Neuromuscular Disorders
Viral and retroviral myositis
Jun. 16, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Myotonic dystrophies represent the most common muscular dystrophy in adults, encompassing complex genetic disorders characterized by progressive muscle weakness with myotonia and multisystemic involvement. Advances in understanding the pathogenic mechanisms have ushered in a new era of targeted therapies, with multiple nucleic acid–based therapeutics now in phase III clinical trials. The field has rapidly progressed from initial observations of RNA toxicity mechanisms to sophisticated therapeutic approaches targeting the genetic underpinnings of the disease, including antisense oligonucleotides, RNA interference, and genome editing technologies.
|
• The myotonic dystrophies are the most common cause of adult-onset muscular dystrophy. | |
|
• Myotonic dystrophy type 1 according to age of onset and symptoms is divided into five forms: congenital, childhood, juvenile, adult, and late-onset. | |
|
Phenotypes of DM1 and DM2 are similar, but there are some important differences, including the presence or absence of congenital form, muscles primarily affected (distal vs. proximal), involved muscle fiber types (type 1 vs. type 2 fibers), and some associated multisystemic phenotypes. | |
|
• For the enormous understanding of the molecular pathogenesis of myotonic dystrophy type 1 and myotonic dystrophy type 2, these diseases are now called “spliceopathies” and are mediated by a primary disorder of RNA rather than proteins. | |
|
• Despite clinical and genetic similarities, myotonic dystrophy type 1 and type 2 are distinct disorders requiring different diagnostic and management strategies. | |
|
• Multiple therapeutic modalities have emerged for patients with myotonic dystrophy, including small molecules, nucleic acid–based therapies, and genome engineering approaches now in the clinical trial phase. | |
|
• Effective management significantly reduces the morbidity and mortality of patients. |
Myotonic dystrophies represent a group of dominantly inherited, multisystem (eye, heart, brain, endocrine, gastrointestinal tract, uterus, skin) diseases that share the core features of myotonia, muscle weakness, and early onset cataracts (before 50 years of age). Clinicians considered myotonic dystrophy to be a single disease until 1909 when Steinert and colleagues first clearly described the “classic” form of myotonic dystrophy, which was called Steinert disease (84). The gene defect responsible for myotonic dystrophy described by Steinert was discovered in 1992 and was found to be caused by expansion of a CTG repeat in the 3’ untranslated region of myotonic dystrophy protein kinase gene (DMPK), a gene located on chromosome 19q13.3 (OMIM 605377), encoding a protein kinase (24; 64; 128). After the discovery of this gene defect, DNA testing revealed a group of patients with dominantly inherited myotonia, proximal more than distal weakness, and cataracts; these patients were previously diagnosed as having myotonic dystrophy of Steinert but lacked the gene defect responsible for this disease. Subsequent clinical studies of kindreds with patients having these characteristics led to new diagnostic labels for these patients: myotonic dystrophy type 2 (241), proximal myotonic myopathy (PROMM) (198; 144), or proximal myotonic dystrophy (PDM) (248). Later studies demonstrated that many of the families identified as having myotonic dystrophy type 2, PROMM, or PDM had a single disorder that results from an unstable tetranucleotide CCTG repeat expansion in intron 1 of the nucleic acid-binding protein (CNBP) gene (previously known as zinc finger 9 gene, ZNF9) on chromosome 3q21 (OMIM #116955) (194; 121).
Myotonic dystrophy of Steinert, the classical form of myotonic dystrophy that results from an unstable trinucleotide repeat expansion on chromosome 19q13.3, was termed myotonic dystrophy type 1. Patients with the clinical picture of myotonic dystrophy type 2, PROMM, or PDM who have positive DNA testing for the unstable tetranucleotide repeat expansion on chromosome 3q21 were classified as having myotonic dystrophy type 2. Reliability of DNA testing to establish or to exclude the diagnosis of myotonic dystrophy type 1 and type 2 is close to 100% (253). This chapter focuses on myotonic dystrophy type 1 and myotonic dystrophy type 2. The clinical spectrum for both myotonic dystrophy type 1 and myotonic dystrophy type 2 remains a work in progress due to the fact that it has been possible to identify these disorders only recently, specifically with DNA testing. At present, much more information is available on the natural history of myotonic dystrophy type 1 than myotonic dystrophy type 2, but knowledge of myotonic dystrophy type 2 will increase at a rapid pace over the next several years.
Myotonic dystrophy type 1 (myotonic dystrophy of Steinert). Myotonic dystrophy type 1 is clinically classified into five distinct forms based on age of onset and clinical presentation: congenital, infantile (symptoms occurring during the first decade), juvenile (onset between 10 and 20 years), adult-onset (typically third decade), and late-onset (minimal symptoms, often presenting only with cataracts, baldness, or cardiac conduction abnormalities). This classification system, developed by the OMMYD (Outcome Measures in Myotonic Dystrophy) consortium, provides a framework for understanding disease progression and planning therapeutic interventions across the lifespan.
Adult-onset myotonic dystrophy type 1. Adult-onset myotonic dystrophy type 1 can present in the third decade with grip myotonia, slurring of speech, trouble manipulating items, foot slapping, or weak ankles. Premature male pattern baldness also occurs, but it is not usually reported. In cases presenting in the third decade, weakness of the face muscles (inability to bury eyelashes), the long flexors of the fingers (flexor profundus muscles), the intrinsic hand muscles, and the dorsiflexors of the feet (tibialis anterior, extensor hallucis longus, and extensor digitorum longus muscles) are typically present along with myotonia. An MRI study of 20 different upper and lower leg muscles of patients with myotonic dystrophy type 1 has shown abnormal values for muscle fat fraction, muscle volume, and T2 water relaxation time reflecting putative edema (91). Fat fraction correlated with the 6-minute walk test and muscular impairment rating scale.
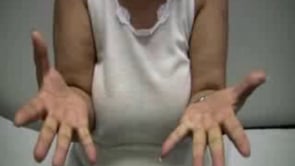
Myotonia occurs following a powerful isotonic contraction of the fingers or percussion of the thenar and forearm extensor muscles, which tends to be directly proportional to the severity of myotonia (122).
On clinical evaluation, especially of the thenar and forearm extensor muscles, percussion is more sensitive in detecting minimal degrees of myotonia than is grip testing. Percussion of the tongue can also reveal myotonia, but it is a cumbersome diagnostic test with no obvious advantage over percussion of hand and forearm muscles.
Myotonia, along with muscle weakness, usually contributes to the difficulties with speech, swallowing, respiration, and smooth muscle dysfunction (which include abnormal intestinal motility and uterine dysfunction). It is interesting to note that myotonia is not apparent clinically or on electrodiagnostic testing early in life in myotonic dystrophy type 1 patients. Myotonia is also difficult to detect on clinical examination in the late stages of disease, especially in wasted, weakened muscles. In a study conducted on a large population of 144 myotonic dystrophy type 1 patients, leukocyte CTG repeat length resulted to be statistically correlated with both myotonia and grip strength, which are two major primary neuromuscular symptoms (94). Patients with myotonic dystrophy type 1 who present in the third decade usually develop progressive dysarthria, difficulty swallowing, gastric regurgitation, hypogonadism, insulin resistance, deficient release of growth hormone, hypersomnia, neuropsychological and cognitive alterations, sleep apnea, decline in forced vital capacity, and cardiac conduction disturbances (84). Lifespan may be shortened. Respiratory failure, pneumonitis, and cardiac conduction disturbances are the usual causes of death (49; 133; 84; 263).
Respiratory failure is the most common mechanism of death in patients with myotonic dystrophy due not only to progressive skeletal muscle dysfunction but also to central dysregulation of the control of breathing (191; 87). A brief overview of major sleep disorders in patients with myotonic dystrophy was provided in one review: sleep disorders breathing (SDB) with the both central and obstructive sleep apneas (CSA, OSA), excessive daytime sleepiness (EDS), sleep-related movement disorders, and poor sleep quality. Possible pathogenesis and outline management were described (234). A large cohort of patients with myotonic dystrophy type 1 has been widely characterized in the phenotype to assess prevalence and identify predictors of restrictive respiratory syndrome. Twenty-one adult patients with myotonic dystrophy type 1 and matched controls underwent detailed investigations with spirometry, manometry, and diaphragm ultrasound, and in addition, surface EMG of the diaphragm and oblique abdominal was performed following cortical and posterior cervical magnetic stimulation of the phrenic nerves or magnetic stimulation of lower thoracic nerve roots (89). The results of this original study showed that in myotonic dystrophy type 1, respiratory muscle weakness relates to clinical disease severity and involves respiratory and probably expiratory muscle strength. Axonal phonic nerve pathology may contribute to diaphragm dysfunction (89). A high prevalence of restrictive syndrome has been found mainly due to respiratory muscle weakness, and most of the patients showed indication to noninvasive ventilation. Data suggest that optimization of respiratory therapeutic management, particularly regarding launching of noninvasive ventilation, might help to reduce the rate of deaths due to respiratory complications in myotonic dystrophy type 1 (205). Noninvasive ventilation has been found to significantly improve ventilation and oxygenation starting from the first night of treatment, and follow-up revealed stable normoxia and normocapnia without deterioration of sleep outcomes for up to 52 months (232). Trunk muscles in patients with myotonic dystrophy type 1 show significant higher levels of fat infiltration and reduced muscle size compared to age and gender matched controls. Fat infiltration is associated with reduced muscle strength, mobility, balance, and lung function, although muscle size is associated with reduced muscle strength and lung function. These findings are of importance for clinical management of the disease (231).
The heart is commonly involved in myotonic dystrophy type 1 patients, and cardiac disorders are also an important cause of premature death in these patients (262). Patients with myotonic dystrophy type 1 typically manifest conduction defects, ventricular dysfunctions, and supraventricular and ventricular arrhythmias (262). Sudden death prevention is central and relies on annual follow-up and prophylactic permanent pacing in patients with conduction defect on ECG and/or infra-Hisian blocks on electrophysiological study. Implantable cardiac defibrillator therapy may be indicated in patients with ventricular tachyarrhythmia (263). The first case of a myotonic dystrophy type 1 patient with type 1 Brugada ECG pattern has been reported (178), suggesting that the combination of ECG abnormalities commonly seen in both myotonic dystrophy type 1 and Brugada syndrome may share a common pathophysiologic pathway, perhaps due to the loss-of-function of the sodium channel SCN5A (63; 178). Overt cardiomyopathy is relatively rare, although cardiac MRI studies suggest that more subtle changes in myocardial trabeculation and mass are more common than previously recognized (42). Physicians dealing with myotonic dystrophy type 1 may take into consideration that cardiac MRI in patients without apparent cardiac disease may show increase in cardiac extracellular volume and decrease in strain as signs of early cardiac pathology (06).
Sleep may also be impaired in myotonic dystrophy type 1 patients. Excessive daytime sleepiness is seen in 33.1% of these patients and tends to be proportional to the amount of muscular impairment. In addition, patients with myotonic dystrophy type 1 often have longer sleep periods (more than 10 hours), excessive daytime sleep, difficulty falling asleep, and somnolence after meals; this is similar to patients with idiopathic hypersomnia (111). Periodic limb movements in myotonic dystrophy type 1 are also frequently associated (202). In a large cohort of Canadian patients with myotonic dystrophy type 1 followed for 9 years, the predicting factors of daytime sleepiness and fatigue were modifiable factors as BMI, psychological distress, hypothyroidism, and sleep habits (112).
Severe vitamin D deficiency is common in myotonic dystrophy type 1 and it is associated with secondary hyperparathyroidism, and primary hyperparathyroidism, though rare, may occur. Therefore, greater attention should be given to vitamin D status in order to administer an appropriate replacement therapy (166). Moreover, patients with myotonic dystrophy type 1 have a higher incidence of hypercalcemia compared to the general population (93) and higher fragility fractures in myotonic dystrophy type 1 versus myotonic dystrophy type 2, whereas hip osteopenia was more prominent in myotonic dystrophy type 2 (167).
Early onset posterior subscapular cataract (less than 50 years of age) is considered a characteristic feature of myotonic dystrophy type 1 and is known to be a key feature for timely diagnosis (261). Early detection of Christmas tree cataract also constitutes a common ophthalmologic finding in patients with myotonic dystrophy type 1 (162). Occurrence of cataracts before the age of 50 years should alert the clinician to consider myotonic dystrophy. Retinal degenerative changes have also been documented pathologically (13). New findings of retinal changes by spectral domain optical coherence tomography (OCT) in a cohort of patients with myotonic dystrophy type 1 have been described (04). They are consistent with typical retinal pigment epithelium changes and abnormalities of the vitreoretinal interface, particularly epiretinal membranes, resulting in central macular thickness. Both inner and outer retinal alterations are associated with increasing age, suggesting a premature aging of the retina in myotonic dystrophy type 1.
Gastrointestinal manifestations are common in myotonic dystrophy type 1 patients, affecting their quality of life, with a relatively high frequency of gallbladder removal occurring at a younger age compared to normal population. In a study that analyzed the progression of gastrointestinal manifestations, the most common changes reported by patients with myotonic dystrophy type 1 were new reports of stomach ulcers and trouble swallowing (92). In these patients the greater risk of a gastrointestinal manifestation has been associated with higher body mass index and longer disease duration. A study conducted on a cohort of 152 patients with myotonic dystrophy type 1 revealed that 32% of the study population reported fecal incontinence, which has changed their lifestyle (188). In one study, a gender-related prevalence and severity of gastrointestinal manifestations was documented in myotonic females versus males, who showed high serum GPT and γGT levels (177).
Patients with myotonic dystrophy type 1 have a clear age-related decline of cognitive functions as demonstrated through detailed neuropsychological studies, linguistic levels, praxis evaluations, and executive task evaluations. Cognitive deficits may include a variable combination of global cognitive impairment with involvement across different domains, including social cognition, memory, visuospatial functioning, and recognition of emotions conveyed by facial expression and body postures (157; 26; 117). The level of decline does not tend to correlate with either the number of CTG repeats or the severity of muscle weakness (147; 276; 68). These studies give support for proposals on a possible degenerative brain process (146).
Advanced MRI studies demonstrated across the brain a widespread white matter disruption and a multifocal gray matter volume loss by using various single MRI techniques, including diffusion tensor imaging and voxel-based morphometry, with correlations found between corresponding quantitative MRI parameters and triplet expansion, neuropsychological tests, and the severity of muscular involvement (11; 266; 146; 279; 277; 34; 278; 226; 218; 283; 254). Abnormal patterns of brain connectivity have also been reported in patients with myotonic dystrophy type 1 and have been demonstrated to account for patients’ personality traits (228; 224; 223). A work on 31 patients with myotonic dystrophy type 1 shows that in myotonic dystrophy type 1 a prominent deficit of decision-making, which is critical for succeeding in social and professional life, might be related to increased connectivity between ventral tegmental area (VTA) and brain areas critically involved in the reward/punishment system and social cognition. These findings suggest a potential dopaminergic function as potential target for pharmacological and nonpharmacological interventions in myotonic dystrophy type 1 (227). An abnormal cortical thickness associated with deficits in social cognition was found by MRI-3T in 30 patients with myotonic dystrophy type 1. This study confirms the presence of widespread brain changes associated with cognitive impairment in patients with myotonic dystrophy type 1 (225). Volumetric analysis by neuroimaging has revealed a reduced intracranial volume in myotonic dystrophy type 1 subjects compared with controls, and some morphological differences observed are associated with CTG repeat length, indicating plausible links to key myotonic dystrophy type 1 symptoms including cognitive deficits and excessive daytime somnolence (255).
Nevertheless, the mechanistic links between the genetic abnormalities, the pathomolecular mechanisms, and CNS involvement remain elusive at the moment. The identification of adequate biomarkers of brain involvement in myotonic dystrophy type 1 is of great importance as CNS outcomes measures are necessary for upcoming gene therapy clinical trials (67; 66; 137; 76). In one study, it has been observed that identification of outcome measures with good specificity for brain involvement in myotonic dystrophy type 1 is challenging because complex cognitive assessments may be compromised by peripheral muscle weakness, and self-reported questionnaires may be influenced by mood and insight. This highlights the need for further large, longitudinal studies to identify and validate objective measures, which may include imaging biomarkers and cognitive measures not influenced by motor speed (82). A paper shows a distinct pattern of brain atrophy and its progression over time of a decade in pediatric and adult onset in 21 patients with myotonic dystrophy type 1 (nine with pediatric onset, 12 with adult-late onset). These findings indicate a possible neurodevelopment origin of brain abnormalities in myotonic dystrophy type 1, along with the existence of additional neurodegenerative process (110).
Congenital myotonic dystrophy type 1. Myotonic dystrophy type 1 can also present with severe symptoms in newborns. This presentation is termed congenital myotonic dystrophy. Babies typically have generalized weakness and hypotonia, respiratory failure or insufficiency, feeding difficulty, and clubfoot deformity. Children with congenital myotonic dystrophy have 25% mortality rate in the first year if their disease is severe enough to warrant prolonged ventilation (27). Based on current information, congenital myotonic dystrophy occurs only in myotonic dystrophy type 1 and does not develop in myotonic dystrophy type 2. No clinical electrophysiologic myotonia is apparent in infants with congenital myotonic dystrophy. Children with congenital myotonic dystrophy demonstrate impaired orofacial functioning that affects communication and swallowing (19). A study shows a positive correlation between methylation, particularly upstream of the CTG repeat, and maternal inheritance in congenital myotonic dystrophy-affected individuals, indicating that DMPK methylation may account for the maternal bias for congenital myotonic dystrophy transmission (16; 113). Careful evaluation of the mother is helpful in establishing the diagnosis and demonstrating the presence of myotonia (percussion and grip) and weakness (long flexors of the fingers and dorsiflexors of the feet). Electromyographic study of the mother is also useful. Without a careful evaluation of the mother, the underlying cause of the illness in the infant may be missed, and an opportunity for future preventive therapy in both the infant and the mother will be lost. The cause of the hypotonia, feeding difficulty, and respiratory problems can be erroneously ascribed solely to perinatal factors, such as germinal matrix hemorrhage or eventration of the diaphragm, both of which occur in congenital myotonic dystrophy (84).
Babies with congenital myotonic dystrophy type 1 who survive the newborn period (and those who have less severe weakness as infants and elude diagnosis) typically have intellectual disability, learning disabilities, behavior problems, toilet training delay, and slowed motor development. In one study, detailing brain magnetic resonance imaging findings in neonates and children with congenital myotonic dystrophy type 1 and white matter abnormalities have even been found in the neonatal period (171). Fortunately, these children usually have improvement in all these limitations throughout childhood and during their early teens. Motor function improves during the first decade, is most pronounced during the first 6 years, reaches a plateau during adolescence, and starts to deteriorate in the beginning of the second decade (107). Unlike other neuromuscular diseases, older children (3 to 13 years old) with congenital myotonic dystrophy type 1 have a muscle mass closer to age-matched controls, consistent with the clinical profile of increasing strength in childhood (35). Later in the second or in the third decade patients with congenital myotonic dystrophy type 1 begin to show progressive weakness. A decrease in muscle strength has been observed to be more pronounced in the distal than in the proximal muscle groups (107). Superimposed on the skeletal muscle and brain manifestations that persist from congenital myotonic dystrophy type 1, patients also develop a clinical picture resembling that of typical patients with myotonic dystrophy type 1. A preliminary study found a prevalence of 25.8% congenital myotonic dystrophy pediatric patients with cardiac abnormalities (229).
Infantile and juvenile myotonic dystrophy type 1. Patients with symptoms occurring during the first decade will be assigned to infantile myotonic dystrophy type 1 whereas children with onset between 10 and 20 years of age will be classified as a juvenile form of myotonic dystrophy type 1 (57). Neurocognitive dysfunction is a hallmark of the childhood forms of myotonic dystrophy type 1 (infantile and juvenile onset forms), with age of onset after 1 year. This dysfunction is comprehensive of neuropsychiatric problems, such as predominantly inactive subtype of attention deficit hyperactive disorders or autism spectrum disorders (10; 56; 09). As with the congenital type, children with childhood-onset myotonic dystrophy type 1 (infantile and juvenile forms) will develop muscle symptoms at an older age, causing physical disabilities comparable to the severe adult-onset type 1 disease (58; 57). The juvenile form with age of onset after 11 years of age is characterized by school and behavioral problems and is often underrecognized. These childhood forms have to be considered as a CNS disease rather than a muscular or systemic disease (58). A neuroimaging study of brain in children and adolescents suggests a relationship between white matter damage and working memory (277).
Comprehensive studies of pediatric myotonic dystrophy type 1 have revealed that the clinical spectrum in childhood differs substantially from adults, with congenital myotonic dystrophy type 1 showing more severe health issues than childhood-onset/juvenile patients across multiple domains. Children with congenital and childhood-onset myotonic dystrophy type 1 demonstrate significant difficulties with intellectual function, fine motor skills, gastrointestinal function, and neuromuscular function, though the severity varies by age of onset. These findings emphasize the need for pediatric-focused, multidisciplinary management approaches that address the unique constellation of symptoms in younger patients (99; 115).
Late-onset myotonic dystrophy type 1. Myotonic dystrophy type 1 has the most variable clinical features of the myotonic dystrophies. It can present late in life with only cataracts, baldness, or occasionally heart block (late-onset oligosymptomatic form). These complaints are often attributed to aging or coronary disease. Muscle weakness in such cases is often mild or absent, and diagnosis of myotonic dystrophy type 1 may not be considered.
Gender is an unrecognized factor influencing myotonic dystrophy type 1 clinical profile and severity of the disease, however, in a cross-sectional analysis of main multiorgan clinical parameters in 1409 adult patients with myotonic dystrophy type 1 it has been reported that gender may impact on myotonic dystrophy type 1 phenotype and severity with worse socioeconomic consequences of the disease and higher morbidity and mortality in males (55). Men more frequently had (1) severe muscular disability with marked myotonia, muscle weakness, cardiac, and respiratory involvement; (2) developmental abnormalities with facial dysmorphism and cognitive impairment inferred from low educational levels and work in specialized environments; and (3) lonely life. Alternatively, women more frequently had cataracts, dysphagia, digestive tract dysfunction, incontinence, thyroid disorder, and obesity. Most differences were out of proportion to those observed in the general population. Compared to women, males were more affected in their social and economic life. In addition, they were more frequently hospitalized for cardiac problems and had a higher mortality rate. This study indicates that gender should be considered in the design of both stratified medical management and clinical trials.
Myotonic dystrophy type 2 (PROMM; proximal myotonic myopathy). There are no distinct clinical subgroups in myotonic dystrophy type 2, and clinical presentation comprises a continuum ranging from early adult-onset severe forms to very late-onset mild forms that are difficult to differentiate from normal aging. Only two cases of neonatal forms have been reported so far in the literature: one of these patients had reduced intrauterine movements and muscle hypotonia after birth (108), and the second had only congenital talipes equinovarus without any other clinical sign (196). At present, there is no evidence of a congenital or childhood form of myotonic dystrophy type 2 (47). Contrary to myotonic dystrophy type 1, in myotonic dystrophy type 2, anticipation has been described only with clinical criteria in a few families, but no longer CCTG repeat expansions in patients with earlier age at onset have been observed (217; 47; 219). Myotonic dystrophy type 2 typically presents in adulthood and has variable manifestations such as early onset cataracts (less than 50 years of age), various grip myotonias, thigh muscle stiffness, muscle pain, and weakness (in hip flexors, hip extensors, or long flexors of the fingers) (198; 241; 199; 144; 48; 135; 155; 47). These complaints often appear between 20 and 50 years of age. Posterior subscapular cataract before 50 years of age is a characteristic feature of myotonic dystrophy type 2, and early onset cataract can be a presenting feature of the disease, preceding all other symptoms (163). Pain is a common as well as a highly relevant problem for many patients with myotonic dystrophy type 2, with an estimated lifetime prevalence of 76% and a negative effect on quality of life (257). Patients and their care providers ascribe the symptoms to overuse of muscles, “pinched nerves,” “sciatica,” arthritis, or fibromyalgia. In comparison to other chronic muscle disorder patients, patients with proximal myotonic dystrophy type 2 more frequently describe a pain that is sometimes reported to be exercise-related, temperature-modulated, and palpation-induced (74). Younger patients may complain of stiffness or weakness when running up steps, whereas they infrequently complain of cramps. The muscle pain in myotonic dystrophy type 2 has no consistent relationship to exercise or to the severity of myotonia found on clinical examination. The pain, which tends to come and go without obvious cause, usually fluctuates in intensity and distribution over the limbs. It can last for days to weeks. This pain seems qualitatively different from the muscle and musculoskeletal pain that occurs in patients with myotonic dystrophy type 1. In a study on qualitative as well as quantitative aspects of pain in patients with myotonic dystrophy type 2, it has been observed that mechanical hyperalgesia is the main finding present in the rectus femoris, trapezius, and thenar, suggestive of at least a peripheral mechanism of pain (257). Pain appears to be most often located symmetrically in the proximal limbs (257). Myotonic dystrophy type 2 scored significantly lower than myotonic dystrophy type 1 on the bodily pain scale, indicating more body pain in myotonic dystrophy type 2. This finding has a high disease impact on physical as well as on mental health functioning (243), and on professional performance (235). A transcriptomic analysis performed on 12 muscle biopsy specimens obtained from patients with myotonic dystrophy type 2 has identified 14 muscle genes significantly up- or downregulated in myalgic patients compared to nonmyalgic myotonic dystrophy type 2 patients. These data support the idea that molecular changes in the muscles of patients with myotonic dystrophy type 2 are associated with muscle pain and suggest that muscle-specific molecular pathways might play a significant role in myalgia (151). Identifying the molecular source of pain pathophysiology should greatly facilitate the development of mechanism-based therapeutic strategies to treat musculoskeletal pain.
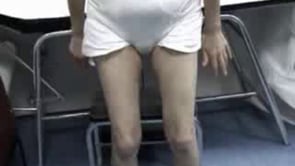
Early in the presentation of myotonic dystrophy type 2, there is only mild weakness of hip extension, thigh flexion, and finger flexion. Myotonia of grip and thigh muscle stiffness varies from minimal to moderate severity over days to weeks. Direct percussion of forearm extensor and thenar muscles is the most sensitive clinical test for myotonia in myotonic dystrophy type 2. Myotonia of grip is sometimes prominent and often has a jerky quality that seems to differ from that in myotonic dystrophy type 1 and the nondystrophic myotonias. Myotonia is often less apparent in myotonic dystrophy type 2 than in patients with myotonic dystrophy type 1.
In cases of late onset myotonic dystrophy type 2, myotonia may appear only on electromyographic testing after examination of several muscles (199; 47). Facial weakness is mild in myotonic dystrophy type 2 as is muscle wasting in the face and limbs. Weakness of neck flexors is frequent. Trouble arising from a squat is common, especially as the disease progresses.
Calf muscle hypertrophy occasionally is prominent (198; 241; 199).
Other manifestations, such as excessive sweating, hypogonadism, glucose intolerance, cardiac conductions disturbances, cognitive alterations, and neuropsychological alterations, may also occur and worsen over time (48; 142; 47; 143). Sleep complaints and breathing disorders are also frequent in myotonic dystrophy type 2 (203).
The cardiac manifestations of patients with myotonic dystrophy type 2 have been found to cause sudden death. In a population of 297 German myotonic dystrophy type 2 patients, four had cardiac sudden death before the age of 45. Of these four patients, three were asymptomatic, all had histologically proven dilated cardiomyopathy, and two had conduction system fibrosis (221).
A study on frequency and progression of cardiac and muscle involvement in a large cohort of patients with myotonic dystrophy type 2 demonstrated that the frequency and severity of cardiac involvement and muscle weakness are reduced in myotonic dystrophy type 2 compared to myotonic dystrophy type 1 and that progression is slower and less severe (212). Nevertheless, careful cardiac evaluation is recommended to identify patients at risk for potential cardiac major arrhythmia. A retrospective study comprised of 62 adult patients with myotonic dystrophy type 2 showed that cardiac conduction and rhythm defects are relatively rare in myotonic dystrophy type 2, although diastolic dysfunction is common, suggesting that regular ECG and echocardiography screening is needed in these patients (179). Cardiovascular magnetic resonance demonstrates that in patients with myotonic dystrophy type 2, subclinical myocardial injury was already detectable in preserved left ventricular ejection fraction. Moreover, extracellular volume was also increased in regions with no focal fibrosis and myocardial fibrosis was related to conduction abnormalities (216).
Patients with both myotonic dystrophy type 1 and type 2 have lower scores on frontal lobe functioning tests compared to controls and have an increased prevalence of avoidant personality disorders (143). In a study that analyzed personality patterns in a cohort of myotonic dystrophy type 1 and type 2 patients, no significant personality impairments have been observed in patients with myotonic dystrophy type 2, and the most common clinical symptoms observed in these patients were anxiety and somatization (168). In patients with type 2 disease, conventional brain MRI findings can be entirely normal. However, in advanced stages or more severe cases, diffuse white-matter changes can be present although be less pronounced than or different to that in myotonic dystrophy type 1 (201; 146). It has been reported that the main transcranial sonography finding in patients with myotonic dystrophy type 2 is brainstem raphe hypoechogenicity, which is associated with fatigue and excessive daytime sleepiness. In addition, substantia nigra hyperechogenicity and increased diameter of the third ventricle has been observed (192). A specific type of “avoidant” personality and a significant impairment in frontal lobe function (especially limited ability to perform executive functions) have been observed in myotonic dystrophy type 1 and type 2 patients, although these abnormalities were milder in patients with myotonic dystrophy type 2 (143). Similar observations have been reported in a study performed in a larger cohort of patients with myotonic dystrophy type 2 (182). There are clinical, neuropsychological, and neuroimaging data that support the hypothesis of central nervous system involvement also in myotonic dystrophy type 2 (140). In conclusion, cognitive manifestations are evident in myotonic dystrophy type 2 but milder when compared to patients with myotonic dystrophy type 1 (184).
As in myotonic dystrophy type 1, gastrointestinal manifestations are common in myotonic dystrophy type 2 patients, affecting their quality of life. A study on progression of gastrointestinal manifestations in these patients reports that during the 5 years of follow-up, the most common changes are the development of trouble swallowing and constipation and that female patients demonstrate a greater risk of a gastrointestinal manifestation (92). A relatively high frequency of cholecystectomy on average before 45 years of age is also reported (92).
It has been reported that hearing impairment is a frequent symptom in patients with myotonic dystrophy type 2 and that the sensorineural hearing impairment is located in the cochlea (256). This suggests it is important to perform audiometry when hearing impairment is suspected in order to propose early hearing rehabilitation with hearing aids when indicated.
A significant association between myotonic dystrophy type 2 and autoimmune diseases has been established, with autoimmune diseases present in up to 30% of patients with myotonic dystrophy type 2 (181). The most commonly associated autoimmune conditions include Hashimoto thyroiditis (18.1% prevalence), rheumatoid arthritis, diabetes mellitus type 1, systemic lupus erythematosus, Sjogren disease, localized scleroderma, psoriasis, and celiac disease. Mechanistic studies have revealed that expanded CCTG repeats lead to chronic endoplasmic reticulum stress through accumulation of aberrant RNA and toxic peptide products from repeat-associated non-ATG (RAN) translation. This chronic stress promotes mitochondrial DNA release into the cytoplasm, which is subsequently detected by the cGAS/STING pathway, triggering a type I interferon response that predisposes to autoimmunity (204). This discovery provides a mechanistic rationale for the increased prevalence of autoimmune diseases in myotonic dystrophy type 2 and suggests that screening for autoimmune conditions should be part of routine myotonic dystrophy type 2 management. The presence of autoimmune diseases and antinuclear antibodies may also lead to underdiagnosis of myotonic dystrophy type 2, as muscle symptoms may be attributed to inflammatory conditions.
It has been observed that metabolic syndrome is common in patients with myotonic dystrophy type 2 but not more frequent than in healthy subjects. However, treatment of metabolic disturbances may reduce cardiovascular complications and improve quality of life in patients with myotonic dystrophy type 2.
Body composition assessed by DEXA (dual-energy x-ray absorptiometry) reveals that patients with myotonic dystrophy type 1 and type 2 have similar total body mass, bone mineral content, fat mass, and lean tissue mass. Patients with myotonic dystrophy type 2 have less visceral fat deposition than those affected by myotonic dystrophy type 1. Also, right rib bone mineral density was lower in patients with myotonic dystrophy type 2 (180).
In a study conducted on a large cohort of 307 genetically confirmed myotonic dystrophy type 2 patients, a profound gender and age influence on the phenotype has emerged, emphasizing that female gender and aging may be associated with a higher disease burden (148). Indeed, it appears that with aging, there is a tendency towards the worsening of weakness, whereas myalgia and myotonia tend to decrease. Females seem to be more severely affected than men as they show more frequently muscle weakness, multisystem involvement, and need of using walking aids. This study suggests that these age- and gender-specific differences should be considered in diagnostics, management, and future clinical studies of myotonic dystrophy type 2.
Studies have identified parent-of-origin effects in myotonic dystrophy type 2 that influence disease presentation. Maternal inheritance of the myotonic dystrophy type 2 mutation correlates with earlier symptom onset, particularly within the first 3 decades of life, and increases the risk of cataracts and cardiovascular events as the initial myotonic dystrophy type 2 manifestations. This finding contrasts with the traditional view that myotonic dystrophy type 2 lacks significant anticipation phenomena and suggests that genetic counseling should consider parental inheritance patterns when discussing disease risks and expected clinical course (75).
In a large cohort of Serbian patients, 299 had clinical suspicion of myotonic dystrophy type 2 and 69 had genetic confirmation of myotonic dystrophy type 2. The following parameters were significant predictors for diagnosis of myotonic dystrophy type 2: presenile cataracts, myotonia on needle EMG, hand tremor, positive family history, and calf Hypertrophy (98). It has been also recommended to screen for various autoimmune disorders, such as Hashimoto thyroiditis, rheumatoid arthritis, systemic lupus, Sjogren disease, and localized scleroderma, in myotonic dystrophy type 2 (181). Approximately one quarter of patients with myotonic dystrophy type 2 have a significant cognitive impairment that interferes with their everyday functioning (185).
Myotonic dystrophy type 1 (myotonic dystrophy of Steinert). As a general rule patients having onset of symptoms in childhood and early adulthood develop more severe manifestations of myotonic dystrophy type 1 and have larger CTG repeat expansions in their leucocyte DNA (84). Myotonia is usually the first clinical sign in myotonic dystrophy type 1, and it usually becomes symptomatic before distal weakness becomes a problem. Progression of muscle weakness (facial, pharyngeal, and distal limb muscles) occurs gradually, typically over 2 or more decades. But some individuals develop more rapid muscle wasting in the presence of chronic medical problems, such as recurrent gastrointestinal dysfunction, diabetes, or prolonged infection. In one study, the average annual strength decline using manual muscle testing was .95% (134). Mildly affected individuals may have only minimal or no clinical myotonia and weakness and do not develop symptoms until late middle age. Early onset cataracts, baldness, or cardiac conduction disturbance may be their only signs of myotonic dystrophy type 1.
The life-threatening complications in myotonic dystrophy type 1 are primarily respiratory (respiratory failure) and cardiac (heart block, other serious conduction disturbances) (49; 133). These serious complications can occur in patients with and without severe muscle wasting and weakness. Sudden cardiac events appear to be related to age of the patient, duration of disease, and male gender, but not to CTG repeat expansion (207). However, in one study, the size of the CTG expansion has been associated with a worse long-term outcome, including a higher incidence of sudden and total death (41). Respiratory insufficiency is often precipitated by pneumonia or aspiration.
The predisposing factors involved in cardiac arrhythmia are less clear. There is agreement that cardiac complications in myotonic dystrophy type 1 develop more frequently in patients with more severe neuromuscular symptoms. One investigation found lower mean left ventricular contraction velocities in myotonic dystrophy type 1 patients with more severe neuromuscular involvement (65). There is conflicting evidence on the importance of CTG repeat length with regard to cardiac conduction, arrhythmias, and survival in patients with myotonic dystrophy type 1 (272). Reports show a correlation between conduction disturbance and the size of the CTG repeat (80). In particular, larger CTG expansion in the blood of patients with myotonic dystrophy type 1 is associated with the development of conduction system defect, left ventricular dysfunction, and supraventricular arrhythmias (41). Moreover, motor function and CTG repeat length have been found to be significantly correlated with left ventricular diastolic dysfunction in patients with myotonic dystrophy type 1 (164). Significantly higher levels of both serum cTnT and cTnI have been observed in patients with myotonic dystrophy type 1 and type 2 compared with controls, suggesting that these factors might represent a helpful serum biomarker to "predict" cardiac risk in myotonic dystrophy disease (251; 83). Moreover, patients with myotonic dystrophy type 1 and type 2 having ECG abnormalities show NT-proBNP serum levels higher than those seen in patients with normal ECG, suggesting that NT-pro-BNP levels may be considered to be used clinically to identify myotonic dystrophy patients at increased risk of developing myocardial conduction abnormalities (250). Although left ventricular dysfunction is a major prognostic determinant in myotonic dystrophy type 1, patients with preserved left ventricular ejection fraction exhibit significantly altered left ventricular global longitudinal strain as compared with controls (71). This parameter could be a predictive factor of sudden cardiac death, and myotonic dystrophy type 1 patients with impaired longitudinal strain could benefit from improved and appropriate therapeutic management. Ischemic stroke may occur in patients with myotonic dystrophy type 1 and is associated with atrial fibrillation (282). A high prevalence of myocardial fibrosis was demonstrated in a cardiovascular magnetic resonance (CMR) study on 52 myotonic dystrophy type 1 patients; however, no association between cardiovascular magnetic resonance myocardial fibrosis with late gadolinium enhancement (CMR-LGE) and surface conduction abnormality has been found (33). A study of 18 patients with myotonic dystrophy type 1 found a significant increase in global and septal cardiac extracellular volume on cardiac magnetic resonance imaging in patients with myotonic dystrophy type 1, suggesting the presence of increased cardiovascular risk, mainly due to cardiac fibrosis, even in the absence of overt cardiac pathology at other common cardiovascular exams, and of increased risk of early septal fibrosis, with important implications on the risk for fatal arrhythmias (01). In addition, women with myotonic dystrophy type 1 seem to be more prone to myocardial fibrosis. Physicians dealing with myotonic dystrophy type 1 may consider cardiac magnetic resonance imaging as a screening tool for the early identification of patients with increased cardiovascular risk.
Myotonic dystrophy type 2 (proximal myotonic myopathy). Overall, the prognosis for patients with myotonic dystrophy type 2 is more favorable than for individuals with myotonic dystrophy type 1 (199; 155; 47). Patients usually have a slower, less severe, and less widespread progression of muscle weakness and less muscle wasting. There does not seem to be a more severe phenotype associated with the homozygotic form of this disease (219). As in myotonic dystrophy type 1, patients with myotonic dystrophy type 2 who have an earlier onset of symptoms have an earlier onset of myotonia and weakness (217). The natural history of myotonic dystrophy type 2 remains to be fully defined, but present information indicates that most patients have a normal lifespan. Respiratory failure, hypersomnia, and recurrent aspiration or pneumonia are not common in myotonic dystrophy type 2 (244). Cardiac conduction disturbances occur (155), but they are less frequent compared to myotonic dystrophy type 1 (141; 221). An investigation using a variety of standard tests of autonomic function (response to Valsalva maneuver, deep breathing, change in posture, grip, analysis of heart rate variability) reveals no major abnormalities in patients with myotonic dystrophy type 2 myopathy (62).
Myotonic dystrophy type 1 (myotonic dystrophy of Steinert). A 41-year-old man presented to our clinic for difficulty with manipulating objects and with gait problems that had started about 4 years before the evaluation. More specifically, he complained of falls from tripping on the ground (for example on carpet edges). Moreover, he reported that since he was 16 years old, he noticed stiffness in his hands after grip that was worsening over the years. His mother died due to sudden death at the age of 50 years. Past history and review of systems were remarkable for diabetes and excessive daytime sleepiness. He underwent a study that revealed the presence of sleep-disordered breathing; therefore, he started to use assisted nocturnal ventilation with a partial benefit on daytime sleepiness. His neurologic examination revealed baldness, mild facial weakness, and tongue myotonia. Moreover, there was evidence of weakness of neck, deep finger, and of the feet muscles. Action and percussion myotonia was present in both hands with a warmup phenomenon. Electromyography showed myotonic discharges in abductor pollicis brevis and subsequent genetic analysis confirmed the abnormal expansion of CTG repeats (range 600 to 800) in the 19q13.3 DMPK gene. A check of the major system involved in myotonic dystrophy type 1 allowed the diagnosis of an initial cataract and a bundle branch block. He started therapy with mexiletine 200 mg three times a day with improvement of stiffness, and he began to use ankle-foot orthoses to help gait and prevent falls. In the follow-up, an episode of loss of consciousness occurred. The cardiologic evaluation and the electrocardiographic monitoring showed an impairment of conduction parameters; thus, the patient underwent an artificial pacemaker implantation with improvement of the symptoms.
Myotonic dystrophy type 2 (proximal myotonic myopathy). A 59-year-old woman with no known family history of neuromuscular disease presented to the clinic for a progressive impairment of walking. She said that she walked “dragging” her legs and she found it difficult to stand up from a chair. She did not complain of any symptoms in the upper limbs except for a mild and proximal stiffness in both the arms, more severe on the right side. Her symptoms started almost 30 years earlier and they worsened after she had been pregnant. She first experienced difficulty climbing up stairs and getting on the bus unless she had something to hold on to. Later she had several falls and complained that it was difficult to walk. The clinical neurologic examination showed proximal weakness but no action nor percussion myotonia were detected. Past history and systems review were remarkable for cholecystectomy and appendicectomy, bilateral cervical lymphadenopathy of unknown origin, cataract in both eyes, thyroid nodules, struma, two episodes of loss of consciousness, and several lipothymic episodes, idiopathic artery hypertension, and right bundle branch block. Family history showed an early onset cataract and difficulty climbing stairs in her father. She had never presented to a neurologist until 57 years of age when blood tests performed after a treatment with atorvastatin showed high CPK levels of serum creatine phosphokinase. Abnormal values persisted after the treatment was discontinued. She underwent an electromyography that showed myotonic discharges in both upper and lower limbs and a motor polyneuropathy. Because genetic analysis was negative for myotonic dystrophy type 1 a muscle biopsy was performed. The biopsy showed a myopathic pattern with preferential type II fibers atrophy, abnormal number of central nuclei centralization, and several nuclear clumps; the fluorescence in situ hybridization with (CAGG)5 probe on muscle biopsy sections detected nuclear foci confirming the diagnosis of myotonic dystrophy type 2. Subsequent genetic analysis showed an abnormal tetranucleotide CCTG expansion (range 5000 to 6000).
Tissue-specific pathogenesis and differences between myotonic dystrophy types 1 and 2. Although both myotonic dystrophy types 1 and 2 share the core RNA toxicity mechanism, important differences exist. In myotonic dystrophy type 2, the RNA-binding protein RBFOX1 specifically binds to CCUG but not CUG repeats, competing with MBNL1 for CCUG binding and partially releasing MBNL1 from sequestration. This may explain the generally milder phenotype observed in myotonic dystrophy type 2 compared to myotonic dystrophy type 1 (222; 200).
Studies have also highlighted significant tissue-specific differences in pathogenesis. In brain tissue, astrocytes exhibit more severe RNA toxicity and spliceopathy than neurons, with extensive missplicing affecting genes regulating cell adhesion, cytoskeleton, and morphogenesis (54). This finding challenges the traditional neuron-centric view of myotonic dystrophy brain pathology and suggests that glial dysfunction may play a central role.
Myotonic dystrophy type 1 results from an unstable CTG repeat expansion in the 3’ non-coding region of the gene for a serine and threonine kinase (myotonic dystrophy type 1 protein kinase, DMPK) on chromosome 19q13.3 (24; 64; 128). The cause of the unstable CTG repeat expansion is unknown; however, it is thought to occur during gametogenesis and is more extensive when coming from a female carrier (53). CCG, CTC, and GGC repetitions interspersed within the 3′ or 5’ end of the CTG expansion have been reported in myotonic dystrophy type 1 patients with a prevalence of 3% to 5% (23; 187). Direct observation of de novo interruptions across one or several generations in myotonic dystrophy type 1 families have been reported (23; 22; 187; 45). Interruptions have also been detected at the 5’ end of the CTG array (22). Interruptions within the DMPK expanded alleles might have a stabilizing effect on the mutational dynamics and can modulate the severity of symptoms in patients with myotonic dystrophy type 1 (23; 22; 187; 45). A study has provided firm evidence that various types and patterns of repeat interruptions confer stability to DMPK expansions in somatic cells, predisposing patients with myotonic dystrophy type 1 to develop disease later than average. These data support the assumption that repeat interruptions can act as a genetic modifier of myotonic dystrophy type 1 phenotype (186; 145). Because patients with variant repeats may have unusually mild symptoms, identification of these individuals has important implications for genetic counselling and patient stratification in clinical trials. In one study, two myotonic dystrophy type 1 families with apparent contractions and no worsening of myotonic dystrophy type 1 symptoms in two and three successive maternal transmissions have been described (246). In these families, patients with multiple CCG interruptions or a new unique CAG interruption at the 5′ end of the CTG repeat expansion have been detected, suggesting that these two types of interruptions are associated with successive maternal CTG repeat contractions and low somatic instability. Several observations raise the possibility that environmental or external factors may influence theevelopent of pathologic enlargement of CTG repeats at the myotonic dystrophy type 1 locus (189; 281).
In conclusion, studies on patients with variant repeats indicate their stabilizing effect on DMPK expansion because no congenital cases have been described and the age of onset is later than expected (183).
Normal individuals have five to 37 CTG repeats in leucocyte DNA. Repeat lengths of 38 to 50 are considered premutation alleles, whereas 51 to 100 repeats are protomutations, both of which show increased instability toward expansions. Carriers of premutations or protomutations present no or few mild symptoms, such as cataracts. Research suggests that premutations in the upper normal range of CTG sizes become pathologically enlarged within a few generations (132; 02). Patients with myotonic dystrophy type 1 have a wide range of CTG repeat sizes; more severely affected individuals have repeat sizes in the thousands (84). Late onset cases may have minimal skeletal muscle manifestations of myotonic dystrophy type 1 and these individuals typically have small repeat expansions (less than 100 CTG repeats) (84). Those patients with onset in the second or third decades (early middle-life) in general have more prominent symptoms on initial examination and have larger expansion sizes that extend over a wide range. Infants with congenital myotonic dystrophy typically have expansions of more than 800 CTG repeats (84). Strict correlation between genotype and phenotype appeared to be unreliable because there is overlap in the CTG repeat enlargements between groups with classical adult onset and childhood onset myotonic dystrophy type 1 (84). Moreover, the dynamic nature of the expansion makes correlation of genotype and phenotype even more challenging because, over the years, the CTG repeat size in circulating leucocytes from the same individual increases (84). One study attempted to correlate CTG repeat length with progressive myotonic dystrophy type 1 phenotypes; CTG repeat tract length has been measured and screened for interrupting variant repeats in 192 participants from a well-characterized Canadian cohort. Using statistical models that include confounding factors, the analysis has revealed a strong correlation between myotonic dystrophy type 1 genotype and respiratory function and skeletal muscle power (161). To determine the suitability of saliva DNA as a source for myotonic dystrophy type 1 genotyping, small pool-PCR has been used to perform a detailed quantitative study of the somatic mutational dynamics of the CTG repeat in saliva and blood DNA from 40 myotonic dystrophy type 1 patients. The modal allele length in saliva resulted only moderately higher in saliva, and data indicate that saliva constitutes an accessible, noninvasive, and suitable DNA sample source for performing genetic studies in myotonic dystrophy type 1 (44).
Tissue mosaicism adds a further challenge to genotype-phenotype correlations. The repeat size is larger in DNA from skeletal and cardiac muscle compared to that from leucocytes (84), and there is no reliable way to predict the size of the repeat in a given target tissue based on its size in circulating leucocytes. However, it is tempting to propose that the tendency of the myotonic dystrophy type 1 gene to expand and its greater size in certain target tissues exert a determining influence in the variation in clinical severity and in the distribution of manifestations that occurs in individual patients with myotonic dystrophy type 1. However, the general rule remains that the size of the repeat expansion in circulating leucocyte DNA correlates with rate of progression and severity of muscle manifestations. One example of the relationship between CTG repeat size and disease severity in myotonic dystrophy is the phenomenon of anticipation, best exemplified by congenital myotonic dystrophy (84). Anticipation is the earlier onset of more severe clinical manifestations in offspring of affected individuals. In congenital myotonic dystrophy type 1 there is a dramatic enlargement in the size of the CTG repeat with severe symptoms in the infant. Evidence suggests that CTG repeat enlargement in the myotonic dystrophy type 1 gene occurs to a greater degree in the eggs than in the sperm of affected individuals, and that this accounts for the almost exclusive restriction of cases of congenital myotonic dystrophy to children of mothers with myotonic dystrophy type 1 (84). However, the size of the CTG repeat usually increases progressively in successive generations in the eggs of females and the sperm of males who carry the myotonic dystrophy type 1 mutation. Studies of male mice in a transgenic mouse model of myotonic dystrophy type 1 (CTG expansions of 300 to 338 repeats) have shown that germinal expansions occur in spermatogonia and that the length of the CTG repeat does not change between spermatogonia and mature spermatozoa. The size of the CTG repeat expansions in sperm increases with age and probably results from the accumulation of expansions over lifetime as spermatogonia undergo mitotic divisions or as DNA repair mechanisms operate repeatedly on these cells. Further study of these mice indicate that the function of the DNA mismatch repair enzyme MSH2 (muts (E coli) homolog 2; colon cancer gene) is necessary to have the repeat enlargement and suggests that the mechanism underlying the CTG repeat instability is a meiosis-independent event (215). The important idea these results convey is that the mechanisms responsible for instability in germ line and somatic tissues may be identical. Given the important role that DNA mismatch repair genes play in mediating expansions in mouse models, modifier gene effects with 13 DNA mismatch gene polymorphisms (one each in MSH2, PMS2, MSH6, and MLH1; and nine in MSH3) have been tested. The data obtained suggest that MSH3 is a key player in generating somatic variation in patients with myotonic dystrophy type 1 and further highlight MSH3 as a potential therapeutic target (149).
Myotonic dystrophy type 2 results from an unstable tetranucleotide repeat expansion, CCTG in intron 1 of the nucleic acid-binding protein (CNBP) gene (previously known as zinc finger 9 gene, ZNF9) on chromosome 3q21 (194; 121; 15; 120). The cause for the unstable expansion is unknown. In contrast to the (CTG)n repeat in myotonic dystrophy type 1, in myotonic dystrophy type 2/proximal myotonic myopathy the (CCTG)n repeat is a part of the complex repetitive motif (TG)n(TCTG)n(CCTG)n, and the (CCTG)n repeat tract is generally interrupted in healthy range alleles by one or more GCTG, TCTG, or ACTG motifs, whereas it is typically uninterrupted in the expanded alleles (121; 15; 14).
The size of the (CCTG)n repeat Is below 30 repeats in normal individuals, whereas the range of expansion sizes in patients with myotonic dystrophy type 2 is huge (14). The smallest reported mutations vary between 55 and 75 CCTG (121; 14) and the largest expansions have been measured to be about 11,000 repeats (121). The expanded myotonic dystrophy type 2 alleles show marked somatic instability, with significant increase in length over time (121; 47), thus, the threshold size of the disease-causing mutation remains to be determined. The size of the CCTG repeat appears to increase over time in the same individual, and, like myotonic dystrophy type 1, this is a dynamic gene defect (47). These two genetic findings complicate the correlation between genotype and phenotype. The gene mutation responsible for myotonic dystrophy type 2 appears to have arisen from a Northern European founder (15; 120), but single-kindred Afghan (219) and Japanese (208) cases have been described. Both mutations are believed to have occurred after migration out of Africa, between 120,000 and 60,000 years ago. The age of the myotonic dystrophy type 2 founder mutation has been estimated at 4000 to 12000 years (about 200 to 540 generations) (15). The molecular pathomechanism leading to the manifestations of myotonic dystrophy type 2 is felt to be similar to that in myotonic dystrophy type 1 and relates to a toxic effect of the abnormally expanded RNA that accumulates in the muscle nuclei (130; 193; 245; 129; 32).
RNA-mediated toxicity and spliceopathy: the central pathogenic mechanism. The myotonic dystrophies represent prototypical RNA-dominant disorders in which pathogenesis stems from a toxic gain-of-function mechanism mediated by mutant RNA transcripts rather than loss-of-protein function (156; 200). The expanded CUG (myotonic dystrophy type 1) and CCUG (myotonic dystrophy type 2) RNA repeats form stable secondary hairpin structures that accumulate as ribonuclear foci in the nuclei of affected tissues (156). These RNA aggregates exert their pathogenic effects through sequestration of critical RNA-binding proteins, most notably the muscleblind-like (MBNL) family of splicing regulators.
It is now clear that the gain-of-function RNA mechanism is the predominant cause of myotonic dystrophy pathogenesis in which the CUG and CCUG repeats alter cellular function of several RNA-binding proteins. It has been demonstrated that mutant CUG and CCUG RNAs are very stable due to a deficiency of RNA helicase p68 (101). The expanded CUG and CCUG RNA form hairpins, imperfect double-stranded structures that lead to dysregulation of two important RNA-binding proteins: muscleblind like 1 (MBNL1) and CUG-binding protein 1 (CUGBP1), which are antagonist regulators of alternative splicing of various genes (239; 46). Data demonstrate that MBNL1-containing foci in myotonic dystrophy type 2 cells also sequester snRNPs and hnRNPs, splicing factors involved in the early phases of transcript processing (60; 174), thus, strengthening the hypothesis that a general alteration of pre-mRNA posttranscriptional pathway could be at the basis of the multifactorial phenotype of myotonic dystrophy type 2 patients. In myotonic dystrophies, the downregulation of MBNL1, due to its sequestration in mutant RNA foci, and the upregulation of CUGBP1 result in abnormal expression of embryonic isoforms in adult tissues. The alteration of pre-mRNA processing strengthens the hypothesis of a spliceopathy that leads to an expression of isoforms inadequate for a particular tissue or developmental stage (160; 138). The sequestration of MBNL proteins by expanded RNA repeats leads to their functional depletion and dysregulates their RNA targets. This is further compounded by the upregulation of CUG-BP and ETR-3-like factors (CELF) family proteins, particularly CELF1, through PKC-mediated hyperphosphorylation and stabilization (109; 200). The resulting imbalance between MBNL and CELF proteins causes a pathognomonic reversion from adult to embryonic alternative splicing patterns, leading to expression of fetal protein isoforms in adult tissues – the hallmark "spliceopathy" of myotonic dystrophies (127; 156).
This spliceopathy affects hundreds of transcripts critical for normal cellular function. Key affected targets include the insulin receptor (INSR), chloride channel 1 (CLCN1), cardiac troponin T (TNNT2), dystrophin (DMD), and sodium channel SCN5A, directly linking the molecular defects to clinical manifestations such as insulin resistance, myotonia, cardiac conduction abnormalities, and muscle weakness (238; 156).
Beyond spliceopathy: expanding pathogenic mechanisms. Investigations have revealed that RNA toxicity in myotonic dystrophies extends beyond alternative splicing dysregulation. The pathogenic mechanisms now encompass the following.
Dysregulated signal transduction pathways. The expanded RNA disrupts multiple kinase networks, including PKC, GSK3β, AKT, AMPK, and PKR, leading to altered protein translation, impaired autophagy, and activation of inflammatory pathways, such as NF-κB signaling and IL-6 upregulation (156).
Cellular senescence activation. Multiple studies have demonstrated that myotonic dystrophy cells exhibit features of premature cellular senescence. Expanded CUG RNA induces mitochondrial dysfunction, leading to excess reactive oxygen species production, DNA damage, and activation of cell cycle checkpoint regulators (p53, p21, p16) and senescence-associated secretory phenotype markers (72; 86; 156).
microRNA dysregulation. miRNAs are small, noncoding RNA modulating gene expression at the posttranscriptional level, and their expression and intracellular distribution are deregulated in many human diseases, including muscular dystrophies (59; 77; 69; 173; 78). Both in myotonic dystrophy type 1 and in myotonic dystrophy type 2 it has been demonstrated that the highly regulated pathways of miRNA are altered in skeletal muscle, potentially contributing to myotonic dystrophy pathogenetic mechanisms (69; 173; 78). The misregulation of miR-1 observed in the hearts of people with myotonic dystrophy types 1 and 2 may contribute to the cardiac dysfunctions observed in patients through increased expression of extracellular matrix proteins (195). The significant reduction of miR-1 in myotonic dystrophy cardiac muscle appears to be caused by the expression of expanded CUG repeats and subsequent MBNL1 nuclear sequestration (195). Interestingly, several miRNAs have been found deregulated in peripheral blood plasma from patients affected by myotonic dystrophy type 1 (175; 176; 105; 106). The levels of these miRNAs, alone or in combination, correlate with both skeletal muscle strength and creatine kinase (176). However, in a study on the expression of 175 known serum miRNAs in myotonic dystrophy type 1 blood samples, none of the miRNA analyzed show consistent differences in serum expression between patients and controls and, thus, do not result in useful serum biomarkers for myotonic dystrophy type 1 (61). A deregulation of microRNA in skeletal muscle and plasma from patients with myotonic dystrophy type 2 has been also reported (78; 176). The identification of minimally invasive analytical biomarkers for myotonic dystrophies and the established potential of circulating miRNAs as prognostic and diagnostic biomarkers are particularly important to monitor myotonic dystrophies progression and the effectiveness of new drug treatments. Although it seems clear that miRNA pathway is disrupted in myotonic dystrophies, the functional implications of this dysregulation require further investigation. To identify “functional” miRNAs actually engaged in mRNA/target inhibitions relevant for myotonic dystrophy type 1 disease mechanisms, the RISC-associated RNAs have been analyzed in muscle biopsies from patients with myotonic dystrophy type 1. Among the 24 miRNA/mRNA correlations identified, functionally relevant miRNA/mRNA interactions have been identified in skeletal muscles, and in particular, the dysfunction of couple miR-29c/ASB2 has been demonstrated (28). miR-23b and miR-218 have been identified as downregulators of MBNL1 and MBNL2 in HeLa cells. Antagonists of these miRNAs enhance MBNL protein levels, rescue pathogenic missplicing events in myotonic dystrophy type 1 myoblasts, and improve splicing alterations, histopathology, and myotonia in the HSALR mice. These data provide evidence for therapeutic blocking of the miRNAs that control muscleblind-like protein expression in myotonic dystrophy (36). A pilot study indicates that another class of noncoding RNAs, circular RNAs (circRNAs), are dysregulated in myotonic dystrophy type 1 skeletal muscle. Out of nine tested, four transcripts showed an increased circular fraction (CDYL, HIPK3, RTN4_03, and ZNF609), and their circular fraction values correlated with skeletal muscle strength and with splicing biomarkers of disease severity, and they displayed higher values in more severely affected patients. Increased circular fractions of RTN4_03 and ZNF609 have also been observed in differentiated myogenic cell lines from patients with myotonic dystrophy type 1 (260).
Repeat-associated non-ATG (RAN) translation. A novel molecular mechanism that may contribute to the pathogenesis of myotonic dystrophies has been described by Zu and collaborators (285). RNA transcripts containing expanded CAG or CUG repeats can be translated in the absence of a starting ATG, and this noncanonical translation, called repeat associated non-ATG translation (RAN-translation), occurs across expanded repeats in all reading frames to produce potentially toxic homopolymeric proteins (169; 285). RAN-translation results in the accumulation of polyglutamine (polyGln) nuclear aggregates in myotonic dystrophy type 1 mouse tissues and human cardiac myocytes, leukocytes, and myoblasts not detectable in control tissues (285). RAN-translation products appear to be toxic to cells and may contribute to myotonic dystrophy type 1 pathology. It has been demonstrated that RAN-translation also occurs across transcripts containing the myotonic dystrophy type 2 CCUG or CAGG expansion mutation, producing tetra-repeat expansion proteins with a repeating Leu-Pro-Ala-Cys (LPAC) or Gln-Ala-Gly-Arg (QAGR) motif (284). Both LPAC and QAGR RAN proteins accumulate in myotonic dystrophy type 2 human autopsy brains in distinct patterns. For LPAC, cytoplasmic aggregates are found in neurons, astrocytes, and glia in the gray matter regions of the brain. In contrast, QAGR RAN protein accumulation, which is nuclear, is found primarily in oligodendrocytes located in white matter regions of the brain. Moreover, it has been evidenced that RAN protein accumulation can be modulated by MBNL1 levels and that nuclear sequestration of CCUG, CUG, or CAG RNAs decrease steady-state levels of RAN proteins (284). These data suggest that RAN-translation may be common to both myotonic dystrophy type 1 and type 2 and that RAN proteins may be responsible for some of the CNS features of myotonic dystrophies.
Stress granule dysfunction. Altered response to cellular stress and abnormal clearance of stress granules caused by MBNL1/CELF1 disruption represent additional pathogenic elements contributing to transcriptome deregulation and cellular dysfunction (81; 200).
Epigenetic modifications. It has been hypothesized that DNA methylation contributes to disease development and progression and explains part of the phenotypic variability reported in myotonic dystrophy type 1 (126; 16). It has been observed that DNA methylation at the DMPK gene locus contributes to variability of both muscular strengths and respiratory profiles in myotonic dystrophy type 1 and that these associations are independent of the CTG repeat length. These results, thus, provide evidence that measuring DNA methylation might help to predict progression of the disease and to establish a more reliable prognosis for those patients (116).
To help understand the molecular mechanism and facilitate research into myotonic dystrophy type 1, 120 RNASeq transcriptomes have been generated from skeletal and heart muscle derived from healthy and myotonic dystrophy type 1 biopsies and autopsies. Splicing and gene expression, tissue-specific changes in RNA processing, and uncovered transcriptome changes have been analyzed. Moreover, a web resource has been created at to be maximally accessible to investigators who would like to rapidly browse RNAseq read coverage across the genome and access previously computed gene expression (265). The web resource can be accessed at the following site: http://DMseq.org .
Another open question in the field of myotonic dystrophies is to clarify the pathomechanisms underlying the phenotypic differences between myotonic dystrophy type 1 and type 2. Clinical signs in myotonic dystrophy type 1 and type 2 are similar, but there are some distinguishing features: myotonic dystrophy type 2 is generally less severe and lacks a prevalent congenital form. This suggests that other cellular and molecular pathways are involved besides the shared toxic-RNA gain of function hypothesized. An important step forward in understanding the differences between myotonic dystrophy type 1 and type 2 has been made. Indeed, rbFOX1 has been reported as a novel RNA binding protein that specifically binds to expanded CCUG repeats, but not to expanded CUG repeats. rbFOX1 is enriched in skeletal muscle, heart, and brain and is involved in the regulation of various aspects of mRNA metabolism. In the study, it has been demonstrated that rbFOX1 co-localizes with CCUG RNA foci in muscle cells and skeletal muscle tissues of individuals with myotonic dystrophy type 2, but not with CUG RNA foci in myotonic dystrophy type 1 samples (222). Interestingly, rbFOX1 competes with MBNL1 for binding to CCUG expanded repeats, and its overexpression partly releases MBNL1 from sequestration within CCUG RNA foci in muscle cells. Furthermore, expression of rbFOX1 corrects alternative splicing alterations and rescues muscle atrophy, climbing, and flying defects caused by expression of expanded CCUG repeats in a Drosophila model of myotonic dystrophy type 2 (222).
Several studies have revealed a role for CNBP in myotonic dystrophy type 2. CNBP deletion in several animal models results in severe brain and muscle phenotypes and other abnormalities similar to those seen in myotonic dystrophy type 2 (39; 03; 40; 269). These defects can be rescued by reintroduction of wild-type levels of CNBP, suggesting that a loss of CNBP function likely contributes to myotonic dystrophy type 2. Two reports using cell models describe a reduction of the rate of protein translation in myotonic dystrophy type 2 muscle cells due to a decrease of CNBP protein levels in myotonic dystrophy type 2 myoblasts and adult muscle (96) and due to the interaction of CCUG repeats with cytoplasmic multiprotein complexes, which dysregulate translation and degradation of proteins in patients (209). Sammons and colleagues report that CNBP activity is reduced in myotonic dystrophy type 2 human myoblasts leading to a decrease in CNBP activation of IRES-mediated translation of the human ODC and suggest that CNBP activity may contribute to myotonic dystrophy type 2 phenotype (211). Moreover, the reduction of CNBP expression has been reported in myotonic dystrophy type 2 muscle biopsies but not in myotonic dystrophy type 1, thus, explaining some of the phenotypic disparities between both types of myotonic dystrophies (29). Taken together, these data suggest that myotonic dystrophy type 2 pathology may be due to a combination of an RNA gain of function and CNBP loss of function.
The role of CUGBP1 in myotonic dystrophy type 2 is particularly intriguing, with contradictory results being reported (119; 172; 209; 29). Cardani and colleagues demonstrated that this protein is overexpressed in muscle biopsies from patients affected by the adult classical form of myotonic dystrophy type 1 but not in muscle from myotonic dystrophy type 2 patients, suggesting that sequestration of MBNL1 evidently has a central role in splicing misregulation in both types of myotonic dystrophies, whereas CUGBP1 overexpression might be an additional pathogenic mechanism in myotonic dystrophy type 1 not shared by myotonic dystrophy type 2 (29). However, it has been shown that that MBNL1 overexpression in a mouse model of RNA toxicity (DM200) is not effective in reversing myotonic dystrophy type 1 phenotypes such as myotonia and cardiac conduction abnormalities. Also, the mice do not show improvement in function assays such as grip strength or treadmill running, and MBNL1 overexpression notably increases muscle histopathology and results in variable rescue of a number of splicing targets (280).
A novel pathogenic mechanism identified in myotonic dystrophy type 2 involves chronic endoplasmic reticulum stress caused by expanded CCTG repeats and RAN translation products. This chronic stress leads to mitochondrial DNA release into the cytoplasm, which is detected by the cGAS/STING pathway, triggering a type I interferon response that predisposes to autoimmunity. This mechanism provides a molecular explanation for the increased prevalence of autoimmune diseases observed in patients with myotonic dystrophy type 2 (204; 200).
Myotonic dystrophy type 1 and 2 are degenerative neuromuscular disorders characterized by progressive skeletal muscle weakness and atrophy. However, the cause for the muscle weakness and wasting in myotonic dystrophies is still unclear. Patients with myotonic dystrophy type 2, in contrast to patients with classic myotonic dystrophy type 1, usually have only mild muscle wasting. However, there is an uncommon adult-onset variant of myotonic dystrophy type 2 termed proximal myotonic dystrophy, which causes severe wasting of proximal arm and thigh muscles as the illness progresses (248). Moreover, there is still no mechanistic explanation for the histopathological features characteristic of myotonic dystrophies, which include fiber atrophy-hypertrophy, increased number of central nuclei, and presence of fibers with nuclear clumps. It is known that most muscle symptoms can be explained by pathomechanistic effects of toxic RNA and spliceopathy. However, aberrations in DNA replication and transcription of the myotonic dystrophy loci or in protein translation and proteome homeostasis could also affect the control of proliferation and differentiation of muscle progenitor cells or the maintenance and physiological integrity of muscle fibers during a patient’s lifetime (08). Vihola and collaborators investigated the molecular basis of muscle weakness and wasting and the differences in muscle phenotype between myotonic dystrophy type 1 and type 2. They identified differences in muscle gene expression and splicing between myotonic dystrophy type 1 and type 2 patients. In particular, the aberrant splicing isoform of TNNT3 is twice as frequent in myotonic dystrophy type 2 compared to myotonic dystrophy type 1. Moreover, in myotonic dystrophy type 1 and type 2, a different protein expression pattern has been found in the highly atrophic fibers (258). Potential molecular mechanisms underlying skeletal muscle loss has been studied in a skeletal muscle-specific mouse model of myotonic dystrophy type 1 (CUG960) showing progressive skeletal muscle wasting. RNA-seq and protein array analysis indicate that the balance between anabolic and catabolic pathways that normally regulate muscle mass may be disrupted by deregulation of PDGFRβ signaling and PI3K/AKT and AMPK pathways. Renna and colleagues reported that IRS1-Akt/PKB and Ras-ERK pathway are impaired in myotonic dystrophy skeletal muscle, leading to a lower activation of mTOR and to an increase in MuRF1 and Atrogin-1/MAFbx expression, possibly explaining myotonic dystrophy skeletal muscle fiber atrophy (197). Similar changes has been detected in skeletal muscle of patients with myotonic dystrophy type 1 (150). A role of disruption of PI3K/AKT pathway in myotonic dystrophy type 1 muscle pathology has also been reported by Timchenko and collaborators (101; 268). Using the HSALR mouse model of myotonic dystrophy type 1, they observed an increase of active GSK3β in skeletal muscle of mice, prior to the development of skeletal muscle weakness. Inhibition of GSK3β reduced muscle weakness and myotonia, corrected atrophy, normal fiber size, and reduced central nuclei (101; 268). Correction of GSK3β with small molecule inhibitor of GSK3, Tideglusib, not only normalizes the GSK3β-CUGBP1 pathway but also reduces the mutant DMPK mRNA in myoblasts from patients with adult and congenital myotonic dystrophy type 1. Moreover, the correction of this pathway with Tideglusib increases postnatal survival and improves growth and neuromotor activity of DMSXL mice. Concerning myotonic dystrophy type 2, skeletal muscle phenotype has been studied in heterozygous Cnbp KO mice and in human muscle samples (267). The study demonstrates that CNBP protein expression is reduced in cytoplasm of myotonic dystrophy type 2 muscle fibers, and it is predominantly localized at membrane level where its interaction with α-dystroglycan is increased compared to controls. These findings suggest that alterations of CNBP in myotonic dystrophy type 2 might cause muscle atrophy, not only via misregulation of mRNA but also via protein-protein interactions with membrane proteins affecting myofiber membrane function (267).
Brain function is compromised in myotonic dystrophies, but the underlying mechanisms are not fully understood. By using transgenic mouse models, it has been found that the major pathological changes in the myotonic dystrophy brain result from disruption of the MBNL-2-mediated developmental splicing program (38). A study on myotonic dystrophy type 1 brain mechanisms conducted on DMSXL mouse model and human patients demonstrates glial molecular abnormalities affecting neuronal activity through neuroglial miscommunication (230). Moreover, a global proteomics approach revealed downregulation of the GLT1 glutamate transporter, providing exciting therapeutic perspectives through the modulation of GLT1 levels and glutamate signalling (230).
Future studies using IPSC-derived patient cells should provide additional insights into cellular pathways affected in myotonic dystrophy type 1 and myotonic dystrophy type 2 (233).
These cell systems will also be useful for testing the effects of various gene editing approaches including use of CRISPR/Cas 9 (clustered regularly interspaced short palindromic repeats) (125).
Myotonic dystrophy type 1 is the most prevalent form of adult muscular dystrophy. Although traditionally estimated at 5 to 20 per 100,000 (84), recent population-based studies using genetic screening have revealed substantially higher prevalence rates. Newborn screening programs have identified pathogenic CTG expansions in approximately one in 2100 individuals, whereas genome-wide analyses have reported frequencies as high as one in 1786, indicating that the actual prevalence of myotonic dystrophy type 1 may be significantly underestimated (100; 97). Certain regions maintain exceptionally high prevalence, such as Saguenay-Lac St Jean region of Quebec, Canada (162 per 100,000), Norbotten in Northern Sweden, the Basque region of Spain, and Istria region of Croatia (84).
Myotonic dystrophy type 2 appears to have a lower prevalence than myotonic dystrophy type 1 and primarily affects populations with a Northern European heritage (120). For myotonic dystrophy type 2, there are currently no established prevalence estimates; myotonic dystrophy type 2 is generally thought to be rarer than myotonic dystrophy type 1, but large-scale population studies to confirm this have not been performed. In Germany, 267 mutation-verified molecular diagnoses were made between 2003 and 2005 compared with 277 myotonic dystrophy type 1 diagnoses within the same period. These data suggest that myotonic dystrophy type 2 appears to be more frequent than previously thought, with most patients with myotonic dystrophy type 2 currently undiagnosed with symptoms frequently occurring in the elderly population (236). A large-scale registry study showed an almost equal representation of patients with myotonic dystrophy type 1 and myotonic dystrophy type 2 in Finland, Germany, Poland, Czech/Slovakia, and Serbia (270). However, many patients in older generations with myotonic dystrophy type 1 or type 2 with milder symptoms are clearly undiagnosed. It is noteworthy that recessive mutations in the chloride channel gene CLCN1, which have a high frequency in the general population, can act as modifiers in patients with myotonic dystrophy type 2 disease by amplification of their myotonia (237; 30; 170). Meola’s group has identified patients with myotonic dystrophy type 2 presenting an atypical phenotype characterized by early and severe myotonia without mutation on the CLCN1 gene but with mutations on SCN4A gene (25; 139; 21). Thus, both CLCN1 and SCN4A mutations may contribute to exaggerate the myotonia in myotonic dystrophy type 2 (139).
Myotonic dystrophy type 1 (myotonic dystrophy of Steinert). Genetic counseling is part of the multidisciplinary management of myotonic dystrophy patients. Myotonic dystrophy is an autosomal dominant disease with almost complete penetrance. Therefore, in 50% of cases the proband's offspring inherit the mutation and develop features of the disease. Moreover, due to instability of the CTG repeat expansion in the disease, the anticipation phenomena are described, ie, the offspring can show more severe phenotype than parents. A major role of genetic counseling is reproductive counseling, especially in the evaluation of the risk of having a child with congenital myotonic dystrophy. The estimated risk of a congenital myotonic dystrophy pregnancy is about 10% to 40% (104). Some authors found a major risk if the mother has multisystem involvement or if her symptoms start before 30 years of age (271; 104). Martorell and colleagues found 12 asymptomatic females with congenital myotonic dystrophy children (131). Cobo and collaborators found the risk of having a congenital myotonic dystrophy child to be 59% if the repeat size is greater than 300 and 10% if it is smaller (43).
Prenatal testing. It is possible to make a prenatal diagnosis determining the size of CTG repeat expansion from fetal cells obtained by amniocenteses (16th week) or through chorionic villus sampling (12th week). It is important to have molecular confirmation of myotonic dystrophy type 1 in both parents before performing the procedure (131). It is not possible to predict exactly the phenotype of the fetus. Normally if an expansion over 1000 is found there is a high probability of congenital myotonic dystrophy.
Preimplantation genetic diagnosis. Preimplantation genetic testing of in vitro-fertilized embryos followed by selective transfer to the mother’s uterus of unaffected embryos is a feasible solution to increase the chances of an unaffected pregnancy. Standard repeat-spanning or flanking PCR to detect the normal DMPK allele of the affected parent is commonly employed in preimplantation genetic testing for myotonic dystrophy type 1 due to reliability issues in detecting the expanded allele, especially when performed directly on the limited genetic material of single cells (50). A robust strategy for preimplantation genetic testing for myotonic dystrophy type 1 that can be applied to virtually any at-risk couple has been proposed (118). This strategy combines detection of the CTG repeat expansion by bidirectional TP-PCR with linked haplotype analysis generated from a dodecaplex marker panel after whole-genome amplification. The aim of this strategy is to provide direct detection of the expansion mutation when present, while concurrently providing haplotype based diagnostic confirmation for virtually any preimplantation genetic test case. The use of multiple microsatellite markers also mitigates the risk of ambiguous haplotype phasing arising from insufficient informative markers.
Myotonic dystrophy type 2 (proximal myotonic myopathy). In myotonic dystrophy type 2, as in myotonic dystrophy type 1, it is possible to make prenatal diagnosis through the same procedures. However, it is seldom requested due to the absence of the congenital form and the less severe phenotype presented in myotonic dystrophy type 2.
Myotonic dystrophy type 1 and myotonic dystrophy type 2 share a similar pathogenetic mechanism and for several years the presence of common features in muscular and multisystemic involvement have been emphasized. Nowadays, advances in understanding myotonic dystrophy type 2 allow us to find clinical differences between the 2 disorders.
It seems that several factors other than splicing alteration modulate the clinical features of the diseases.
One of the most debated arguments is the presence, or not, of anticipation in myotonic dystrophy type 2. In fact, it is well known that in myotonic dystrophy type 1 CTG expansion may increase in length during meiosis and mitosis leading to an earlier and more severe phenotype in successive generation. Indeed, in myotonic dystrophy type 1 onset and disease severity correlate with repeat length. However, in myotonic dystrophy type 2 there is not a clear correlation between repeat size and disease severity, and no evidence of an anticipation phenomenon has been demonstrated (217; 47; 219). Only a debated case of congenital myotonic dystrophy type 2 has been described so far with an expansion size similar between son and mother (108).
The clinical spectrum presents slight but important differences between the two diseases.
Muscle weakness is the main feature of both diseases, but it shows a different pattern of muscles involvement. Adult-onset myotonic dystrophy type 1 starts like a distal myopathy (deep finger flexor, wrist extensor, ankle dorsiflexor) that involves also facial and neck flexor muscle, whereas in myotonic dystrophy type 2 the pelvic girdle is one of the first areas where weakness is noticed. Moreover, muscular atrophy is more severe in myotonic dystrophy type 1 than in myotonic dystrophy type 2 except in proximal myotonic dystrophy variant where marked atrophy, and absent of clinical myotonia, it can mimic spinal muscle atrophy (248; 206).
Clinical myotonia is more severe in myotonic dystrophy type 1 than in myotonic dystrophy type 2 and electrical myotonia presents different features between the 2 forms, as an easier evocability and the presence of waxing-waning discharges in myotonic dystrophy type 1, compared to more difficult evocability and the waning discharges in myotonic dystrophy type 2 (123).
From a histopathological point of view, previous studies showed similar pattern characterized by fibers size variation, nuclear internalization, and nuclear clumps. Over the years several authors have identified a more characteristic pattern in myotonic dystrophy type 2 compared to myotonic dystrophy type 1, which consists of a prevalent type 2 fibers atrophy and nuclear internalization (259; 18; 190).
Multisystemic involvement as well as muscular features present slight differences between the two forms. At a cardiac level patients with myotonic dystrophy type 1 are more often affected by conductive disturbances, whereas patients with myotonic dystrophy type 2 have a prominent left ventricular dysfunction (264). Meola and colleagues showed that patients with myotonic dystrophy type 2 have a more favorable prognosis than patients with myotonic dystrophy type 1 (141) whereas Wahbi and colleagues found an overall risk of myotonic dystrophy type 2 comparable to myotonic dystrophy type 1 (264).
The clinical spectrum of endocrine involvement presents differences between the two forms in terms of a higher prevalence of hypogonadism in myotonic dystrophy type 1 and insulin resistance in myotonic dystrophy type 2.
A dysexecutive syndrome caused by frontal lobe dysfunction is a frequent feature of adult myotonic dystrophies although it seems to be less prominent and less severe in patients with myotonic dystrophy type 2 (140).
Nondystrophic myotonias caused by sodium or chloride channelopathies are distinguished from myotonic dystrophy type 1 and myotonic dystrophy type 2 based on normal muscle strength and by lack of muscle wasting or other systems involvement. Having the patient squeeze his or her eyes closed four to five times consecutively is helpful in identifying paradoxical myotonia. Paradoxical myotonia is myotonia that worsens with repeated muscle contractions. This type of myotonia is not characteristic of the myotonic dystrophies. Paradoxical myotonia favors a diagnosis of sodium channel myotonia caused by a mutation in the skeletal muscle sodium channel (152; 153).
One typical presentation for patients with undiagnosed myotonic dystrophy is to complain of muscle weakness, muscle stiffness, or muscle pain without obvious diagnostic findings on clinical examination.
The gold standard for establishing the diagnoses of myotonic dystrophy type 1 and myotonic dystrophy type 2/proximal myotonic myopathy is to demonstrate the presence of abnormal expansions of CTG repeats in the 19q13.3 DMPK gene and of CCTG repeats in the 3q21 zinc finger protein 9, in intron 1of the CCHC-type zinc finger nucleic acid binding protein (ZNF9/CNBP) gene involved with myotonic dystrophy type 2. Best practice guidelines and recommendations on the molecular diagnosis of myotonic dystrophy type 1 and type 2 have been published (102). Standard leucocyte DNA testing is available for myotonic dystrophy type 1 (253). However, the observation of patients with myotonic dystrophy type 1 presenting interrupted allele has practical consequences for myotonic dystrophy type 1 molecular genetic test. Indeed, variant repeats with extreme GC contents yield false negatives in both repeat primed PCR and standard PCR based approaches to diagnostics. For this reason bidirectional triplet primed PCR (TP-PCR) should be included in the routine diagnostic protocol used for myotonic dystrophy type 1 testing because it is very sensitive to detect myotonic dystrophy type 1 expansions presenting variant repeats (214; 51). A commercial kit based on the bidirectional TP-PCR approach, the FastDM1TM DMPK sizing kit, has been validated to be used in diagnostic myotonic dystrophy type 1 testing (114).
Leucocyte DNA testing is also available for myotonic dystrophy type 2, but previous DNA analysis for diagnosing myotonic dystrophy type 2 and proximal myotonic myopathy may have missed as many as 20% of affected individuals (47). As for myotonic dystrophy type 1, a new ready to use genetic test has been validated to identify the myotonic dystrophy type 2 disease, with the advantage to reduce errors that can be introduced using homemade reagents (252). However, the myotonic dystrophy type 2 diagnostic odyssey is complicated by the difficulties to develop an accurate, robust, and cost-effective method for a routine molecular assay (139).
A more practical tool for myotonic dystrophy type 2 diagnosis than the complex genotyping procedure is via in situ hybridization detection of nuclear accumulations of CCUG-containing RNA in myotonic dystrophy type 2 muscle biopsy using specific probes (32; 210). Moreover, because MBNL1 is sequestered by mutant RNA foci, it is possible to visualize the nuclear accumulation of MBNL1 by immunofluorescence on muscle sections. However, although MBNL1 represents a histopathological marker of myotonic dystrophies, it does not allow one to distinguish between myotonic dystrophy type 1 and myotonic dystrophy type 2 (31). Another tool to investigate muscle weakness and wasting is muscle imaging with MRI. In type 1 disease, initial changes are seen in the soleus and medial gastrocnemius; these muscles in the lower legs are completely replaced by fatty infiltration (degenerative changes). It has been reported that MRI of tibialis anterior could be a "surrogate measure" in myotonic dystrophy type 1 (136). The MRI techniques will help to identify asymptomatic patients or patients with only minimal clinical abnormalities; information that clarifies the natural history and prognosis of the disease; and a pharmacologic action in this muscle group. Finally, another advantage that the MRI provides is its ability to assess each individual muscle group and demonstrate subtle changes in muscle structure. In type 2 disease, early muscular changes develop in the anterior vastus group of thigh muscles, with relative sparing of the rectus femoris (249).
Myotonic dystrophy type 1 (myotonic dystrophy of Steinert). The management of myotonic dystrophy type 1 requires a comprehensive, multidisciplinary approach given the multisystemic nature of the disorder. The intricate nature of the condition necessitates collaboration with cardiologists, pulmonologists, ophthalmologists, physiatrists, gastroenterologists, endocrinologists, and geneticists to ensure comprehensive and optimal clinical care (200). Attending to treatable multisystemic features and symptomatic aspects not only enhances patients' quality of life but also extends their lifespan, as diminished life expectancy in myotonic dystrophy type 1 is primarily due to respiratory failure, cardiac rhythm disturbances, and an elevated risk of malignancies (85; 200).
Multidisciplinary care framework. Annual multidisciplinary team evaluations are recommended and are composed of a primary care clinician, neuromuscular specialist, physical therapist, speech therapist, neuropsychologist, pulmonologist, cardiologist, and genetic counselor (85). The 2018 international consensus-based care recommendations, developed by 66 experienced clinicians, provide standardized care protocols for 19 discrete body systems and considerations (12).
Musculoskeletal management. The basic approach to treatment remains symptomatic, utilizing bracing with ankle-foot orthoses or high-top shoes, motorized scooters, and wheelchairs, as needed. A multidisciplinary approach to prevent falls should be implemented as patients with myotonic dystrophy type 1 are 10 times more likely to fall or stumble compared to healthy cohorts (274). For severe, disabling myotonia impacting daily activities, mexiletine (150 to 200 mg three times daily) is recommended as first-choice anti-myotonic therapy, with prior and regular ECG monitoring (200). Data from randomized, double-blind, crossover trials in 60 patients with myotonic dystrophy type 1 confirmed that mexiletine at these dosages is effective, well-tolerated, and safe (124; 88).
Regular low or moderate aerobic exercises have been found to be both safe and effective in improving overall fitness (159). Behavioral interventions targeting physical activity can increase lower extremity muscle cross-sectional area, preferentially in healthy-appearing muscle (90). Other anti-myotonic medications, including phenytoin, acetazolamide, clomipramine, imipramine, and taurine, may provide benefit in select cases (247).
Cardiac management. Annual ECG monitoring is essential, with cardiologist referral recommended for symptomatic patients or those with abnormal findings. A 24-hour Holter ECG should be obtained for symptomatic subjects, and echocardiography is recommended at diagnosis and every 3 to 5 years, or more frequently if initial findings are abnormal (200). The use of antiarrhythmic drugs requires caution, and implantable cardioverter defibrillators should be considered for ventricular tachyarrhythmias, with anticoagulants for atrial fibrillation/flutter.
Despite advances in understanding cardiac involvement, gaps in knowledge limit optimal rhythm management approaches. The benefit of prophylactic pacemakers or implantable cardioverter defibrillators remains uncertain as sudden death has been observed even in patients with these devices, justifying consideration of randomized trials for prophylactic device implants in high-risk patients (79).
Respiratory management. There is currently no consensus on standardized respiratory diagnostic and management protocols, though physicians should carefully assess respiratory symptoms and perform minimal lung function screening (213; 240). Baseline and annual respiratory examination with pulmonary function tests are recommended, monitoring for recurrent respiratory infections in patients with dysphagia, impaired cough, and lung clearance (200).
Evidence strongly supports early implementation of noninvasive ventilation as studies demonstrate significant improvement in ventilation and oxygenation from the first night of treatment, with sustained benefits, including stable normoxia and normocapnia maintained for up to 52 months (205; 232). Optimization of respiratory therapeutic management, particularly timely initiation of noninvasive ventilation, may help reduce the rate of deaths due to respiratory complications, which remain the most common cause of mortality in patients with myotonic dystrophy type 1. Vaccination against SARS-CoV-2, influenza, and pneumonia is recommended (200).
Central nervous system and cognitive management. Patients and caregivers should be informed about myotonic dystrophy type 1 as a brain disorder, with routine assessment for cognitive, behavioral, or psychiatric impairments and neuropsychological assessment post-diagnosis (200). Referral to mental health specialists should be considered when appropriate. Excessive daytime sleepiness deserves aggressive treatment as it may lead to loss of job productivity and social isolation. First-line treatments include modafinil, methylphenidate, or caffeine/theobromine, with regular monitoring using fatigue and sleep scale scores (85; 200).
Cognitive behavioral therapy should be considered for severely fatigued patients. Data from the OPTIMISTIC study demonstrate that cognitive behavioral therapy focusing on addressing reduced patient initiative, increasing physical activity, optimizing social interaction, regulating sleep-wake patterns, coping with pain, and addressing beliefs about fatigue significantly increased patients' capacity for activity and participation compared to standard care alone (158).
Gastrointestinal management. Screening for dysphagia, dysphonia, and weight loss is essential, with dietary modifications and "safe swallowing techniques" introduced as needed. Gastrostomy should be considered for severe cases (200). First-choice therapy includes dietary modifications (small meal size, lower fat content), treatment of gastroesophageal reflux (elevate head of bed, avoid late evening meals), and pharmacotherapy for persistent symptoms. Life-threatening complications like aspiration and pseudo-obstruction require monitoring, with specialist referral for acute abdominal symptoms.
Endocrine and metabolic management. Hormonal status, lipid profile, and glucose metabolism should be assessed at baseline, with annual monitoring of HbA1c and fasting serum glucose. The American Diabetes Association guidelines should be followed for glucose metabolism disorder management, with metformin preferred for impaired glucose metabolism (200). The effects of metformin on muscle function have shown promise, with improved mobility and gait abilities in patients with myotonic dystrophy type 1 (17).
Thyroid hormone status and lipid profile should be assessed every 3 years, prioritizing cholesterol-lowering agents other than statins (eg, ezetimibe). Patients with gonadal dysfunction or fertility issues should be referred to subspecialists, with detailed physical examination for gonadal atrophy or cryptorchidism in male patients (200).
Ophthalmologic management. Annual ophthalmologist check-ups with slit lamp examination are recommended. Surgical treatment for cataracts or severe ptosis should be considered if symptoms interfere with daily activities. Optical coherence tomography may help avoid misdiagnosis of treatable visual impairment conditions (200). Cases of recurrent capsular opacification requiring multiple capsulorhexis have been described in postoperative patients with myotonic dystrophy type 1 (73).
Cancer surveillance. Patients with myotonic dystrophy type 1 have increased malignancy risk, requiring strict adherence to general cancer screening guidelines, especially for ovarian, endometrial, colon, brain, skin, and thyroid neoplasms that are more frequent in myotonic dystrophy type 1 compared to the general population (200). In a cross-sectional study, 12.4% of patients with myotonic dystrophy type 1 reported at least one benign tumor, and 6.2% reported at least one malignant tumor, with risk as high as 7-fold for some cancers (07; 20).
Palliative and end-of-life care. End-of-life management should involve palliative care services to maximize quality of life and address caregiver burden. Guidelines emphasize the importance of early palliative care introduction, particularly when significant swallowing and cardiorespiratory complications arise, as these are major contributors to mortality (275; 200). A traffic light system for triaging patients based on condition severity is recommended, with referral to palliative care specialists at appropriate stages. Advance care planning is crucial, allowing patients to record preferences for end-of-life care, with regular review to ensure patient preferences are respected.
Genetic counseling and family management. Genetic counseling remains an essential component of myotonic dystrophy type 1 management, though many affected individuals have limited interest in counseling, likely relating to personality characteristics and avoidant behavior that often develops in myotonic dystrophy type 1 (84; 143). The 2019 consensus-based recommendations for congenital and childhood-onset myotonic dystrophy type 1 provide standardized care protocols for pediatric patients, recognizing the distinct needs of this population (99).
Myotonic dystrophy type 2 (proximal myotonic myopathy). The management of myotonic dystrophy type 2 shares fundamental similarities with myotonic dystrophy type 1 but requires specific considerations for its distinct clinical features and associated comorbidities. Although there is generally less need for supportive care, such as bracing, scooters, or wheelchairs, due to the milder phenotype, a multidisciplinary approach remains essential given the multisystemic nature of the disorder (220; 200).
Autoimmune disease screening and management. A critical and recently recognized aspect of myotonic dystrophy type 2 management is the systematic screening for autoimmune diseases, which occurs in up to 30% of patients with myotonic dystrophy type 2—a significantly higher rate than in the general population or even in patients with myotonic dystrophy type 1 (181; 200). The most commonly associated autoimmune conditions include Hashimoto thyroiditis (18.1% prevalence), rheumatoid arthritis, diabetes mellitus type 1, systemic lupus erythematosus, Sjogren disease, localized scleroderma, psoriasis, and celiac disease.
The mechanistic basis for this association has been elucidated: expanded CCTG repeats lead to chronic endoplasmic reticulum stress through accumulation of aberrant RNA and toxic peptide products from repeat-associated non-ATG (RAN) translation. This chronic stress promotes mitochondrial DNA release into the cytoplasm, which is subsequently detected by the cGAS/STING pathway, triggering a type I interferon response that predisposes to autoimmunity (204). Regular screening for antinuclear antibodies and specific autoimmune markers is recommended, as the presence of autoimmune diseases and antinuclear antibodies may lead to underdiagnosis of myotonic dystrophy type 2, with muscle symptoms potentially attributed to inflammatory conditions (181).
Cardiac and respiratory management. Cataracts require regular monitoring, and serial ECG monitoring is necessary to detect covert arrhythmias. Although cardiac rhythm disturbances are less frequent than in myotonic dystrophy type 1, significant abnormalities do occur and can include dilated cardiomyopathy leading to sudden death (155; 47; 219). Annual ECG monitoring with cardiologist referral for symptomatic patients or abnormal findings is recommended. A 24-hour Holter ECG should be considered for symptomatic subjects, and echocardiography is recommended at diagnosis and every 3 to 5 years (200).
Respiratory complications are less common than in myotonic dystrophy type 1, with respiratory failure, hypersomnia, and recurrent aspiration being infrequent (244). However, baseline and annual respiratory examination with pulmonary function tests should still be performed, with monitoring for respiratory infections in patients with dysphagia.
Myotonia and pain management. Myotonia tends to be less marked and troublesome in myotonic dystrophy type 2 than in myotonic dystrophy type 1, but when muscle stiffness is frequent and persistent, anti-myotonia therapy with mexiletine (150 to 200 mg three times daily) can be helpful, requiring prior and regular ECG monitoring (200).
A particularly challenging and characteristic problem in myotonic dystrophy type 2 is the peculiar muscle pain, which has a lifetime prevalence of 76% and significantly impacts quality of life. This pain is commonly described as abdominal, musculoskeletal, and exercise-related, often aggravated by cold temperatures. The pain tends to be located symmetrically in the proximal limbs and can be associated with mechanical hyperalgesia affecting the rectus femoris, trapezius, and thenar muscles (256; 200).
Although the exact mechanism remains unclear, transcriptomic analysis has identified 14 muscle genes that are significantly dysregulated in patients with myalgic versus non-myalgic myotonic dystrophy type 2, suggesting that muscle-specific molecular pathways contribute to pain pathophysiology (151). Treatment approaches include carbamazepine or mexiletine along with nonsteroidal anti-inflammatory medications. Additional options that have been explored include gabapentin, low-dose thyroid replacement, steroids, and tricyclic antidepressants, though none are consistently effective (200). Patients with severe pain may require opiates on a regular basis for relief.
Endocrine and metabolic management. Hypogonadism and insulin resistance require monitoring similar to myotonic dystrophy type 1. Metabolic syndrome is common in patients with myotonic dystrophy type 2, and treatment of metabolic disturbances may reduce cardiovascular complications and improve quality of life. Hormonal status, lipid profile, and glucose metabolism should be assessed at baseline, with annual HbA1c and fasting glucose monitoring. Metformin is preferred for impaired glucose metabolism, following American Diabetes Association guidelines (200).
Thyroid hormone status should be assessed every 3 years, with particular attention to screening for Hashimoto thyroiditis given its high prevalence. Lipid profiles should be monitored regularly, prioritizing cholesterol-lowering agents other than statins (eg, ezetimibe).
Cognitive and neuropsychological management. Cognitive difficulties occur in myotonic dystrophy type 2, though they are less severe than in myotonic dystrophy type 1. Approximately 25% of patients with myotonic dystrophy type 2 have significant cognitive impairment that interferes with everyday functioning. The changes appear to be associated with decreased cerebral blood flow to frontal and anterior temporal lobes and decreased brain volume (37; 05; 62). A specific "avoidant" personality pattern and significant impairment in frontal lobe function, especially limited ability to perform executive functions, have been observed, though these abnormalities are milder than in myotonic dystrophy type 1 (143; 182).
Management should include routine assessment for cognitive, behavioral, or psychiatric impairments, with neuropsychological assessment when indicated. Excessive daytime sleepiness can be treated with modafinil, methylphenidate, or caffeine/theobromine. Transcranial sonography studies have identified brainstem abnormalities in myotonic dystrophy type 2, with substantia nigra hyperechogenicity related to fatigue and excessive daytime sleepiness (200).
Hearing and sensory management. Hearing impairment is a frequent symptom in patients with myotonic dystrophy type 2, with sensorineural hearing loss located in the cochlea. Audiometry should be performed when hearing impairment is suspected to facilitate early hearing rehabilitation with hearing aids when indicated (256).
Specialized care guidelines. The 2019 consensus-based care recommendations developed by international myotonic dystrophy type 2 experts provide comprehensive guidance for managing the wide spectrum of disease manifestations (220). These evidence-based protocols address multisystemic involvement and emphasize the importance of early detection and treatment of associated conditions, particularly autoimmune diseases that may otherwise lead to delayed or missed myotonic dystrophy type 2 diagnosis.
Prognosis and long-term management. Overall, the prognosis for patients with myotonic dystrophy type 2 remains more favorable than for myotonic dystrophy type 1, with slower, less severe progression of muscle weakness, less muscle wasting, and typically normal lifespan. However, the multisystemic aspects of the disease require ongoing multidisciplinary care, including state-of-the-art cardiac and brain imaging to detect and treat complications earlier. Regular screening for autoimmune conditions has become a crucial component of comprehensive myotonic dystrophy type 2 care.
Emerging treatments. Although no curative treatments have been approved for myotonic dystrophy, the landscape of emerging therapies has evolved dramatically in recent years. The pursuit of effective therapies for myotonic dystrophy type 1 and myotonic dystrophy type 2 has been marked by unprecedented progress across multiple therapeutic modalities. Current treatment approaches focus on three primary categories: small molecules, nucleic acid-based therapies, and genome/transcriptome engineering technologies.
Antisense oligonucleotides. Antisense oligonucleotides represent one of the most promising therapeutic approaches for myotonic dystrophy type 1, offering high target specificity by binding to mutant RNA via complementary sequences.
|
DYNE-101 (ACHIEVE trial) | |
|
• Mechanism: Antisense oligonucleotide targeting DMPK mRNA for RNase H-mediated degradation | |
|
• Delivery: Conjugated with transferrin receptor 1 (TfR1)-binding antibody fragments for enhanced muscle targeting | |
|
• Clinical status: Phase I/II trial (NCT05481879) | |
|
• Results: Demonstrated robust reduction of toxic DMPK RNA, reversal of myotonia, and favorable safety profile in preclinical studies | |
|
IONIS-DMPKRx (Baliforsen) | |
|
• Mechanism: Antisense oligonucleotide reducing DMPK RNA levels | |
|
• Clinical status: Phase I/IIa completed | |
|
• Outcomes: Generally well tolerated, but skeletal muscle drug concentrations were below therapeutic levels, leading to study discontinuation | |
|
PGN-EDODM1 | |
|
• Mechanism: Peptide-conjugated antisense oligonucleotides preventing sequestration of splicing factors | |
|
• Clinical status: Phase I trial (NCT06204809) | |
|
• Innovation: Enhanced delivery through peptide conjugation | |
|
VX-670 | |
|
• Mechanism: Endosomal escape vehicle oligonucleotide preventing sequestration of splicing factors | |
|
• Clinical status: Phase I/II trial (NCT06185764) | |
|
• Advantage: Improved cellular uptake and stability | |
|
AOC 1001 (delpacibart etedesiran) - MARINA trial | ||
|
• Mechanism: siRNA targeting DMPK mRNA conjugated with monoclonal antibody against transferrin receptor 1 | ||
|
• Clinical status: Phase III trial (NCT06411288) | ||
|
• Developer: Avidity Biosciences | ||
|
• Key achievements: | ||
|
-- Received orphan drug status and fast-track designation | ||
|
-- Mid-study results showed significant DMPK reduction and enhanced alternative splicing | ||
|
-- Clinical improvements observed at higher dose levels | ||
|
• Extended study: MARINA-OLE for continued treatment over 2 years (NCT05479981) | ||
|
ARO-DM1 | ||
|
• Mechanism: RNA interference targeting DMPK mRNA | ||
|
• Clinical status: Phase I/IIa trial (NCT06138743) | ||
|
ATX-01 (ArthemiR™ trial) | |
|
• Mechanism: Anti-miR oligonucleotide targeting miR-23b to enhance endogenous MBNL1 expression | |
|
• Clinical status: Phase I/IIa trial (NCT06300307) | |
|
• Developer: ARTHEx Biotech | |
|
• Regulatory status: Received orphan drug designation from U.S. and European authorities | |
|
• Rationale: Compensates for functional MBNL1 depletion by sequestration | |
Small molecule therapeutics. Small molecule approaches leverage existing pharmacokinetic and pharmacodynamic profiles, facilitating rapid translation to clinical applications.
|
Mexiletine | |
|
• Mechanism: Sodium channel blocker reducing myotonia | |
|
• Clinical status: Phase II completed, phase III ongoing | |
|
• Results: Demonstrated positive effects on handgrip myotonia in ambulatory patients; also shown to downregulate DMPK mRNA levels | |
|
Metformin | |
|
• Mechanism: Biguanide antidiabetic drug activating AMPK pathway | |
|
• Clinical status: Phase III trial (NCT05532813) | |
|
• Results: Statistically significant improvements in mobility and mechanical power during gait in over 50% of patients; increased walking distance in 6-minute walk test in per-protocol population | |
|
Tideglusib | |
|
• Mechanism: GSK3β inhibitor reducing toxic DMPK RNA and normalizing CUGBP1 downstream targets | |
|
• Clinical status: Phase II/III trials (NCT03692312, NCT05004129) | |
|
• Developer: AMO Pharma | |
|
• Results: Improved muscle weakness, myotonia, and central nervous system symptoms in most patients | |
|
CRISPR-Cas9 approaches | |
|
• Mechanism: Direct targeting and removal of pathogenic CTG expansions | |
|
• Potential: Offers potential cure by addressing disease root cause | |
|
• Delivery: Vectorized AAV administration for enhanced targeting | |
|
Genethon strategy | |
|
• Approach: CRISPR-Cas9 from Staphylococcus aureus targeting DMPK CTG expansions | |
|
• Status: Preclinical development | |
|
LocanaBio (PIN-dCas9) | |
|
• Mechanism: AAV9-vectors encoding PIN-dCas9 with guide RNA targeting CUG repeats | |
|
• Status: Lead selection and optimization phase | |
|
• Results: Dose-dependent reduction of toxic RNA foci, MBNL1 redistribution, rescued missplicing, reduced myotonia, and muscle weakness recovery in myotonic dystrophy type 1 patient cells and mouse models | |
Although therapeutic development has predominantly focused on myotonic dystrophy type 1, the shared pathogenic mechanisms between myotonic dystrophy types 1 and 2 suggest considerable therapeutic cross-applicability. Both conditions involve RNA toxicity through sequestration of MBNL proteins, indicating that many small molecule approaches targeting downstream pathways may benefit both patient populations. However, specific considerations for myotonic dystrophy type 2 include the need for CCTG repeat-targeting strategies analogous to myotonic dystrophy type 1's CTG targeting and the development of CNBP-specific antisense approaches. The distinct tetranucleotide repeat structure in myotonic dystrophy type 2 requires tailored nucleic acid-based therapies, whereas metabolic modulators like metformin may prove particularly relevant given myotonic dystrophy type 2's associated insulin resistance. Despite these challenges, the advancing therapeutic pipeline for myotonic dystrophy type 1 provides a foundation for future myotonic dystrophy type 2 treatment development.
Overall, the emergence of multiple therapeutic modalities represents unprecedented hope for patients with myotonic dystrophy. The integration of small molecules, nucleic acid-based therapies, and genome engineering approaches offers a comprehensive strategy for addressing this complex genetic disorder. Although challenges remain in delivery, off-target effects, and immune responses, the robust clinical trial pipeline and innovative therapeutic strategies signal a promising future for disease-modifying treatments in both myotonic dystrophy types 1 and 2.
Myotonic dystrophy type 1 (myotonic dystrophy of Steinert). Increased pregnancy and delivery-related risks require a multidisciplinary approach involving obstetrician, anesthesiologist, neonatologist, geneticist, midwife, and physiotherapist. Women with myotonic dystrophy type 1 have an increased risk of miscarriage, preterm delivery, respiratory insufficiency (especially in the third trimester), and failed labor during delivery. They fatigue rapidly during labor and are at risk of postpartum hemorrhage. Extreme care should be taken with analgesics and sedating anesthetic drugs (200).
Myotonic dystrophy type 2 (proximal myotonic myopathy). Similar to myotonic dystrophy type 1, pregnancy-related risks require multidisciplinary management. Women with myotonic dystrophy type 2 have higher rates of fertility issues, urinary tract infections, and delivery complications, including preterm labor and stillbirth (200). Some women may experience their first myotonic symptoms during pregnancy, with potential reversibility after delivery, though more data are needed.
Myotonic dystrophy type 1 (myotonic dystrophy of Steinert). Patients with myotonic dystrophy type 1 have increased sensitivity to sedative medications, especially barbiturates and opiates, and have paradoxical reactions to depolarizing muscle relaxants (84; 273). There is an increased risk of postoperative apnea following general anesthesia (84; 152), and it is prudent to monitor patients with oximetry for 24 hours after general anesthesia to assure full recovery of their baseline respiratory function. Patients who have significantly low forced vital capacity measurement prior to general anesthesia are at increased risk of hypoventilation and postoperative atelectasis (84; 154). In selected cases, the anesthesiologist and surgeon may choose to delay extubation and maintain ventilator support of the patient, especially in surgeries that require several days of postoperative pain control with opiates. Aggressive pulmonary physical therapy, use of assisted cough device treatments, nasal bilevel positive airway pressure, and careful postoperative monitoring are often needed, and coordination of care with pulmonary medicine and respiratory therapy consultants is necessary.
There is also an increased risk of cardiac arrhythmia following general anesthesia in patients with myotonic dystrophy type 1 (84). Cardiology consultation is useful if arrhythmia develops to advise about treatment because patients with myotonic dystrophy type 1 sometimes display an unexpected, undesirable reaction to certain medications (84).
Myotonic dystrophy type 2 (proximal myotonic myopathy). One study of a large number of individuals with myotonic dystrophy type 2 has found no significant problems with the ability of patients to tolerate general anesthesia (47). In a report of a large German patient cohort, the overall frequency of severe complications was 0.6% (two of 340). The overall lower risk seems to be predominantly related to the minor respiratory involvement in myotonic dystrophy type 2 than in myotonic dystrophy type 1 (103).
Thirty-one years have passed since the (CTG)n repeat expansion mutation was discovered in patients with myotonic dystrophy type 1, and 22 years ago the (CCTG)n mutation was identified in type 2 disease. Emerging data indicate that molecular pathomechanisms are much more complex than could have been envisioned when the respective mutations were just identified. RNA toxicity clearly has a major role, yet spliceopathy alone does not seem to fully account for all aspects of the multisystemic phenotype in myotonic dystrophies. Other pathomechanisms consistent with the toxic RNA model probably entail regulation of gene expression and translation and various cellular stress pathways and extend beyond the nucleus to the cytoplasm. Nevertheless, it is important to emphasize that despite clinical and genetic similarities, myotonic dystrophy type 1 and type 2 are distinct disorders requiring different diagnostic and management strategies.
Although treatment of myotonic dystrophy type 1 and myotonic dystrophy type 2 is currently limited to supportive therapies, research over the past 20 years has advanced quickly from initial observations of the disease mechanisms of myotonic dystrophy type 1 and myotonic dystrophy type 2 and has ushered in a new era of RNA targeted treatments (165).
Preclinical experimental therapies for myotonic dystrophy involve animal models and have focused on the use of antisense oligonucleotides (ASOs), small interfering molecules, ribozymes, and engineered small nuclear RNAs (70). IONIS-DMPKRx trials started in 2015 on human patients affected by myotonic dystrophy type 1. The phase 2 study was completed in June 2016 (NCT02312011). It was a blinded, placebo-controlled study of 48 patients with myotonic dystrophy type 1. Study medication (baliforsen) was given subcutaneously once weekly over 6 weeks. The study was safe even at higher dosage. However, the drug failed to achieve the target tissue concentration of antisense oligonucleotide to be necessary to knockdown DMPK mutant allele. However, additional strengths of the study included the feasibility to conduct the trial over eight clinical sites, the feasibility to perform two multiple small needle muscle biopsies in the same patient, and the use of validated measures of muscle strength and function (242). In October 2018, the AMO Pharma Limited sponsored a randomized, double-blind study to evaluate the efficacy and safety of tideglusib versus placebo for the treatment of children and adolescents (aged 6 to 16 years) with congenital and childhood myotonic dystrophy type 1. A phase 2 study on 16 patients of AMO-02 was safe and well tolerated (NCT02858908). This study suggests a potential treatment for congenital and childhood onset type 1 myotonic dystrophy (95). Another targeted therapeutic trial underway is a phase 1/2 study of AOC1001 (RNA therapeutic antibody oligonucleotide conjugate) in adult patients with myotonic dystrophy type 1 (NCT05027269). AOC1001 is an RNA based molecular or a DMPK siRNA conjugated to a humanized antibody targeting human transferrin receptor 1 (TfR1). The antibody targets muscles for delivery of siRNA into the cytoplasm and nucleus where it mediates DMPK mRNA degradation. Results of this study, presented at AAN 2023, show improvement of multiple functional assessments, including measures of myotonia, mobility, and strength; amelioration of splicing changes; and a favorable safety and tolerability profile with mild or moderate adverse events (242).
The future holds great promise for advances in translational research in myotonic dystrophy type 1 and myotonic dystrophy type 2. The teamwork will expedite the development of targeted therapies and improve the lives of patients and their families (153).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Giovanni Meola MD PhD
Dr. Meola of the University of Milan, Casa di Cura Igea has no relevant financial relationships to disclose.
See Profile
Elena Abati MD PhD
Dr. Abati of Ospedale Maggiore Policlinico, Milan, Italy, has no relevant financial relationships to disclose.
See Profile
Nicholas E Johnson MD MSCI FAAN
Dr. Johnson of Virginia Commonwealth University received consulting fees and/or research grants from AMO Pharma, Avidity, Dyne, Novartis, Pepgen, Sanofi Genzyme, Sarepta Therapeutics, Takeda, and Vertex, consulting fees and stock options from Juvena, and honorariums from Biogen Idec and Fulcrum Therapeutics as a drug safety monitoring board member.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuromuscular Disorders
Jun. 16, 2026
Neuromuscular Disorders
May. 27, 2026
Neuromuscular Disorders
May. 21, 2026
Developmental Malformations
May. 08, 2026
Neuromuscular Disorders
Apr. 23, 2026
Neuromuscular Disorders
Apr. 23, 2026
Neuromuscular Disorders
Apr. 23, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026