Neuromuscular Disorders
Viral and retroviral myositis
Jun. 16, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
A diagnosis of skeletal muscle channelopathy can be rewarding for both patients and clinicians, as most symptoms respond well to pharmacological treatment. This article on periodic paralysis and the nondystrophic myotonias (skeletal muscle channelopathies) introduces readers to their clinical presentations and current understanding of their pathophysiology and provides a practical guide for investigating and treating them. Updates include significant new developments in our understanding of the clinical presentations of muscle channelopathies and randomized controlled trial data for pharmacological therapies.
|
• Episodes of periodic paralysis typically involve generalized muscle weakness with hypotonia and areflexia, but in adults, bulbar and respiratory muscles and cognition are generally spared. | |
|
• Bulbar and respiratory muscles are commonly involved in children, especially those with SCN4A gene-related disorders. | |
|
• Severe neonatal episodic laryngospasm is a severe phenotype of sodium channel myotonia presenting in infancy. | |
|
• Genetic testing is now the mainstay of diagnosing these syndromes in most patients. | |
|
• Measuring serum potassium levels, recognizing precipitating factors, and identifying the clinical features of myotonia, cardiac arrhythmia, or distinctive craniofacial and skeletal features help distinguish the different forms of inherited periodic paralysis. | |
|
• Serum potassium levels can remain within the normal range during the attacks of both hypokalemic periodic paralysis and hyperkalemic periodic paralysis. | |
|
• The cardiac complications of Andersen-Tawil syndrome are greater than previously appreciated, and patients require yearly cardiac review, including Holter monitoring. | |
|
• Acetazolamide is most commonly used as prophylactic therapy for periodic paralysis, although clinical trial data on efficacy are still lacking, and some genotypes may be exacerbated by using it. | |
|
• In randomized, double-blind, placebo-controlled trials, dichlorphenamide appears promising in ameliorating symptoms in patients with hypokalemic and hyperkalemic periodic paralysis but can be difficult to obtain and is a high-cost drug. | |
|
• Sodium channel blockers remain the mainstay of treating myotonia, but mexiletine and lamotrigine benefit from trial evidence of efficacy. | |
|
• Mexiletine is now licensed for treating those over age 18 years with nondystrophic myotonia but at a high cost. | |
|
• Lamotrigine performs well against mexiletine in a head-to-head crossover study. Although prescription is off-license, it remains an affordable and readily available alternative to mexiletine. |
Periodic paralysis (110) and the related myotonic disorders, paramyotonia congenita (22) and myotonia congenita (101), were first described over 100 years ago. During the early part of the twentieth century, an association was recognized between episodic weakness and low serum potassium levels (10), and later, with elevated serum potassium levels (adynamia episodica hereditaria) (28). The association between cardiac arrhythmias and periodic paralysis has been noted by several investigators (51). Andersen and colleagues described a triad of periodic paralysis, ventricular ectopy, and characteristic physical features (04), a phenotype later refined by Tawil and colleagues (86). This syndrome is variously referred to as Andersen syndrome, Andersen-Tawil syndrome, or long QT syndrome type 7 (LQT7).
With molecular technology, mutations have been identified in multiple sarcolemmal voltage-gated ion channel genes: sodium, SCN4A gene (hyperkalemic periodic paralysis, paramyotonia congenita, sodium channel myotonia, and, less commonly, hypokalemic periodic paralysis); potassium, KCNJ2 gene (Andersen-Tawil syndrome); calcium, CACNA1S gene (hypokalemic periodic paralysis); and chloride channels, CLCN1 gene (myotonia congenita) (16).
Significant advances in understanding the pathophysiology of these disorders have directly informed pharmacological treatment. Next-generation sequencing and other methodologies (eg, MLPA) show that these clinical phenotypes are due to mutations in the described genes in most patients. The outlier is that in a notable proportion of patients with Andersen-Tawil syndrome, no genetic mutation is identified in KCNJ2.
|
• Hypokalaemic periodic paralysis is flaccid muscle weakness in the presence of lowered serum potassium. | |
|
• Hyperkalaemic periodic paralysis is flaccid muscle weakness in the presence of raised serum potassium. | |
|
• Periodic paralysis in the presence of dysmorphic features or fixed cardiac conduction abnormalities may indicate Andersen-Tawil syndrome. | |
|
• Thyrotoxic periodic paralysis is much more common in males (especially Asian males) than in females. |
The sine qua non for the diagnosis of most periodic paralyses is recurrent attacks of weakness (Table 1). Typically, the weakness is generalized and involves the arms and legs, sparing bulbar and respiratory muscles. Focal weakness of isolated muscles or individual limbs has been described. During attacks, affected muscles are in a state of sustained depolarization and are electrically inexcitable (14). This is reflected clinically in weakness, hypotonia, and areflexia. Attacks may last minutes, hours, or even days. Episodes are often precipitated by specific triggers (eg, exercise or diet) or are associated with abnormal serum potassium levels.
Measuring serum potassium levels, recognizing precipitating factors, and identifying the clinical features of myotonia, cardiac arrhythmia, or distinctive craniofacial and skeletal features help distinguish the different forms of inherited periodic paralysis (94).
In nondystrophic myotonias, myotonia is the major symptom and is typically either improved or “warms up” with exercise (myotonia congenita) or worsened by exercise (paradoxical myotonia of paramyotonia congenita) (27).
|
|
Primary symptom |
Additional symptoms |
Common Triggers |
Typical duration of paralytic episodes |
Diurnal pattern of paralytic episodes |
Examination |
Diagnostic “clues” |
|
Hyperkalaemic periodic paralysis |
Episodic paralysis or weakness |
Myotonia Myalgia Fatigue |
Foods or drugs that increase serum potassium Rest after exercise Cold environment |
Mins to hours |
Any time of day |
May be normal if asymptomatic Muscle hypertrophy (due to myotonia) Myotonia Proximal myopathy may occur |
Myotonia |
|
Hypokalaemic periodic paralysis |
Episodic paralysis or weakness |
Myalgia Fatigue |
Foods or drugs that lower serum potassium Rest after exercise |
Hours to days |
Typically early morning when waking |
May be normal if asymptomatic Proximal myopathy may occur |
Wakes with quadraparesis |
|
Andersen-Tawil syndrome |
Episodic paralysis or weakness |
Palpitations Pre-syncope Syncope Myalgia Fatigue |
Commonly, food or drugs that lower serum potassium Occasionally, food or drugs that increase serum potassium Rest after exercise |
Hours to days |
Any time of day or early morning waking |
Dysmorphic features, eg, short stature, micrognathia, low-set ears, clinodactyly, syndactyly |
Cardiac history Abnormal ECG Dysmorphic features |
Hypokalemic periodic paralysis. In hypokalemic periodic paralysis, episodes of weakness are associated with decreased serum potassium levels, sometimes dramatically decreased (eg, lower than 2.0 mEq/L). Inheritance is autosomal dominant, although reduced penetrance may mask any obvious family history. De novo cases are also relatively common. Approximately 60% of patients have mutations in the gene coding for the alpha-subunit of the dihydropyridine-binding voltage-sensitive L-type calcium channel CACNA1S, and some authors designate this as hypokalemic periodic paralysis type 1. Ten percent to twenty percent of families not linked to the CACNA1S gene locus have mutations in the gene coding for the skeletal muscle sodium channel SCN4A, which some authors designate as hypokalemic periodic paralysis type 2. The SCN4A gene is quite ubiquitous, with mutations causing numerous clinical phenotypes. However, of the mutations in the CACNA1S and SCN4A genes that cause hypokalemic periodic paralysis, nearly all occur in distinct regions of the channels known as the voltage sensors (57).
Familial or primary hypokalemic periodic paralysis symptoms usually begin around the age of puberty (94). Men are more severely affected than women. Attacks may be triggered by rest after intense exercise, carbohydrate-rich meals, insulin, a high-sodium diet, emotional stress, alcohol consumption, concomitant illness (eg, viral), or sometimes exposure to cold. A typical attack lasts for hours or, less often, days. Some patients report their strength improves steadily over a period of weeks before fully returning to normal. The attacks may occur repeatedly: daily, weekly, monthly, or less often. They typically occur during the night or in the early morning on waking. Episodes of weakness are not accompanied by sensory, cognitive, or cardiac symptoms (although if potassium derangement is profound, secondary cardiac arrhythmia or palpitations may occur).
In the fourth or fifth decades, slowly progressive and chronic proximal leg weakness can occur (36). Serum CK is often normal but may be elevated, typically in the region of a few 100s to 1000IU/L. A muscle biopsy will include typical myopathic features of abnormal increase in central nuclei, variation of fiber size, and vacuoles. However, there are no specific findings, and a muscle biopsy is unnecessary for diagnosis. Patients with sodium channel mutations (hypokalemic periodic paralysis type 2) may exhibit an earlier age of onset. Myotonia does not typically occur in hypokalemic periodic paralysis but is described in rare cases of SCN4A-associated hypokalemic periodic paralysis.
Thyrotoxic periodic paralysis. Thyrotoxic periodic paralysis is not a Mendelian disorder, but it can mimic hypokalemic periodic paralysis clinically. In contrast with familial periodic paralysis, the initial episode of thyrotoxic periodic paralysis typically occurs later in persons aged 20 to 40 years and predominantly (although not exclusively) in Asian males (90). Pediatric cases are rare, but in one series, just over 4% of those presenting with TPP did so between the ages of 10 and 19 years (48). The clinical manifestations of thyrotoxic periodic paralysis and hypokalemia are indistinguishable from those of familial hypokalemic periodic paralysis, except for elevated blood levels of thyroid hormones and depressed thyroid-stimulating hormone. Symptoms will resolve with effective treatment of thyrotoxicosis, so it is essential to consider this diagnosis in anyone presenting with a first episode of flaccid muscle weakness in association with low serum potassium. Symptoms and signs of hyperthyroidism should be looked for, but may be subtle or absent. Although spontaneous resolution of attacks occurs in a few hours to 2 days, even without potassium chloride supplementation, cardiac arrhythmias and respiratory failure are possible life-threatening complications. Potassium supplementation is warranted when severe weakness and ECG abnormalities are present due to hypokalemia. These patients may be sensitive to rebound hyperkalemia, and careful potassium monitoring is advised (117).
The Na-K ATPase pump is integral to maintaining extracellular potassium homeostasis, alongside sarcolemmal potassium channels. Thyrotoxicosis increases the activity of the Na-K ATPase pump, contributing to hypokalemia (90). Variants in the KCNJ18 gene, which encodes a skeletal muscle-specific inwardly rectifying potassium channel Kir2.6, have been reported in some patients with thyrotoxic periodic paralysis (83) and also in sporadic cases of hypokalemic periodic paralysis (19). Kir2.6 exerts negative inhibition on wild-type Kir2.6 and Kir2.1, another Kir channel expressed in skeletal muscle. It is hypothesized that the decreased potassium current from hypofunction of Kir2.6 predisposes the sarcolemma to hypokalemic-induced depolarization, which leads to Na+ channel inactivation and inactivation of muscles. TPP is a non-Mendelian disorder, and it is a crucial observation that symptoms only present if a patient is thyrotoxic. The relatively high prevalence of KCNJ18 variants in healthy controls as well as cases of sporadic periodic paralysis and their unaffected siblings, however, has led to some debate over the significance of these mutations in vivo (43). Multiple studies have shown these variants to be absent from thyrotoxic patients without periodic paralysis, which is notable considering the high mutation rate in the population as a whole. This may support the notion that an individual harboring one of these variants is at risk of TPP should they become thyrotoxic, but it may otherwise have no clinical effect.
Hyperkalemic periodic paralysis. In this less common form of periodic paralysis, episodes of weakness are typically associated with elevated serum potassium levels. Inheritance is autosomal dominant. Most patients have mutations in the gene for the skeletal muscle sodium channel SCN4A. Symptoms begin during early childhood (usually in the first decade). Males and females are equally affected. Attacks may be triggered by rest after intense exercise, immobility, or fasting and may be aborted by carbohydrate-rich meals. The duration of episodes is usually shorter than attacks in hypokalemic periodic paralysis, typically lasting for minutes to hours. They can occur at any time of the day. At the onset of an attack, patients may complain of muscle tightness or nonspecific muscle discomfort. Usually, the frequency or severity of attacks diminishes with age but may be replaced by fixed proximal weakness, as in hypokalemic periodic paralysis. Serum CK may be normal or mildly elevated. A clinical feature distinctive to hyperkalemic periodic paralysis is the presence of myotonia. This may manifest clinically or be detected only on EMG and is typically exacerbated by cold exposure. The presence of myotonic discharges in a patient with otherwise typical periodic paralysis symptoms establishes hyperkalemic periodic paralysis as the likely cause, as this is not (almost never) seen in hypokalemic periodic paralysis.
Andersen-Tawil syndrome. Andersen-Tawil syndrome is an autosomal dominant disorder and the rarest form of periodic paralysis. It is characterized by a triad: periodic paralysis, distinctive craniofacial and skeletal anomalies, and cardiac conduction defects. However, affected individuals may express only one or two of the three components (104).
The episodic weakness is usually associated with hypokalemia (86). The dysmorphic features are highly variable and include short stature, scoliosis, syndactyly, clinodactyly (permanent lateral or medial curvature of a finger or toe), hypertelorism (wide-set eyes), small or prominent ears that are low-set, micrognathia (small chin), broad forehead, and dental abnormalities (eg, delayed tooth eruption or missing teeth) (104). Cardiac manifestations vary from an asymptomatic long QT syndrome to life-threatening ventricular tachyarrhythmia requiring an implantable defibrillator (61). ECG may exhibit either a prolonged QTc or QU interval with characteristic U-wave morphology (115) or ventricular ectopy, including bigeminy or trigeminy, or it may appear normal. A prolonged Holter monitor, however, will usually demonstrate abnormalities (104). Episodes of muscle weakness usually begin before the age of 10 years or in adolescence but can fluctuate widely in severity. Proximal myopathy may be seen (104). Muscle creatine kinase levels remain normal or slightly elevated. Historically, muscle biopsies have demonstrated tubular aggregates, variability of fiber size, and central nuclei, but muscle biopsy is not recommended as the findings are nonspecific. Yoon and colleagues described a distinct neurocognitive phenotype associated with Andersen-Tawil syndrome, characterized by deficits in abstract reasoning and executive dysfunction (116). Most patients (approximately 70%) have mutations in KCNJ2 on chromosome 17q23, the gene encoding Kir2.1, an inwardly rectifying potassium channel expressed in cardiac and skeletal muscle and brain (71). These cases are designated Andersen-Tawil syndrome type 1. The remaining 30% of patients presumably have some other as-yet-unidentified mutation and are designated Andersen-Tawil syndrome type 2 by some authors.
There are case reports and small series of recurrent KCNJ5 gene variants in Asian populations linked to Andersen-Tawil syndrome and long QT syndrome (107; 41). Despite some in vitro evidence of loss-of-function effect (114; 41), most reported patients had only a cardiac phenotype (107), and in other families, a significant “reduced penetrance” was postulated (35). Other authors have argued that the variant in question is not disease-causing, as its prevalence among the general population is higher than the incidence of long QT (49).
|
• Myotonia is the core feature of the nondystrophic myotonias, which affect skeletal muscle only and have none of the systemic features of myotonic dystrophy. | |
|
• Myotonic dystrophy type I is much more common than nondystrophic myotonias. | |
|
• Young people with Myotonic dystrophy type I may present with myotonia and lack systemic features early in the course of their illness. |
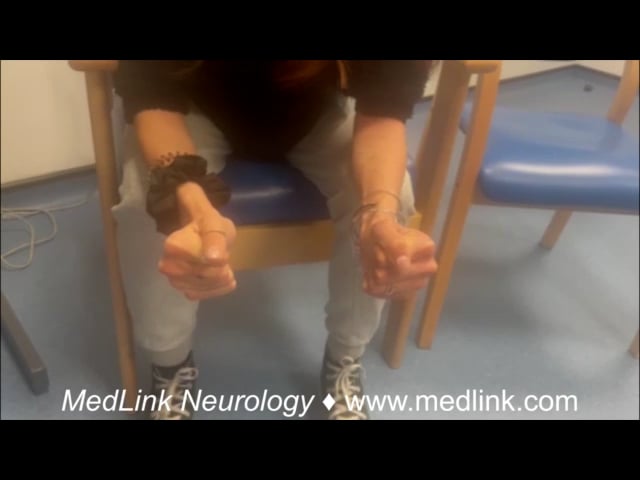
Paramyotonia congenita. Paramyotonia congenita is an autosomal dominant disorder due to mutations in the SCN4A gene that codes for the alpha subunit of the voltage-gated skeletal muscle sodium channel Nav1.4 (74). It is allelic to hyperkalemic periodic paralysis, ie, due to mutations in different locations in the same gene. Myotonia is the primary symptom (Table 2) and is dramatically worsened by cold and exercise or rest after exercise (22). The myotonia worsens with repeated muscle contraction (hence, the term “para”-myotonia or paradoxical myotonia). Myotonia can affect any skeletal muscle, but symptoms are typically most prominent in the muscles of the face, including extraocular muscles, eyelids, the jaw, the tongue, and hands (27). Onset is usually in the first decade. Some patients also experience episodic weakness or paralysis that may take hours to reverse. There is some overlap in phenotype between hyperkalemic periodic paralysis and paramyotonia congenita, but the predominant clinical symptom, ie, myotonia or paralysis, usually drives the diagnosis. Serum CK may be normal or elevated. Generalized muscle hypertrophy is relatively common.
Sodium channel myotonia. Sodium channel myotonia is a term used to describe an autosomal dominant myotonic disorder due to SCN4A gene mutations in which there are usually no accompanying episodes of muscle weakness (54). The distribution of myotonia may be similar to paramyotonia congenita, but rather than paradoxical myotonia, these patients may display the warm-up phenomena that are usually more reminiscent of myotonia congenita. Sometimes, paradoxical worsening and warm-up can be demonstrated in the same patient. Extraocular myotonia is strongly indicative of myotonia due to the SCN4A gene, whether it is sodium channel myotonia or paramyotonia congenita (84; 59). Before the genetic basis of the nondystrophic myotonias was known, many different myotonic presentations that lacked paralysis were described clinically. These included myotonia fluctuans (80), myotonia permanens (47), chronic myotonia (47), and acetazolamide-responsive myotonia (73). When it became clear they were all allelic disorders, the term “sodium channel myotonia” was suggested to simplify the diagnosis for patients with isolated myotonia who have mutations in SCN4A (81).
Severe neonatal episodic laryngospasm. Severe neonatal episodic laryngospasm refers to a severe form of sodium channel myotonia that presents at birth or in early infancy (50). Affected babies experience generalized attacks of myotonia that cause whole-body stiffening (sometimes confused with seizure) but have accompanying respiratory muscle and laryngeal myotonia. They experience respiratory distress of variable severity, with or without stridor (58). In milder cases, this is self-limiting without significant sequelae, but apnea and cyanosis may occur in the more severely affected. Frequency is also highly variable. It may be an isolated event or recurrent, with up to 20 events per day described. Those at the severe end of the spectrum require ventilatory and ICU support (29). Massive muscle hypertrophy can be observed, giving a “Herculian” or “mini athlete” appearance (72). Mild dysmorphic features are also reported. G1306E is a recurrent mutation that seems to make individuals particularly susceptible to respiratory muscle myotonia (45; 18), but severe neonatal episodic laryngospasm is not exclusively associated with this genotype. One series demonstrated that respiratory and bulbar symptoms are generally common in children with sodium channel myotonia compared to adults in whom this is a relatively rare finding (60). Other features becoming increasingly recognized in children include intermittent or variable strabismus (84), joint contractures, scoliosis (113), and short stature (54).
Myotonia congenita. Myotonia congenita is due to mutations in the CLCN1 gene that codes for the skeletal muscle chloride channel CLC-1. Myotonia congenita may be either autosomal dominant (Thomsen disease) (101) or recessive (Becker disease) (08), the latter form being the more common. These two conditions are distinguished by the mode of inheritance and, to some degree the, severity of the symptoms. Unlike in paramyotonia congenita, the myotonia improves with repeated contractions (known as a “warm-up” phenomenon).
The distribution of muscle involvement is also in some respects “opposite” to paramyotonia congenita, with the leg muscles being more commonly and severely affected than others; however, like paramyotonia congenita, any skeletal muscle can be affected (27). The clinical features of dominant and recessive forms are generally similar, but there are some differences. Myotonia in the recessive form is often considered more severe and disabling, but the phenotype is variable even within affected family members. Age of onset is typically between 4 and 12 years of age. Some authors report the dominant form to present earlier within the first few years of life, whereas others have found no significant difference in the age of onset (24). A transient muscle weakness that improves rapidly with repetition is commonly described in the recessive form and rarely in the dominant. Muscle hypertrophy is common. Characteristically, hypertrophy producing large calves is highlighted, but as with paramyotonia congenita, it is often generalized. With advancing age, a proximal myopathy can occur. CK may be normal or elevated (67).
|
|
Primary symptom |
Additional symptoms |
Common triggers |
Typical pattern of muscle involvement |
Examination |
Diagnostic “clue” |
|
Myotonia congenita |
Myotonia with warm-up phenomena |
Myalgia Fatigue Episodic muscle weakness that improves with repetitive action |
Initiation of movement or sudden change in speed or direction Cold environment or extremes of temperature (both hot and cold) |
Legs more than hands and face |
Muscle hypertrophy Often prominent in calves but can be generalized Myotonia--eye closure, grip, or gait with warm-up demonstrated Muscle weakness that rapidly improves with repetition Proximal myopathy may occur |
Falls |
|
Paramyotonia congenita |
Paradoxical myotonia made worse by repetitive action |
Myalgia Fatigue Episodic muscle weakness or paralysis |
Repetitive movement Striking response to cold environments or extremes of temperature (both hot and cold) |
Hands and face more than legs Extraocular muscles |
Generalized muscle hypertrophy Myotonia--eye closure, grip, or gait with paradoxical worsening demonstrated Proximal myopathy may occur |
Cold is volunteered as the major precipitator Extraocular myotonia |
|
Sodium channel myotonia |
Myotonia with warm-up phenomena or Paradoxical myotonia made worse by repetitive action or Both |
Myalgia Fatigue |
Any of the above |
Hands, face, and legs often to a similar degree |
Generalized muscle hypertrophy Myotonia--eye closure, grip, or gait with either warm-up or paradoxical worsening Proximal myopathy may occur |
The long-term prognosis for these diseases is generally good, with normal life expectancy. Hypokalemic paralytic crises tend to be more severe in men just after puberty and in the early adult years. This can result in significant absences from school and a detrimental effect on educational potential (59). Over subsequent decades, the frequency and severity of attacks tend to remit, although, in some, they may be replaced by fixed progressive limb weakness (37). Whether treating and reducing the frequency or severity of attacks of paralysis would have any role in preventing later myopathy is uncertain. It should be noted that progressive myopathy can be seen in any of the skeletal muscle channelopathies but usually occurs after the age of 40 years, and ambulation is generally maintained, although some patients do require walking aids, including wheelchairs (17). Evidence of myopathy and muscle damage may be evident on an MRI scan prior to the development of clinically significant weakness (105).
In skeletal muscle channelopathies, episodic muscle weakness or myotonia are the core clinical features, but many patients experience disabling muscle pain or fatigue, which is not as well recognized (88; 30). For some, this can be one of their most problematic symptoms (36; 103), and a good management strategy needs to consider these additional clinical features.
Complications of cardiac arrhythmias triggered by abnormal serum levels of potassium during the episodes of weakness in periodic paralysis can be life-threatening if not attended promptly (11). Overcorrection of hypokalemia can also be dangerous (117). In hypokalemic periodic paralysis, potassium is held intramuscularly during an episode of weakness. As the attack subsides, potassium will return to the serum. If this occurs in combination with intravenous potassium replacement, rebound hyperkaliemia can be fatal (02). It is imperative to continue cardiac monitoring and serum potassium level measurements for some hours after restoration of normokalemia to ensure this does not occur. Dysrhythmia or cardiac arrest from anesthetic-induced hyperkalemia (particularly suxamethonium) can also occur, particularly in paramyotonia congenita, sodium channel myotonia, and hyperkalemic periodic paralysis (03) (see also section below on anesthetic considerations).
In Andersen-Tawil syndrome, ventricular arrhythmia can be an inherent part of the disorder due to Kir2.1 channel expression in both cardiac and skeletal muscle. Sudden cardiac death is relatively rare but described (42). Pre-syncope or syncope is a worrying warning sign. It is becoming increasingly apparent, however, that Holter monitoring may reveal prolonged or concerning ventricular arrhythmia, but the patient may experience few or nonconcerning symptoms (104). This indicates that the clinical history of symptoms alone is insufficient to assess cardiac health. With improved cardiac monitoring (eg, at least yearly Holter monitoring in everyone with a diagnosis of Andersen-Tawil syndrome regardless of symptoms), more patients are being identified to require medical treatment or an implanted cardiac device (53; 61; 68).
Two fatalities have been reported in infants diagnosed with severe neonatal episodic laryngospasm (29; 50). Others experienced significant disability and prolonged ICU stays. This is in part due to delayed diagnosis. The myotonia in severe neonatal episodic laryngospasm responds extremely well to treatment with sodium channel blockers (91).
A 56-year-old college professor had attacks of episodic limb weakness that began at the age of 10 years old. The first bout occurred following a football tournament and lasted 12 hours. Subsequently, episodes of weakness occurred after long car rides, on waking in the morning, after large carbohydrate-rich meals, or with rest after physical exertion. Some episodes were mild, lasting several hours, and others resulted in severe limb paralysis and an inability to walk. However, speech, swallowing, and respiration were unaffected, even with profound quadriparesis. His mother had similar episodes. The diagnosis of familial hypokalemic periodic paralysis was made with the documentation of low serum potassium levels (2.5 mEq/L) during spontaneous attacks of muscle weakness. Treatment with potassium supplementation reduced the severity and duration of the attacks. With daily acetazolamide therapy, the attacks reduced significantly in frequency and severity. At the age of 43 years old, despite continued use of acetazolamide, he also noted some difficulty walking upstairs as quickly as he used to. Mild proximal weakness was found on examination, and MRI of his leg muscles demonstrated fatty infiltration throughout the thigh muscles.
The genetic basis of most patients with typical channelopathy phenotypes has been established. This has prompted significant progress in understanding the pathophysiology of these disorders, largely through voltage-lamp studies in mammalian systems. In very simple terms, symptoms of muscle weakness or paralysis arise from the muscle membrane entering a state of prolonged depolarization. Myotonia is reflective of a hyperexcitable muscle membrane that, after an initial stimulated action potential (initial muscle contraction), continues to spontaneously depolarize and produce a train of further action potentials (delayed relaxation).
Hypokalaemic periodic paralysis and Andersen-Tawil syndrome. Most of the body’s potassium is stored within muscle. At modestly low serum potassium levels, potassium channels in the muscle membrane are activated to expel potassium, restoring serum potassium levels but also maintaining the normal excitability of the muscle membrane. Healthy muscle will hyperpolarize with reduced serum potassium levels, but the ability of these ion channels to compensate at more profound levels of hypokalaemia is eventually overwhelmed, and the muscle membrane depolarizes (enters a state of inexcitability), causing muscle weakness or paralysis (14).
In Andersen-Tawil syndrome, Kir 2.1 sarcolemmal potassium channels are inherently defective due to mutations in the KCNJ2 gene, impeding this restorative potassium current. In hypokalemic periodic paralysis, mutations of the CACNA1S and SCN4A genes cause dysfunction of the Cav1.1 and Nav1.4 channels. Their effect on main-channel gating and their respective calcium and sodium currents is relatively modest. However, the mutations nearly all replace positively charged amino acid residues in the channel voltage sensors (57), introducing an anomalous inward cation leak current (96; 93). This inward current impedes the outward compensatory potassium current (40).
The overall effect of impairing outward potassium conductance in Andersen-Tawil syndrome and hypokalemic periodic paralysis is to impair the muscle’s ability to compensate in the presence of low serum potassium. As a result, paradoxical depolarization of the muscle membrane occurs. Affected individuals experience recurrent episodes of muscle weakness with low or relatively low (including physiologically low normal) serum potassium levels that may not ordinarily be low enough to overwhelm compensatory mechanisms in those without a genetic predisposition.
There have been reports of muscle weakness occurring in the presence of both low and high serum potassium in Andersen-Tawil syndrome. Data have demonstrated that the potassium sensitivity depends on the severity of Kir2.1 reduction in function (20). Thus, for moderate loss (10% to 30% of WT), high serum potassium will trigger weakness, and either high or low potassium may trigger weakness if there is severe loss (less than 10%).
Hyperkalemic periodic paralysis and sodium channel myotonias. Early physiological data implicated a potassium-induced alteration in membrane sodium conductance in hyperkalemic periodic paralysis. In vitro studies of muscle from patients with hyperkalemic periodic paralysis demonstrated persistent inward current potentiated by potassium and blocked by tetrodotoxin (indicating that it is likely mediated by persistent sodium influx) (79). Patch clamp analysis of cultured myotubes from an individual with hyperkalemic periodic paralysis revealed elevated levels of extracellular potassium-induced abnormal sodium channel inactivation with prolonged or repetitive openings (15). Therefore, it came as little surprise that linkage studies implicated SCN4A, the gene encoding the alpha-subunit of the skeletal muscle voltage-sensitive sodium channel Skm-1, on chromosome 17q as the causal gene for hyperkalemic periodic paralysis as well as paramyotonia congenita (81).
In general, the phenotypes of both hyperkalemic periodic paralysis and paramyotonia congenita can be explained (at least in part) by defective inactivation of voltage-gated sodium channels alone or in combination with accelerated recovery from inactivation. To summarize these complex data, two regions of the sodium channel are involved in normal inactivation, which may be altered in periodic paralysis or paramyotonia congenita: (1) the intracellular loop between domains III and IV that is thought to act as an inactivation particle, or (2) the docking site that functions as a receptor for the inactivation gate (III-IV loop). Many of the mutations identified occur in locations that interrupt either function; for example, one of the glycine pair (Gly 1306) is substituted in three different mutations that sterically limit movement of the III-IV loop, thus impairing sodium channel inactivation that results in membrane depolarization due to persistent inward sodium current. The exact mechanism by which each mutation causes either a predominantly myotonic phenotype or a paralytic phenotype is probably based on the mutation’s ability to interfere with sodium channel fast inactivation or slow inactivation, causing either slight depolarization that does not inactivate sodium channels but allows recurrent activation (myotonic phenotype) or more severe depolarization that leads to sodium channel inactivation and inexcitability (paralytic phenotype) (13). Slow inactivation of sodium channels is based on conformational changes within the structure of the channel and occurs over a much longer period as compared to fast inactivation (33). Impaired slow sodium inactivation appears to correlate with the hyperkalemic periodic paralysis phenotype, whereas mutations that do not affect slow inactivation but interfere with fast inactivation are associated with nonparalytic, potassium-sensitive myotonic disorders (33; 108). The correlation is, however, incomplete because some hyperkalemic periodic paralysis patients demonstrate electromyographic, and sometimes clinical, myotonia. Furthermore, how hyperkalemia potentiates each phenotype is not clear.
Myotonia congenita. Myotonia congenita has been shown conclusively to be caused by genetic variants that cause a loss of function of the muscle chloride channel CLC-1 (12). The ensuing reduction in chloride current increases sarcolemmal excitability and delays the termination of muscle contraction. Experimental evidence suggests that chloride conductance needs to fall to at least 50% of normal before myotonia occurs. The ClC-1 channel is a homodimer consisting of two subunits, each with its own chloride-selective pore. CLCN-1 gene variants either lead to a reduction in functional channel expression or a shift in the voltage dependence of activation towards depolarizing voltages. Some mutant subunits exert a dominant negative effect on their wild-type subunit partner. Variants with a dominant-negative effect are more likely to be inherited in a dominant manner. Those that don’t are typically associated with a recessive inheritance pattern (99).
The prevalence of skeletal muscle channelopathies is not well defined. One study in the Netherlands calculated minimum point prevalence rates of 2.38/100,000 for skeletal muscle channelopathies as a group, with a breakdown of 1.70/100,000 for the nondystrophic myotonias and 0.69/100,000 for the periodic paralyses (97; 98). This study confirmed myotonia congenita as the most common of the sodium channel myotonias, with hyperkalemic periodic paralysis and Andersen-Tawil syndrome the rarest. Geographical differences are seen, and an earlier study of myotonia congenita alone estimated a prevalence of 9/100,000 in Northern Scandinavia (100). This was mirrored by an estimate of 7.3/100,000 in Northern Finland (07).
• Recognizing and avoiding triggers reduces symptoms. | |
• Dietary modifications can be beneficial. | |
• Prophylactic medications are available to reduce symptom frequency and severity. | |
• Careful cardiac monitoring allows for early identification of conduction abnormalities and prompt treatment where appropriate in Andersen-Tawil syndrome. | |
• Concomitant medication that can exacerbate symptoms should be avoided. |
Clear triggers of symptoms are well established for skeletal muscle channelopathies, and their avoidance can help minimize symptoms. A cold environment can be detrimental in both periodic paralysis and myotonia but is a common trigger to myotonia and, especially, paramyotonia congenita. Warm clothing (eg, extra pairs of socks, thermal underwear) is often useful. Children may request to be excused from outdoor play at school in the winter. A warm environment can, conversely, help to reduce symptom severity, but being “too hot” can also exacerbate symptoms. Other triggering factors, such as rest after activity or concomitant viral illness, are also universally seen. Warm-up before exercise and warm-down after can be beneficial, and a physiotherapist may advise an exercise regime (109).
Hyperkalemic attacks are triggered by anything that raises serum potassium, eg, potassium-sparing diuretics or foods rich in potassium (and can be improved with carbohydrate ingestion), whereas hypokalemic attacks may be induced by anything that lowers serum potassium, eg, starvation or fasting, intravenous dextrose infusions, or carbohydrate ingestion (and can be improved with potassium intake). Swapping fast-release carbohydrates for slow-release can prevent surges in insulin that are more likely to induce hypokalemia (59).
In patients with Andersen-Tawil syndrome, medications that may prolong QTc should be avoided to prevent life-threatening ventricular arrhythmias or sudden cardiac death (70). Corticosteroids have been implicated in provoking paralytic episodes and should be used cautiously (Arzel Hezode et al 2009). This is thought to be due to either the hypokalemic effect via mineralocorticoid activity or direct effects on potassium channel activity (09). Drugs causing hypo or hyperkalemia should be avoided in the relevant periodic paralysis. Myotonia can also be exacerbated by medication, eg, beta-blockers and opiates.
Periodic paralysis. Generally, the differential diagnosis includes secondary causes of periodic paralysis due to hypokalemia or hyperkalemia from renal, adrenal, gastrointestinal, alcohol, and drug-induced causes as well as thyrotoxicosis (34; 01). Thyrotoxic periodic paralysis is associated with hypokalaemia and appears most commonly, although not exclusively, in Asian males (90). Rarely, a cocaine “binge” can induce acute-onset hypokalemic paralysis (66). In these cases, hypokalemia may be induced by intracellular shifts of potassium secondary to the adrenergic effects of cocaine. Excessive glycyrrhizic acid intake in the form of licorice may cause secondary hypokalemic periodic paralysis through the induction of a hypermineralocorticoid state (77).
Nondystrophic myotonia. The main differential for anyone presenting with myotonia is either myotonic dystrophy type 1 or myotonic dystrophy type 2. Myotonic dystrophy type 1 is much more common than any of the nondystrophic myotonias. In young patients or those with milder presentations of myotonic dystrophy type 1, the systemic features may not yet have occurred or may not be obvious, and these presentations can be easily confused with nondystrophic myotonia. Rarer causes of myotonia include Pompe disease (38) and drug use (colchicine). In some genetic myopathies, myotonic potentials may be seen on EMG, but myopathy usually dominates the clinical picture (63).
Periodic paralysis. Guillan-Barré syndrome may be confused with periodic paralysis. Sensory symptoms do not occur in periodic paralysis, which can help to discriminate, but they may also be absent in Guillan-Barré syndrome motor variants. Profound hypokalaemia is not usually observed in Guillan-Barré syndrome (32), but mild hypokalemia may be if the patient has experienced a preceding viral illness, especially gastrointestinal. Occasionally, myasthenia gravis and other neuromuscular transmission disorders may be confused with periodic paralysis. However, ocular and bulbar weakness is typical of myasthenia and rarely occurs in periodic paralysis.
Nondystrophic myotonia. The rare childhood disorders Schwartz-Jampel syndrome (caused by mutations in the basement membrane protein perlecan) and Brody disease (caused by mutations in ATP2A1, leading to abnormal muscle contractures with exercise: “pseudo-myotonia”) can be mistaken for one of the nondystrophic myotonic disorders. This also applies to other paroxysmal conditions that can impair movement (eg, paroxysmal movement disorders), but in these, there is no myotonia on EMG testing.
The diagnosis of periodic paralysis and nondystrophic myotonia is usually confirmed by genetic testing. Advances in the availability, speed, and reduction in the cost of genetic testing mean that in many countries the SCN4A, CACNA1S, KCNJ2, and CLCN1 genes are now sequenced in parallel. It can still take some months for results to be returned, however, and given the available treatment for the majority of secondary causes of periodic paralysis, it is prudent to perform simple blood tests to exclude these other possibilities (eg, renal and thyroid function). In a patient with myotonia, consider testing acid alpha-glucosidase enzyme levels. A careful drug history is also essential for periodic paralysis and nondystrophic myotonias.
Obtaining a raised or lowered potassium level during symptomatic weakness, although helpful diagnostically, can be difficult to obtain. This is because the attack has often subsided or is beginning to subside by the time a patient presents for medical attention. In a patient with profound weakness or quadriparesis due to hypokalemic periodic paralysis, serum potassium would be expected to be below the normal range. With less severe symptoms, caution is needed as the serum potassium may be moving up or down relative to its baseline (pre-attack level) but still may be within normal range.
Attention should be given to craniofacial and skeletal anomalies that suggest the diagnosis of Andersen-Tawil syndrome. An EKG should be performed in all patients presenting with periodic paralysis to identify repolarization abnormalities such as QTc prolongation or a prominent U wave suggestive of Andersen-Tawil syndrome.
Electrodiagnostic evaluation of patients with primary periodic paralysis is usually performed using the long exercise test (62). The overall sensitivity of the test has been estimated at 60% to 80% with high specificity, using a cutoff of a 40% decline in CMAP amplitude from the peak following 5 minutes of exercise (62; 25; 78). A long exercise test can also be useful in documenting the presence of a muscle membrane abnormality when the history is vague or atypical (44). It should be noted, however, that false negatives do occur and that a positive test does not distinguish between primary and secondary forms of periodic paralysis (78).
During the long exercise test, compound muscle action potentials (CMAPs) are recorded from the abductor digiti minimi muscle, supramaximally stimulating the ulnar nerve at the wrist. The subject rests for 10 minutes prior to the test, and then CMAP amplitude and area are recorded every minute for 5 minutes to establish a stable baseline. The fingers are then abducted against resistance for 5 minutes, with brief 3- to 4-second relaxations to avoid ischemia. This is followed by a 40- to 60-minute post-exercise rest period. Supramaximal stimulations are administered every minute during exercise and every 2 minutes during the postexercise period. A reduction in CMAP amplitude of 40% or more from the peak is considered abnormal. It can be a tedious test to perform or tolerate, requires an experienced neurophysiologist, and has a relatively low sensitivity. In many centers, if a patient has a typical history of periodic paralysis, genetic testing will be performed without a long exercise test.
The long exercise test cannot determine a clinical diagnosis of functional neurologic weakness, but results can contribute to the overall clinical assessment. Although a negative test can and does occur in those with genetic forms of periodic paralysis, in one series, this did also relate to frequency of attacks; that is, those with infrequent attacks were more likely to have a negative test, whereas all those reporting daily or weekly episodes of weakness had a positive test (78). Daily attacks of weakness and a negative test may support but not diagnose a clinical impression of functional neurologic disorder, especially if seen alongside other positive discriminators, such as Hoover’s sign.
Characteristic electrodiagnostic findings, along with a clinical history, can help determine the most likely genetic diagnosis to some degree. The presence of clinical or electrical myotonia with a positive long exercise test and characteristic history is virtually diagnostic of hyperkalemic periodic paralysis or paramyotonia congenita due to a sodium channel mutation.
Although genetic tests are now much more readily available, an EMG remains useful in the diagnosis of myotonic disorders (21). In the clinic, it can sometimes be difficult to see myotonia or to be certain that myotonia is causing a delay in hand opening. If there is a positive family history of myotonic disorder, an assumption that described symptoms are due to myotonia can lead to an erroneous diagnosis and treatment. Conversely, uncertainty about clinical examination can delay treatment while genetic tests are awaited. EMG myotonia will also support pathogenicity in cases of genetic variants of uncertain significance. EMG can be particularly useful in cases of severe neonatal episodic laryngospasm because, in the appropriate clinical setting, a demonstration of myotonia can lead to an almost instant diagnosis.
A muscle biopsy is not recommended as part of the diagnostic workup. Myopathic changes can be seen, but they are nonspecific, and diagnosis can usually be reached using a combination of clinical history, examination, neurophysiology, and genetic tests. MRI scanning can be useful to contribute to the suspicion that there is a neuromuscular diagnosis, as it may be abnormal, especially in those having frequent symptoms. No specific diagnostic patterns have yet been identified. A raised CK provides a similar clue (ie, if raised, this indicates a neuromuscular diagnosis but not, of course, which one).
Conservative management of acute attacks of periodic paralysis and nondystrophic myotonia is directed at educating patients about diet and lifestyle changes to minimize triggers (39; 109). Fatigue and pain management also requires attention despite limited data or guidance on the optimal approach. Patients with skeletal muscle channelopathies can also become relatively sedentary because exercise may provoke their symptoms. Guidance from a physiotherapist can help patients avoid secondary complications of weight gain and cardiovascular risk (56). There is also some evidence that immobility or lack of activity may lead to vitamin D deficiency in periodic paralysis (64).
Periodic paralysis. Carbonic anhydrase inhibitors (acetazolamide and dichlorphenamide) have been empirically used for decades to treat periodic paralysis. The short- and long-term effects of dichlorphenamide on attack frequency and quality of life in hyperkalemic and hypokalemic periodic paralysis were studied with two multicentered, randomized, double-blind, placebo-controlled trials lasting 9 weeks followed by a 1-year extension phase (85; 87). The dose for dichlorphenamide in treatment-naive patients was 50 mg twice daily, whereas patients already taking acetazolamide were assigned an equivalent dosage of dichlorphenamide, calculated as 20% of the acetazolamide dosage. In the hypokalemic periodic paralysis trial, the median attack rate, severity-weighted attack rate, and attack duration were significantly reduced with dichlorphenamide. In the hyperkalemic periodic paralysis trial, the median severity-weighted attack rate was significantly reduced in the dichlorphenamide group compared to placebo, although the group differences in median weekly attack rate and duration did not reach significance. The most common side effects were paresthesias, cognitive disorders, dysgeusia, and renal calculi. The maximum recommended dose is 200 mg daily. Despite trial evidence to support efficacy, the manufacture of dichlorphenamide is very limited. It is, therefore, difficult to obtain and is a high-cost drug (55).
No randomized, controlled trials have been performed with acetazolamide in periodic paralysis, although it is routinely used in both hyperkalemic and hypokalemic periodic paralysis. Its use is largely based on anecdotal reports and nonrandomized, single-blind trials (94). A dose of 250 mg two to four times daily or 500 mg twice daily is usually effective in ameliorating or reducing the frequency and severity of attacks. The side-effect profile is similar to that of dichlorphenamide. An increased risk of renal calculi is seen in patients older than 40 years of age compared to the general population in those on long-term treatment. A yearly renal ultrasound may help to prevent complications from undetected nephrolithiasis (99). Acetazolamide remains widely available and low-cost.
Paradoxically, acetazolamide may also increase the frequency and severity of attacks in patients with hypokalemic periodic paralysis due to sodium channel mutations; this is sometimes a clue to the presence of a sodium channel mutation as opposed to the more common calcium channel mutations (95). One cohort study in patients with genetically confirmed hypokalemic periodic paralysis showed 31 of 55 (56%) patients with CACNA1S mutations benefited from acetazolamide compared with only three of 19 (16%) patients with SCN4A mutations (p < 0.002) (58).
Diuretics also have prophylactic benefits in periodic paralysis. Potassium-sparing diuretics are used for hypokalemic periodic paralysis (eg, spironolactone and amiloride), and potassium-wasting diuretics are used for hyperkalemic periodic paralysis (eg, thiazide) (39). They may be tried in combination with acetazolamide or alone.
Mild, nonresistive exercise at attack onset (eg, gentle walking or moving legs on the spot) or a carbohydrate snack may be helpful for acute hyperkalemic episodes. Salbutamol (one to two puffs of 0.1 mg) and other beta-agonists may be effective (94). Acute hypokalemic attacks often respond simply to oral potassium loading over several hours, although a severe crisis may warrant intravenous potassium loading. It is recommended that such intravenous therapies employ saline solutions rather than dextrose in water because the carbohydrate load of dextrose may worsen the weakness. Serum potassium and cardiac monitoring is also mandatory if intravenous replacement is given.
The episodes of muscle weakness in Andersen-Tawil syndrome may also respond well to carbonic anhydrase inhibitors (Gupta et al 2021) or diuretics. Routine prophylactic implantable defibrillator placement is not recommended; however, any patient who develops sustained ventricular tachycardia, Torsade de pointes, or syncope should be evaluated by a cardiologist for possible defibrillator implantation. The role of routine antiarrhythmic therapy in such patients should be managed by a cardiologist (53; 69).
Experimental therapies. Bumetanide has an inhibiting effect on the muscle Na-K-Cl co-transporter. Experimental data from two hypoPP mouse models demonstrated that bumetanide had the ability to prevent loss of muscle force in the presence of hypokalemia and also promote recovery of force once an attack had occurred (111; 112). A randomized, double-blind, placebo-controlled phase II clinical trial administered 2 mg bumetanide to patients with hypoPP who experienced a focal attack of hand weakness during the performance of a long exercise test to determine if bumetanide could abort the attack of weakness (89). It demonstrated no significant adverse events but no significant benefit. A failure to reproduce the effects seen in mice may have been due to small sample size, low bumetanide dose relative to that used in the mouse model, and prolonged immobility of the hand. Further dose escalation studies combining bumetanide with gentle hand movement may be warranted, but at present, there is no use of bumetanide in the clinical setting.
Potassium channel agonists were put forward as a potential treatment for hypokalaemic periodic paralysis more than 30 years ago based on the proposal that they could hyperpolarize the muscle membrane and offset anomalous depolarization caused by genetic variation. More recent preclinical data using mouse models of hypokalaemic periodic paralysis due to both SCN4A and CACNA1S variants have demonstrated that retigabine can prophylactically protect muscle fibers against hypokalaemic-induced loss of force and assist in the recovery of muscle force if administered acutely (75). Retigabine was marketed as an anti-seizure medication but was withdrawn from the market in 2017 due to side effects and limited use. It remains to be seen if new modified potassium channel agonists become available that could offer an alternative treatment for hypokalaemic periodic paralysis.
Nondystrophic myotonia. Sodium channel blockers form the mainstay of treatment for myotonia regardless of genotype. The most commonly used is probably mexiletine, which has large randomized controlled trial evidence to support its efficacy and safety (97; 98). Mexiletine has been licensed specifically for the treatment of nondystrophic myotonia, with the consequence that prescribing for this indication is high-cost. Lamotrigine also has some trial data demonstrating efficacy (05) and remains much more affordable. There are anecdotal data and small series for numerous other options, including carbamazepine, propafenone, flecainide, and phenytoin.
A more recent head-to-head study of lamotrigine versus mexiletine confirmed that both drugs showed significant efficacy in treating myotonia but failed to demonstrate that lamotrigine was noninferior to mexiletine (106). There are pros and cons to choosing either medicine aside from efficacy. Mexiletine is licensed for nondystrophic myotonia, and benefit is generally experienced in a shorter time-frame than seen with lamotrigine. Limitations are that it can be contraindicated in cardiac disease, it is expensive, and its safety profile in pregnancy is largely unknown. Lamotrigine may take longer to titrate to a dose where efficacy is felt by patients, and it is unlicensed for myotonic disorders. This latter point can be of particular concern if considering the rare but serious side effect of Stevens-Johnson syndrome. It does have a good safety profile in pregnancy, however, and remains readily available at a low cost. Although routine cardiac screening is not recommended, the FDA added a warning to lamotrigine’s label regarding its ability to exhibit class IB antiarrhythmic activity (26). Therefore, if there is a cardiac contraindication to using mexiletine, a cardiologist’s opinion is likely needed before commencing lamotrigine.
Most women describe that symptoms worsen during pregnancy (92), although some describe an improvement (23). Medication is generally contraindicated due to either known teratogenic effects (acetazolamide) or lack of data. There have been reports, however, of mexiletine being used safely during pregnancy (82), and there are safety data for lamotrigine from the epilepsy literature. Withdrawal of medication may contribute to the exacerbation of symptoms seen during pregnancy.
Many women with skeletal muscle channelopathies have successful spontaneous vaginal deliveries and shouldn’t automatically be denied this option because they have a rare disease. The symptoms of myotonia or paralysis may prolong labor, however, which needs careful monitoring, and we generally recommend delivery in a hospital where pediatric and anesthetic support is available if needed (56). Another indication for this is for parents who carry SCN4A myotonic mutations, which, if inherited by the baby, may present with postnatal respiratory complications or severe neonatal episodic laryngospasm.
If possible, local or regional anesthesia is preferred to general. If a general anesthetic is given, propofol and nondepolarizing agents are safe, but depolarizing muscle relaxants and suxamethonium should be avoided (102). Both can induce a myotonic crisis (52), and suxamethonium can induce significant hyperkalemia with secondary arrhythmia (03), especially in those with hyperkalemic periodic paralysis or paramyotonia congenita. Unexpected jaw myotonia can make intubation difficult or impossible (52). Management of a myotonic crisis may require intravenous lidocaine. A small jaw in Andersen-Tawil syndrome may also make intubation challenging (68). Other appropriate precautions include minimizing potassium-containing fluid replacement and medications that inhibit potassium excretion in hyperkalemic periodic paralysis and ensuring appropriate potassium repletion (in intravenous saline if required, not in dextrose) in hypokalemic periodic paralysis; in all cases, one should avoid perioperative hypothermia. Early postoperative mobilization is desirable, but feasibility depends on the type of surgery and other mitigating factors. It is unclear whether an increased risk of malignant hyperthermia susceptibility truly exists in patients with periodic paralysis. There have been a handful of case reports (65; 76). Abnormal caffeine contracture tests have been described in excised muscle from patients with inherited myotonia or periodic paralysis, but these tests lack specificity for diagnosing malignant hyperthermia susceptibility in this setting (46).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Emma Matthews MBChB PhD FRCP
Dr. Matthews of St George's University of London has no relevant financial relationships to disclose.
See Profile
Aravindhan Veerapandiyan MD
Dr. Veerapandiyan of University of Arkansas for Medical Sciences has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuromuscular Disorders
Jun. 16, 2026
Neuromuscular Disorders
May. 27, 2026
Neuromuscular Disorders
May. 21, 2026
Developmental Malformations
May. 08, 2026
Neuromuscular Disorders
Apr. 23, 2026
Neuromuscular Disorders
Apr. 23, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026