of cases in which the cause is not known. In idiopathic multiple cranial nerve palsy cases where no cause is discovered despite extensive investigations, corticosteroids may be helpful. A case of cranial polyneuropathy with positive antinuclear antibody, but no established diagnosis, responded to steroids even though there was no change in the titer of antinuclear antibody (61). Marked improvement was reported following two cycles of intravenous immunoglobulin therapy in a child with cranial polyneuropathy involving cranial nerves III-VII and IX-XII after initial, unsuccessful treatment with corticosteroids and acyclovir (67).
Surgical procedures. Although surgical procedures are performed for the correction of sequelae of lesions of some of the individual cranial nerves, patients with multiple cranial nerve palsies are usually considered for surgical procedures only for removal of the causative lesions, such as skull base tumors and rhinocerebral mucormycosis.
In cases of intracranial involvement with rhinocerebral mucormycosis, craniotomy and aggressive debridement are typically necessary procedures in conjunction with systemic antifungal therapy (amphotericin B). In patients with infection limited to the nose and sinuses, endoscopic surgical debridement combined with topical amphotericin B followed by intravenous amphotericin B may be acceptable management for selected patients, with less morbidity than conventional treatments; in a group of 30 patients, the survival rate was 60% overall with this approach (71% in the diabetic group and 40% in the malignancy group) (84).
Treatment outcome depends on the cause of the nerve palsy and the treatment method.
Transitional lower cranial nerve palsies may occur as an adverse effect of the bupivacaine-fentanyl combination used in patients during and following spinal anesthesia (01). This transient complication may occur a few days after the procedure. The delayed onset is difficult to explain but may be due to CSF loss from the lumbar puncture site, causing descent of the brain and stretching of the nerves.
Problems can arise during anesthesia in children with Moebius syndrome. There is a high incidence of difficult or failed intubation, with the potential for problems with aspiration of oral secretions. Premedication to reduce salivary secretion is recommended. Children with Moebius syndrome have a high incidence of other anomalies, including congenital heart disease.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Douglas J Lanska MD MS MSPH
Dr. Lanska of the University of Wisconsin School of Medicine and Public Health has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Peripheral Neuropathies
Jul. 16, 2026
Peripheral Neuropathies
Jul. 14, 2026
Neurobehavioral & Cognitive Disorders
Jun. 17, 2026
Peripheral Neuropathies
Jun. 11, 2026
Peripheral Neuropathies
Jun. 11, 2026
Peripheral Neuropathies
Jun. 11, 2026
Neuro-Oncology
May. 27, 2026
Neuropharmacology & Neurotherapeutics
May. 14, 2026