

Peripheral Neuropathies
Autonomic neuropathy: treatment
Jul. 14, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Neurologic complications of Sjögren disease may involve the central, peripheral, and autonomous nervous systems. The frequency of neurologic involvement is relatively unknown, although the Big Data Sjögren Syndrome Cohort reported a frequency of 6.3% for peripheral neuropathies and 1.9% for central nervous system, according to The European League Against Rheumatism (EULAR) Sjögren's Syndrome Disease Activity Index (ESSDAI) definitions (66). Neurologic involvement often precedes the typical glandular manifestations of Sjögren syndrome and has a wide spectrum of manifestations and underlying neuropathologic mechanisms, making the diagnosis and approach to treatment a difficult challenge that requires a multidisciplinary approach. The diagnosis and treatment of these manifestations must always be coordinated by neurologists. The level of evidence for treatment efficacy of the main drugs used for managing neuroSjogren is limited.
|
• Sjögren disease is a common systemic autoimmune disease, mainly diagnosed in women aged 30 to 50 years old, which is manifested by sicca symptoms and organ-specific systemic involvement. | |
|
• Neurologic symptoms occur in 18% to 45% of patients with Sjögren disease due to involvement of cranial nerves (Bell palsy, trigeminal neuralgia, diplopia), peripheral nerves (sensorimotor neuropathies), and the central nervous system. | |
|
• Other neurologic diseases have to be excluded in patients with Sjögren presenting with neurologic symptoms before considering CNS involvement as specific to Sjögren disease. | |
|
• A high index of suspicion is required given the pleomorphic manifestations and the fact that neurologic symptoms often precede the clinical diagnosis of Sjögren disease. |
In 1933, Henrik Sjögren described the association of keratoconjunctivitis sicca (filamentary keratitis) with arthritis. In 1953, Morgan and Castleman noted the histopathological commonality between the keratitis described by Sjögren and the glandular enlargement described by Mikulicz (52; 50). By 1973, the term “Sjögren syndrome” became widely accepted as these disorders were considered variants of the same process. The name Gougerot-Sjögren syndrome is commonly used in the French literature given that Henry Gougerot first reported, in Paris, the typical symptoms of xerostomia and xerophthalmia due to atrophy of salivary and lachrymal glands.
During initial discussions regarding the organization of the International Symposium on Sjögren Syndrome in Rome (September 2022), two separate projects focused on nomenclature were proposed: one from the United States (Alan Baer) and another from Europe (Manuel Ramos-Casals). These proposals were combined, leading to the formation of an International Task Force with the goal of reaching a consensus on the nomenclature of Sjögren syndrome. The consensus aimed to be grounded in the clinical insights of global experts, current scientific evidence, and the perspectives and experiences of patients.
As a result, the consensus endorses “Sjögren disease” as the official nomenclature to acknowledge distinct pathogenesis and improve clarity in both clinical practice and research (64).
|
• Sjögren disease is a systemic autoimmune disease that affects the exocrine glands, leading to dryness of the main mucosal surfaces, principally in the eyes and mouth. | |
|
• The cause of Sjögren disease is unknown, but genetic and environmental factors seem to play a role. |
Sicca symptoms are one of the most frequent causes of ocular and oral complaints. Sjögren disease may occur in conjunction with other autoimmune disorders, including rheumatoid arthritis and systemic lupus erythematosus. In this setting, it is known as associated Sjögren or Sjögren-overlap syndrome. Moreover, Sjögren disease is a serious disease with an excess of mortality mainly due to hematological cancer. Sjögren disease is more frequent than may be thought, affecting an estimated 2 to 4 million people in the United States and with a prevalence in Europe of 0.6% to 3.3%. Sjögren disease overwhelmingly affects middle-aged women, although children, men and the elderly are also affected.
Systemic involvement in Sjögren disease includes chronic fatigue affecting as many as 50% of patients; arthralgias present in 53%; hematological abnormalities including anemia and leukopenia (33%), increased erythrocyte sedimentation rate (22%), hypergammaglobulinemia (22%); myalgias (22%); and skin lesions such as “burning skin” (18%), cutaneous vasculitis (10%), papular lesions, and annular erythema. Lymphoma may develop in 5% to 7% of patients with Sjögren disease, often associated with cutaneous purpura. Less common manifestations include lung involvement with lymphocytic alveolitis, lymphocytic interstitial pneumonitis, fibrosis, pulmonary pseudolymphoma, and pulmonary hypertension. Other less common manifestations include pericarditis, vascular lesions manifested by Raynaud phenomenon, renal tubular lesions, interstitial nephritis and glomerulonephritis, malabsorption due to lymphocytic infiltrates of the intestine, mild pancreatitis, and hepatitis. Exclusion criteria for the diagnosis of Sjögren disease include infections by HIV, HTLV-1, or hepatitis C virus.
Classification Criteria are updated as new tools are validated. The most recent classification criteria for Sjögren disease were finalized in 2016 and approved by the American College of Rheumatology (ACR) Board of Directors and the EULAR Executive Committee (76). Criteria are based on the following:
First, a patient must either be suspected of having Sjögren disease based on the EULAR Sjögren Syndrome Disease Activity Index (ESSDAI), with at least one domain being positive OR respond positively to at least one of the following questions about symptoms:
|
I. Ocular symptoms: A positive response to at least one of three validated questions: | |
|
1. Have you had daily, persistent, troublesome dry eyes for more than 3 months? | |
|
2. Do you have a recurrent sensation of sand or gravel in the eyes? | |
|
3. Do you use tear substitutes more than three times a day? | |
|
II. Oral symptoms: A positive response to at least one of three validated questions: | |
|
1. Have you had a daily feeling of dry mouth for more than 3 months? | |
|
2. Do you frequently drink liquids to aid in swallowing dry foods? | |
|
III. Ocular signs: Objective evidence of ocular involvement defined as a positive result to at least one of the following two tests: | |
|
1. Schirmer test ≤5 mm/5 min in at least one eye | |
|
2. Ocular Staining Score ≥5 (or van Bijsterveld 1 score ≥4) in at least one eye | |
|
IV. Histopathology: Labial salivary gland with focal lymphocytic 3 sialadenitis and focus score of ≥1 foci/4 mm2. | |
|
V. Salivary gland involvement: Unstimulated whole saliva flow rate ≤0.1 mL/min. | |
|
VI. Autoantibody - presence in the serum of the following autoantibody: | |
|
1. Antibodies to Ro/SS-A | |
|
Exclusion criteria | |
|
(1) History of head and neck radiation treatment | |
|
For Sjögren disease | |
|
The classification of SjD applies to any individual who meets the inclusion criteria,* does not have any of the conditions listed as exclusion criteria,† and has a score of 4 or higher when the weights from the five criteria items below are summed. | |
|
For associated Sjögren disease | |
|
In patients with a potentially associated disease (eg, another well-defined connective tissue disease), the presence of item I or item II plus any two from among items III, IV, and V may be considered as indicative of secondary Sjögren disease. | |
The American College of Rheumatology, in consensus with the Sjögren’s International Collaborative Clinical Alliance (SICCA) investigators, proposed provisional criteria to improve the criteria’s specificity for enrollment in trials for biological agents and other therapies. These criteria require at least two of the following three findings:
|
1. Positive serum anti-SS-A and/or anti-SS-B or positive rheumatoid factors and ANA ≥ 1:320 | |
|
2. Ocular staining score ≥ 3 | |
|
3. Presence of focal lymphocytic sialadenitis with focus score ≥ 1 focus/4mm2 in labial salivary gland biopsies. |
These criteria were found to have a sensitivity of 93% and a specificity of 95% (77).
A number of neurologic manifestations have been reported in about 20% of patients with Sjögren disease (range 6% to 70%) (23; 53). In most cases, neurologic symptoms precede the diagnosis (53). Historically, the most common symptoms include involvement of the peripheral nervous system. The prevalence of peripheral nervous system involvement in Sjögren disease has been reported to range from 2% to over 50%. Bias in study designs, including low numbers of patients and unclearly defined rheumatologic and neurologic diagnoses, as well as changes in classification criteria with the individualization of small fiber neuropathy could explain such variability (17). More recent reports emphasize the involvement of the central nervous system that went underrecognized in the past, often mimicking the clinical symptoms of primary progressive or relapsing-remitting multiple sclerosis. An observational, single center study from Italy found that 5.8% of patients presenting to rheumatology clinic with a diagnosis of Sjögren disease had central nervous system involvement (48) whereas neurologic manifestations in a French cohort were present in 74 of 392 (18.9%) patients, including 63 (16%) with PNS manifestations and 14 (3.6%) with CNS manifestations (19). Table 2 lists some of the most common neurologic manifestations of Sjögren disease.
|
PNS manifestations | ||
|
Neuropathies | ||
|
Painful sensory neuropathy | ||
|
CNS manifestations | ||
|
Focal brain lesions | ||
|
Stroke with motor or sensory deficits, associated with CNS vasculitis | ||
|
Movement disorders and cerebellar syndromes | ||
|
Parkinsonism | ||
|
Diffuse nonfocal symptoms | ||
|
Acute or subacute encephalopathy | ||
|
Spinal cord involvement | ||
|
Transverse myelitis | ||
|
Demyelinating syndromes | ||
|
Optic neuropathy | ||
|
Muscle manifestations | ||
|
Subclinical myositis | ||
|
Myalgias | ||
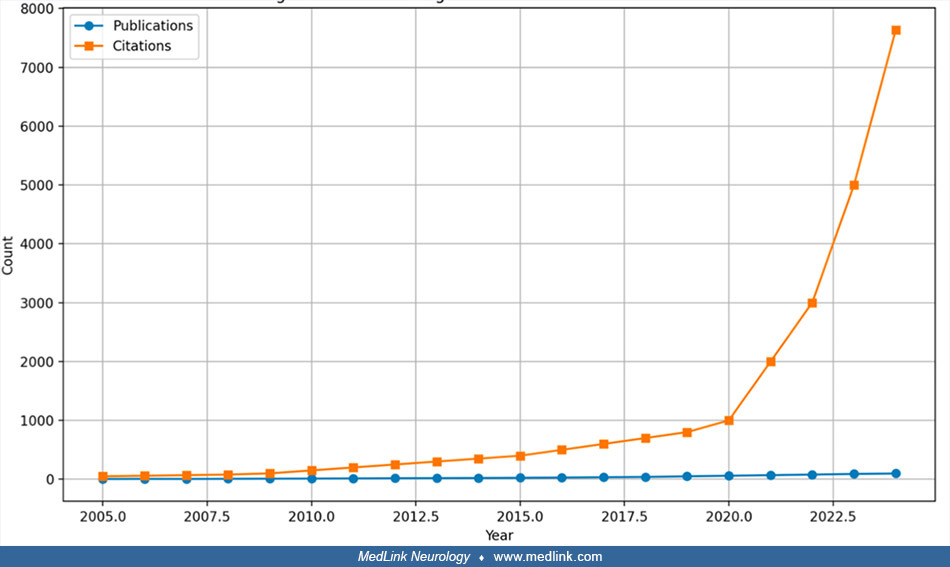
Neurologic manifestations in Sjögren disease can range from mild to severe, often complicating both diagnosis and treatment approaches. A bibliometric review of Akbar and colleagues adopts a comprehensive approach to analyze the research landscape related to these symptoms (02). Prior to 2010, the number of publications was relatively low and inconsistent. However, a significant increase in research activity began around 2015, peaking at 98 publications by September 2023, indicating a growing interest in the neurobiological dimensions of Sjögren disease. Citations for studies on Sjögren disease–related neurologic issues have also seen a steady rise, reaching 7632 in 2023. This pattern highlights the increasing academic and clinical attention to these topics. Early data for 2024 suggest that this upward trend will continue, though citation counts could be underestimated due to incomplete data at this stage. An overview of the annual publication trends related to neurologic symptoms in Sjögren disease is shown in the following figure.
From a geographical standpoint, the United States has emerged as the dominant contributor to research in this field, producing the largest volume of publications and citations, followed by China, the United Kingdom, and Germany. This distribution points to a growing global interest in the neurologic aspects of Sjögren disease, though it also highlights an uneven distribution of research output across different regions. The rising research activity in China signals its increasing involvement in international studies, particularly in the fields of autoimmunity and neuroimmunology. Expanding research efforts in developing regions is essential to addressing geographical disparities in healthcare access and ensuring a broader, more inclusive understanding of Sjögren disease across diverse populations. High-impact publications are vital for enhancing the visibility of research and have a significant influence on the development of clinical guidelines and treatment protocols (02).
Peripheral neuropathies frequently coexist with Sjögren disease. Studies have shown that in numerous patient cohorts with Sjögren disease-related neuropathy, over 90% of individuals experienced neuropathic symptoms before receiving a diagnosis of Sjögren disease. Furthermore, more than a third of patients developed neuropathy before experiencing the hallmark sicca symptoms typically associated with the disease. This presents a diagnostic dilemma; however, identifying the typical neuropathy patterns linked with Sjögren disease is crucial for guiding a more effective diagnostic assessment.
Most studies report that the frequency of peripheral nervous system disease is between 4% and 20% (62). Mori and colleagues reviewed 92 patients (86% women, mean age 60 years) with Sjögren disease and found the following types of neuropathies: sensory ataxia (39%); sensory painful neuropathy (20%); trigeminal neuropathy (17%); multiplex mononeuropathy (12%); multiple cranial neuropathies (5%); autonomic neuropathy (3%); and radiculoneuropathy (4%) (53). Brito-Zerón and colleagues analyzed 563 consecutive patients with suspected peripheral nervous system involvement and found a 10% prevalence of Sjögren disease-related peripheral neuropathy, including axonal sensorimotor polyneuropathy (n=24), pure sensory neuronopathy (n=15), multiplex mononeuropathy (n=15), and demyelinating polyradiculoneuropathy (n=1). Survival was significantly reduced in patients with peripheral neuropathy (especially in those with mononeuropathy multiplex and axonal polyneuropathy) in comparison with the control group (log rank = 0.001) (13).
Dyck described the following nerve fiber lesions in these cases (24). In the ataxic variety, large primary afferent neurons or fibers are selectively affected as suggested by inflammatory infiltrates found in spinal ganglia. In trigeminal neuropathy, all classes of sensory neurons or fibers are involved. In sensory and autonomic neuropathy affecting limb or trunk, all classes of sensory neurons or fibers are affected, but symptoms suggest predominant small fiber sensory involvement with either tactile (allodynia) or thermal hypersensitivity (hyperalgesia), as well as sudomotor abnormalities such as decreased sweating in toes and feet or in an asymmetrical, radicular pattern. These sensory and autonomic neuropathies may occur with minimal or no evidence of sicca symptoms (24).
A total of 199 patients diagnosed with Sjögren disease were included in the study carried out in Turkey, with neurologic involvement observed in 32% of cases. Peripheral nervous system involvement was detected in 23% of patients, with axonal sensorimotor polyneuropathy being the most prevalent subtype at 11%. In a multivariate regression analysis, only articular involvement exhibited a significantly higher risk for developing neurologic complications (odds ratio [OR] 10.01). Additionally, among patients with peripheral nervous system involvement, there was a statistically significant increase in the frequency of anti-Ro/SS-A positivity, low levels of C3, and positivity in Schirmer’s test compared to those without peripheral nervous system involvement (p = 0.032, p = 0.044, and p = 0.029, respectively) (42).
Two large Italian cohort studies of 652 and 1115 patients with Sjögren disease, respectively, reported a prevalence of peripheral nervous system involvement in approximately 4% of cases (10; 62). Among these, the most common phenotype was axonal sensorimotor neuropathy, with a prevalence of 2% (10). These studies also helped define the clinical phenotype of patients with Sjögren disease and peripheral nervous system involvement. Patients with cryoglobulinemic vasculitis were found to have the highest risk of developing peripheral nerve involvement, especially axonal sensorimotor neuropathy (RR: 15.3 [95% CI: 3.9–60.3]; p < 0.001) (62). This association was further supported by the presence of circulating cryoglobulins and laboratory features, such as low C3 and C4, rheumatoid factor positivity, polyclonal hypergammaglobulinemia, and an increased risk of lymphoma. Cryoglobulinemia, and specifically cryoglobulinemic vasculitis, serves as a pathophysiological link between the vasculitic features (such as peripheral neuropathy) and the lymphoproliferative complications observed in Sjögren disease. This dual clinical and immunopathological involvement is well illustrated in a study by Argyropoulou and colleagues, which compared patients with Sjögren disease and cryoglobulinemic vasculitis to those with HCV-related cryoglobulinemia (08). The study showed that although vasculitic peripheral neuropathy is a recognized manifestation of Sjögren disease, it tends to be less severe and less frequent compared to HCV-related cryoglobulinemic vasculitis. In contrast, lymphoproliferative features, such as lymphadenopathy and lymphoma, are more commonly observed in Sjögren disease. Therefore, in patients with suspected Sjögren disease and significant peripheral neuropathy, anti-HCV serologies should always be evaluated.
Axonal neuropathies. Between 17% and 39% of patients with Sjögren disease have minor symptoms of peripheral neuropathy (24). The most common axonal forms are symmetrical sensory neuropathy and sensorimotor neuropathy (23; 24; 53). Vibration perception threshold and thermal perception are sensitive and useful methods of monitoring peripheral nerve function in these patients. The course is generally slowly progressive. Autonomic symptoms are rare. A common complication is carpal tunnel syndrome. Motor and sensory action potentials in the involved nerves are markedly reduced. Sural nerve biopsies have shown vasculitis of vasa nervorum, perivascular cellular infiltration, and necrotizing vasculitis along with focal or multifocal axonal degeneration of large and small myelinated fibers and minor demyelination.
Painful sensory neuropathy (without sensory ataxia). Painful sensory neuropathy can present as acute, subacute, or chronic onset of burning dysesthesias in the toes, feet, or hands, usually in either one limb or over the entire body, including the trunk and the face (53). Sensory loss in a stocking distribution is typically associated with cryoglobulinemic vasculitis (04). Dysautonomia may also be present, although there is no evidence of dorsal column involvement. These patients had preserved motor and vibratory or proprioceptive function, with impairment only in pinprick and temperature sensation on neurologic exam. Sural nerve biopsies show small-fiber loss suggesting predominant impairment of small sensory neurons with preservation of large-diameter sensory neurons (20). The measurements of intraepidermal nerve fiber densities in skin punch biopsy specimens show less than 3.4 fibers/mm, which is consistent with the morphological criteria for small-fiber neuropathy (32).
Painful small-fiber neuropathy. Small-fiber involvement is common in Sjögren disease, occurring either as a “pure,” painful small-fiber neuropathy or in combination with large sensory fiber (LSF) involvement. However, even patients with “mixed” large- and small-fiber sensory neuropathy often have small-fiber neuropathy symptoms (eg, subjective sensory symptoms, mostly painful, including burning, numbness, prickling, paresthesia, and dysesthesia) as a chief complaint, with large-fiber involvement detected only on clinical examination or electrophysiology. The diagnosis of a pure small-fiber neuropathy requires specific histologic or neurophysiologic examinations. The histologic investigation is based on a skin biopsy and the evaluation of the intraepidermal nerve fiber density (IENFD) after immunostaining with anti-neuropeptide (peptide gene product 9.5) antibodies. Among 317 patients with Sjögren disease, Lacout and colleagues found large- and small-fiber neuropathies in 22 (6.9%) and 17 (5.4%) patients, respectively (44). In the study by Seeliger and colleagues, there were no observed differences in patients with small fiber neuropathy who exhibited sensory symptoms (72). However, 24 out of 97 individuals (25%, median age 48.5 years, 75% female) were diagnosed with Sjögren's disease. Additionally, patients with both small fiber neuropathy and Sjögren disease had significantly lower intraepidermal nerve fiber densities (IENFD) (mean 2.6 ± 1.2/mm) compared to patients with idiopathic small fiber neuropathy (mean 3.2 ± 1.5/mm; p = 0.048). Sene and colleagues compared 40 patients with small-fiber neuropathy to 100 without peripheral neuropathy and found that patients with small-fiber neuropathy were older and more frequently had xerostomia and arthralgias and a lower prevalence of serum immunological markers (antinuclear antibodies, anti-Ro, and anti-La, rheumatoid factor, and hypergammaglobulinemia) (35% vs. 62%; p=0.005) (75). Three additional studies reported lower frequencies of anti-Ro antibodies in patients with Sjögren disease presenting with small fiber neuropathy in comparison with those with large fiber neuropathy: 43% versus 55% (20), 39% versus 64% (53), and 5.4% versus 6.9% (p=0.002) (44), respectively. This type of neuropathy is more frequently reported in males and in those without hypergammaglobulinemia or positive autoantibodies.
Sensory ataxic ganglionopathy. This is a distinctive type of sensory neuronopathy with a reported association with ganglion neuronal antibodies in one study (54). The antigenic target of these antibodies is not known, and there are no posterior studies that confirmed this finding. The neuropathology of the sensory neuronopathy is characterized by lesions of the dorsal root ganglia with neuronal loss and presence of mononuclear cell infiltrates (ganglionitis) but without vasculitis (54). Clinically, it is characterized by a profound loss of proprioception and vibratory perception leading to sensory ataxia, with a positive Romberg sign and global areflexia, without motor involvement. The sensory symptoms are usually asymmetrical, segmental, or multifocal, including trigeminal nerve involvement (53). Birnbaum and colleagues described dorsal root ganglion enlargement and T2 hyperintensity in early dorsal root ganglionitis (12), whereas Yoshida and colleagues detected atrophy of the dorsal root ganglion in chronic sensory ataxic ganglionopathy (88).
There is usually autonomic involvement manifested by abnormal pupillary responses including Adie pupils, anisocoria, and oval pupils; orthostatic hypotension decreases in 123I-MIBG cardiac uptake, hypohidrosis, and segmental anhidrosis of the trunk. Sensory potentials are absent, contrasting with normal motor potentials and normal peripheral nerve conduction velocities. Somatosensory evoked potentials are usually absent. Sural nerve biopsies show a selective loss of large, myelinated fibers, with minimal inflammation or vasculitis. Cervical spinal cord MRI may show hyperintense T2-weighted signals corresponding to involvement of the fasciculus cuneatus and fasciculus gracilis in the dorsal columns, proportional to the severity of the damage. This form of neuropathy is chronic and progressive and is usually only minimally responsive to treatment. Some authors have estimated a prevalence of sensory neuronopathy around 5%. Sensory neuronopathy is probably less frequent than painful axonal neuropathy. Although less frequent than other forms of peripheral neuropathies, sensory neuronopathy causes greater handicap. According to Pereira and colleagues, sensory neuronopathy is generally distinguishable from other forms of peripheral neuropathy in Sjögren disease because it tends to be asymmetrical and to predominate in the upper limbs (57). In their series, sensory neuronopathy preceded or coincided with Sjögren disease diagnosis. The most common neurologic findings were ataxia and areflexia followed by paresthesia and pain. Lower limbs were more affected than upper limbs, neurologic deficits were often symmetric, and cranial nerves were affected in three patients. Nine patients had positive ANA (69%) or anti-SS-A (38%). No patient presented with sphincter or autonomic dysfunction. Three differentiated clinical courses have been reported: subacute progression in less than 1 month (7%), late acceleration of pure sensory neuropathy 2 to 4 years after an initial indolent onset (20%), and a very long-term insidious, chronic evolution (73%) with a poor response to treatment, although stabilization of symptomatology for long periods may be observed.
Cranial neuropathies. The most common cranial nerve affected in Sjögren disease is the trigeminal nerve, which is discussed in more detail in the next section. The facial nerve is the most commonly targeted motor nerve and can present bilaterally. Other symptoms include recurrent diplopia due to oculomotor and trochlear nerve involvement, swallowing problems due to compromise of the glossopharyngeal and vagus nerves, and less commonly, simultaneous lesions in multiple cranial nerves (53). Neurosarcoidosis should be included in the differential diagnosis in patients with multiple cranial nerve involvement.
Trigeminal neuropathy. Trigeminal neuropathy is a common complication of Sjögren disease that often presents with bilateral involvement. There is facial numbness, decreased corneal reflexes, and loss of pin prick and soft touch perception in the trigeminal nerve distribution, without motor trigeminal nerve involvement. Immune-mediated damage of Gasserian ganglion neurons is suspected to be the most plausible cause of the neuropathy. Simultaneous sensory neuropathy of the trigeminal, glossopharyngeal, and vagus nerves has been reported.
Hearing loss. Sensorineural hearing loss, due to lesion of the vestibulocochlear nerve manifested by sudden or progressive deafness, has been reported in Sjögren disease. Retamozo and colleagues analyzed the frequency of non-ESSDAI features in The Big Data Sjögren Project Consortium, an international multicenter registry, and found hearing loss in 35 of 6331 patients (0.6%) (66). In some cases, hearing loss was associated with the presence of antiphospholipid antibodies.
Seeliger and colleagues conducted a study on auditory neuropathy in neurologic patients with Sjögren disease (71). The study included 30 patients (57% females, median age 59 years) who showed signs of peripheral polyneuropathy and met the current ACR/EULAR classification criteria for Sjögren disease. Results revealed that 33% of patients with Sjögren disease and polyneuropathy had definite hearing loss. Further pathological test results on audiometric testing were found in other patients, resulting in a total of 80% of patients with hearing dysfunction. The severity of hearing loss, as detected on pure tone audiometry, ranged from mild in most patients (60%) to severe in 10% of patients in the cohort. Interestingly, 15 out of 19 patients with evidence of retrocochlear auditory dysfunction showed pathological auditory brainstem response potentials. Hence, auditory brainstem response potentials are crucial in identifying retrocochlear hearing impairment.
A study using data from Taiwan’s Longitudinal Health Insurance Database and the national registry for catastrophic illnesses evaluated over 20,000 individuals diagnosed with Sjögren disease (86). These patients were compared with nearly 61,000 matched controls. Findings revealed that those with Sjögren disease experienced higher rates of hearing loss (5.6%) and sudden deafness (0.8%) compared to the control group (3.3% and 0.6%, respectively), with both differences being statistically significant. After adjusting for factors such as age, income, geographic region, urbanization level, and comorbidities like diabetes, hypertension, and rheumatoid arthritis, logistic regression analysis showed that Sjögren patients had significantly increased odds of both hearing loss (OR = 1.72) and sudden deafness (OR = 1.37).
Autonomic neuropathy. The peripheral sympathetic nervous system is severely involved in this form of neuropathy. These patients commonly present with sicca syndrome, orthostatic hypotension with syncope, anhidrosis, Adie pupil, neurogenic bladder, erectile dysfunction, as well as gastrointestinal symptoms such as abdominal pain, constipation, or diarrhea (23; 24; 53). In addition to the above, examination reveals reduced cardiac 123I-MIBG uptake, no plasma norepinephrine response after standing, and hypertensive response to minimal doses of norepinephrine (53). These patients usually have elevated titers of nicotinic ganglionic acetylcholine receptor autoantibody, a putative effector of autoimmune cholinergic dysautonomia.
Radiculoneuropathy. A demyelinating polyradiculoneuropathy is the most uncommon peripheral nervous system manifestation in patients with Sjögren disease. Patients present with progressive sensory disturbances in a glove-and-stocking distribution, muscle weakness, and sensory ataxia but without autonomic symptoms. CSF protein is elevated, and nerve conduction studies show reduced conduction velocities and temporal dispersion. Biopsies show histopathological evidence of remyelination. According to Mori and colleagues, the primary lesion in these patients is an inflammatory radiculoneuropathy (53).
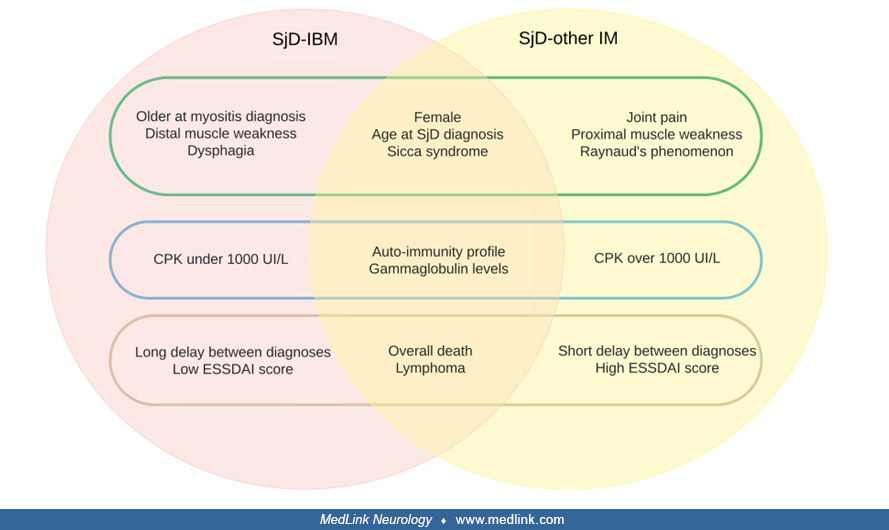
Inflammatory myositis. Myalgias and diffuse musculoskeletal pain, without elevation of muscle enzymes or electromyographic changes, have been reported in 30% to 44% of patients with Sjögren disease; many of them fulfill criteria for fibromyalgia. Fibromyalgia is a diagnosis of exclusion for these patients, and vitamin D deficiency as well as thyroid disease should be ruled out prior to making the diagnosis. Inflammatory myopathies are uncommon in Sjögren disease, reported in 1% to 2% of cases of peripheral nervous system manifestations. The ASSESS cohort analyzed the frequency of myositis (defined according to the 2017 EULAR/ACR criteria) in 395 patients with Sjögren disease (27). Myositis was suspected in 38 patients but was confirmed in only four [1.0% (95% CI: 0.40, 2.6)]. Patients with suspected, but not confirmed, myositis had higher patient-reported scores and more frequent articular and peripheral nervous involvement than others. By contrast, disease duration in patients with confirmed myositis was 3-fold longer than without myositis. Among the four patients with confirmed myositis, two fulfilled criteria for sporadic inclusion body myositis. Muscle biopsy results on this small subset of patients show signs of inflammation in 50% to 72% and evidence of polymyositis (inflammation combined with degeneration and regeneration of muscle fibers) in 47%. Some cases have symmetrical inclusion body myositis diagnosed by electron microscopy. The first study aimed at distinguishing features between inclusion body myositis and other types of inflammatory myopathies in patients with Sjögren disease was conducted by Astouati and collaborators (09). Their findings indicated that individuals without inclusion body myositis showed more frequent systemic (extra-glandular) involvement and higher ESSDAI scores, potentially pointing to a more active form of Sjögren disease. The study’s goal was to outline the clinical profiles of patients with Sjögren disease diagnosed with inclusion body myositis and compare them to those with different inflammatory myopathy subtypes. Researchers retrospectively collected data from 13 medical centers in France, including patients who fulfilled both the ACR/EULAR classification criteria for Sjögren disease and for inflammatory myopathies. Participants were classified as having Sjögren disease–inclusion body myositis if they met inclusion body myositis–specific diagnostic markers, whereas others were labeled as Sjögren disease with alternative forms of inflammatory myopathies. Among the 22 patients with Sjögren disease–inclusion body myositis, the majority were women (86%), with a median age of 54 [38.5–64] at the time of Sjögren disease diagnosis and 62 [46.5–70] at symptom onset of inclusion body myositis. Although glandular and immune-related irregularities were generally present, other systemic manifestations were relatively rare, leading to a moderate disease activity score (ESSDAI 5.5 [1–7.8]) at diagnosis. Common signs of inclusion body myositis included gradually worsening symptoms (59%), asymmetrical (27%) and distal (32%) muscle involvement, and swallowing difficulties (41%), along with modest levels of creatine kinase (386.5 [221.8–670.5] U/L) and CRP (3.0 [3–8.5] mg/L). In 55% of cases, immunosuppressive therapy was reported as effective. In comparison, the group of 50 Sjögren disease patients with other forms of inflammatory myopathy tended to be younger, exhibited more frequent systemic manifestations, had higher ESSDAI scores (11 [3–30]), a shorter time between Sjögren disease diagnosis and the emergence of myositis (0 [–0.5–26]), a greater percentage with creatine kinase levels above 1000 U/L (36%), and showed fewer hallmark inclusion body myositis traits).
Myopathy due to medications such as steroids must be considered in the differential diagnosis. (Abbrev: SjD, Sjogren disease; IBM, inclusion-body myositis. From: Astouati Q, Machet T, Houssais C, et al. Inclusion-body myositis ...
Multiple sclerosis-like manifestations. Alexander and colleagues first reported the frequent occurrence of a relapsing remitting syndrome resembling multiple sclerosis associated with cutaneous vasculitis in patients with Sjögren disease in 1986. All cases had confirmed Sjögren disease by lip biopsy, and all cases met diagnostic criteria for clinically definite multiple sclerosis. Eighty nine percent of the patients had oligoclonal bands in the CSF, but occasionally the bands were lowered with corticosteroid therapy. Fifteen out of 19 patients had positive visual evoked potential testing. Many of these cases presented a combination of optic neuropathy and chronic myelopathy that could be confused with neuromyelitis optica or multiple sclerosis, as well as brainstem and cerebellar symptoms. Sjögren disease and multiple sclerosis share common immunologic dysregulation, as a dominant feature of the immunopathogenesis in Sjogren disease is B cell dysfunction and excessive BAFF (B cell activating factor). Self-reactive B cells as well as BAFF itself can stimulate T cells, which are implicated in the pathogenesis of multiple sclerosis. Peripheral nervous system involvement was seen in 55% of these patients, and perhaps this could help differentiate between multiple sclerosis and CNS Sjögren disease. Neuroimaging studies were carried out by Akasbi and colleagues and disclosed white matter abnormalities in 49% of patients with Sjögren disease with suspected neurologic involvement. White matter abnormalities were classified as vascular pathological changes in 21 patients: 10 had multiple small focal lesions, seven had beginning confluence of lesions, and four had diffuse involvement of the entire region. White matter abnormalities were classified as inflammatory or demyelinating lesions (multiple sclerosis-like) in four patients who fulfilled the MRI Barkhof criteria for multiple sclerosis. Patients with inflammatory or demyelinating lesions were younger and had a lower frequency of hypertension and altered glomerular filtration rate in comparison with patients with vascular lesions. The multivariate age-sex adjusted model identified hypertension and HDL-c levels as independent predictors of white matter abnormalities in patients with Sjögren disease (01). There is no consensus in the medical literature to determine whether a patient has a relapsing remitting demyelinating disease of the CNS as a result of Sjögren disease or if Sjögren disease and multiple sclerosis can present independently. A cross-sectional study of 68 patients with Sjögren disease and 68 healthy controls found no difference in white matter hyperintensity load or distribution between the two groups (36). However, in other autoimmune disorders such as lupus and antiphospholipid syndrome, cerebral white matter hyperintensities are fairly common.
The prevalence of Sjögren disease in patients with multiple sclerosis ranges from 0% to 15% to 17%, although the largest epidemiological study (a population-based study carried out in Taiwan) has estimated a prevalence of 2.4% (26).
Neuromyelitis optica spectrum disorder (NMOSD). The combination of optic neuritis with concurrent myelitis is included in the diagnostic criteria for neuromyelitis optica, also known as Devic disease. More than 95% of neuromyelitis optica cases test positive to the disease-specific serum aquaporin-4 autoantibody. Antinuclear autoantibodies or other systemic autoimmune diseases are often found in patients with neuromyelitis optica, including Sjögren disease, and 7.7% to 12% of patients with neuromyelitis optica have anti-Ro/SS-A or anti-La/SS-B antibodies. In Korea, Kim and colleagues studied eight patients with Sjögren myelopathy and demonstrated that most patients exhibited clinical, radiological, and immunological characteristics of neuromyelitis optica, including positive aquaporin-4 autoantibody testing (41). Min and colleagues studied 12 women with Sjögren disease and recurrent cerebral manifestations (51). MRI showed lesions characteristic of neuromyelitis optica in the third and fourth ventricles and in the posterior limb of the internal capsule, along with cerebral or cerebellar lesions larger than 3 cm in size and with cavity-like formations. Aquaporin-4 autoantibody was positive in six of eight patients tested, and all the seropositive patients showed lesions with increased MRI diffusion, suggestive of vasogenic edema. Birnbaum and colleagues analyzed the relationship between Sjögren disease and neuromyelitis optica and compared frequencies of anti-AQP-4 and Sjögren disease–associated antibodies in 109 patients (11). They found that anti-AQP-4 antibodies were seen exclusively in patients with Sjögren disease with neuromyelitis optica (72.7%) but not in those without neuromyelitis optica (p < 0.01). In contrast, anti-Ro 52, anti-Ro 60, and other autoantibodies were not more prevalent in patients with Sjögren disease with neuromyelitis optica versus those without.
Two large, population-based studies carried out in patients with NMOSD from China and Taiwan have reported a prevalence of Sjögren disease of 7% to 8% (26; 81).
Javed and colleagues tested patients with neuromyelitis optica for concomitant Sjögren disease with labial biopsies. Although less than 20% of patients had symptoms that were suggestive of Sjögren disease, labial biopsy was positive for nine of 12 patients with confirmed neuromyelitis optica and for seven of eight patients with longitudinally extensive transverse myelitis (39). These authors recommend testing for aquaporin-4 autoantibody in all patients with Sjögren myelitis, followed by early aggressive immune therapy in positive patients.
A population-based study using the Korean National Health Insurance database analyzed the risk of autoimmune rheumatic diseases in patients with multiple sclerosis and NMOSD (43). Compared to matched controls, both groups showed a significantly higher risk of developing autoimmune rheumatic diseases. Patients with multiple sclerosis had elevated risks for conditions, such as Behçet disease (HR: 17.24; 95% CI: 4.12 to 72.14), systemic lupus erythematosus (HR: 12.25; 95% CI: 4.12 to 36.44), Sjögren disease (HR: 6.16; 95% CI: 1.80 to 21.04), and seropositive rheumatoid arthritis (HR: 3.32; 95% CI: 1.78 to 6.19). NMOSD patients had even greater risks, especially for Sjögren disease (HR: 82.63; 95% CI: 19.00 to 359.38) and systemic lupus erythematosus (HR: 30.85; 95% CI: 6.23 to 152.80). The findings suggest a strong association between these neuroinflammatory disorders and subsequent development of autoimmune rheumatic diseases.
A study of 155 patients with NMOSD found that nearly half also had Sjögren disease. Those with both conditions showed more severe symptoms, experienced earlier relapses, and reached disability faster. Key predictors of coexisting Sjögren disease included older age, higher Expanded Disability Status Scale (EDSS) score, low white blood cell counts, and positive rheumatoid factor, anti-SSA (Ro), and anti-SSB (La) antibodies. Survival curve analysis revealed that the EDSS score in the NMOSD patients with Sjögren disease was associated with clinical relapse, and these patients reached an EDSS score of 4.0 earlier than those without Sjögren disease. A predictive tool (nomogram) based on these factors accurately identified patients at higher risk for having both diseases (83).
Spinal cord involvement. Following an initial report, at least 60 cases of spinal cord involvement in Sjögren disease have been reported, indicating that this neurologic complication is not uncommon (22; 60; 41). Furthermore, patients with primary progressive multiple sclerosis have a prevalence of Sjögren disease (16.6%) that is much higher than expected in the general population (1% to 5%).
Acute transverse myelitis is the most frequent form of spinal cord involvement in Sjögren disease. The symptoms of acute transverse myelitis develop abruptly, usually with severe neck and thoracic back pain followed by sensory and motor deficits below the level of the lesion. In Sjögren disease, this form of myelopathy is thought to be due to vasculitis and carries a high morbidity and mortality.
Spinal cord involvement may also present as a progressive myelitis. A diagnosis of Sjögren myelopathy requires a high index of suspicion and should be considered especially in women over 45 years of age with progressive spastic paraparesis and abnormalities on spinal cord MRI, even with negative antiextractable nuclear antigen antibodies (Ro/SS-A or La/SS-B). The presence of autoantibodies against fodrin also helps in differentiating myelopathy in Sjögren disease from primary progressive multiple sclerosis. A positive test has 70% sensitivity, 86.7% specificity, 63.6% positive predictive value, and 89.6% negative predictive value.
Less common is the combined involvement of pyramidal tracts and anterior horn cells resembling amyotrophic lateral sclerosis. Postmortem study of two cases confirmed in one patient a lower motor neuron syndrome combined with flaccid bladder and rectum, and in the other patient, unilateral hearing loss, sensory neuronopathy, Adie pupils, upper motor neuron signs, and autopsy-proven anterior horn cell degeneration (40). These cases demonstrate the wide multisystem neuronal involvement that may occur in Sjögren disease. A similar syndrome called pseudo-amyotrophic lateral sclerosis has been reported in patients with HTLV-1 infection.
Focal encephalic manifestations. Focal manifestations may present acutely, with stroke-like features such as aphasia, hemiplegia, or numbness, consistent with a focal vasculitis (23). Intracerebral or subarachnoid hemorrhage may signal the presence of vasculitis. Recurrent or relapsing CNS symptoms may mimic multiple sclerosis as discussed above.
The pooled prevalence of cerebellar ataxia in Sjögren disease is estimated to be 1.5% (95% CI: 0.3%–6.8%) (45). The subacute development of cerebellar ataxia and tremor may be caused by demyelination. In one patient, an MRI showed T2-hyperintensities in the cerebellar white matter and the pons; severe necrotic lesions were found in the cerebellar white matter bilaterally, with several foci of perivenous demyelination in the periphery of the lesions and similar demyelinated areas in the pons; there was minimal granulomatous angiitis (37). Yang and colleagues analyzed the clinical features of cerebellar involvement in 13 patients with Sjögren disease (85). Nine (69.2%) patients went to the clinic because of ataxia, and Sjögren disease was not suspected until accidental screening for autoantibodies. Dysarthria (7, 59.8%), limb tremor (4, 30.8%), and nystagmus (2, 15.4%) were the other symptoms related to the cerebellum. Of the patients, 81.8% (9/11) had abnormal cerebrospinal fluid findings, and 11 patients (84.6%) had cerebellar atrophy on brain MRI. Anti-Ro/SSA antibody was positive in 12 (92.3%) patients and anti-La/SSB in 6 (46.2%) patients.
Tetsuka and colleagues conducted a literature review to assess the clinical characteristics, diagnostic methods, and therapeutic strategies used for patients with Sjögren disease who had cerebellar degeneration (80). They found that all patients in the literature review, as well as their own patient, were positive for anti-Ro/SSA antibodies, although some patients were negative for anti-La/SSB antibodies. The average age of onset was 48.9 ± 19.4 years, and the male to female ratio was overwhelmingly female, with three men and 11 women. Cerebellar atrophy was observed with brain MRI in the majority of patients as the disease progressed (12/14 cases). Three main symptoms suggested cerebellar disturbances: ataxia (14/14 cases), nystagmus (6/14 cases), and dysarthria (10/14 cases). In some cases, cerebellar degeneration was the first manifestation of Sjögren disease. Anti-Ro/SSA antibodies were observed in both serum and CSF, but anti-Ro/SSA antibodies were negative in the CSF of patients with Sjögren disease without CNS involvement. Cerebellar atrophy was observed, and sequelae remained in the majority of patients. Autopsy findings indicated a selective loss of Purkinje cells.
Other focal neurologic manifestations described in Sjögren disease include internuclear ophthalmoplegia, nystagmus, dystonia, athetosis, and intention tremor; or aseptic meningitis with confusion, cerebellar involvement, and spastic tetraparesis. A rigid form of parkinsonism with preponderant akinesia but without tremor and resistance to L-DOPA treatment has been described. There are also rare cases of generalized chorea. Focal and generalized seizures may occur during relapses of cerebral vasculitis.
Sjögren disease may also present as limbic encephalitis. A case series of three patients describes limbic encephalitis occurring after the diagnosis of Sjögren disease was made, with or without other neurologic complications such as motor axonal neuropathy (21). Antineuronal antibodies were tested and were negative, thus, suggesting Sjögren disease was the cause of these symptoms.
Osmotic demyelination syndrome. Osmotic demyelination syndrome is a rare noninflammatory demyelinating disorder of the CNS that primarily affects the white matter tracts of the basilar pons (central pontine myelinolysis) and, in some cases, extrapontine regions, such as the basal ganglia. In a literature review of patients with both Sjögren disease and renal tubular acidosis (SS-RTA), Sandhya found multiple cases of osmotic demyelination syndrome (69). This is intriguing because osmotic demyelination syndrome is generally considered a noninflammatory disease and has not been associated with autoimmune disorders, including Sjögren disease. In response, Sandhya and colleagues performed a detailed literature review of osmotic demyelination syndrome in patients with Sjögren disease and noted the characteristics of these patients, including the presence of renal tubular acidosis (70). The authors compared the demyelinating autoimmune CNS disorder most closely associated with Sjögren disease, neuromyelitis optica spectrum disorders (NMOSD), to osmotic demyelination syndrome. They found that both of these inflammatory and noninflammatory demyelinating disorders of the CNS are often accompanied by Sjögren disease.
The authors identified 15 patients (all women, median age 40 years) with osmotic demyelination syndrome and comorbid renal tubular acidosis. These patients showed heterogeneous clinical manifestations and outcomes. The most common symptom was quadriparesis, seen in 14 of the 15 patients. Eleven of the 15 patients had one of the following features, either alone or in combination: worsening of the sensorium, extensor plantar response, dysphagia/dysarthria, and facial palsy. Ocular palsy was seen in only four of the 15 patients and was a late manifestation. One patient who had extensive long-segment myelitis and subsequent osmotic demyelination syndrome died, but most patients recovered without significant sequelae. None had hyponatremia, whereas all patients had hypokalemia and/or hypernatremia. The authors suggest that hypokalemia causing nephrogenic diabetes insipidus followed by a rapid rise in sodium and the resultant osmotic stress could potentially explain the occurrence of osmotic demyelination syndrome in SS-RTA. They recommend that when a patient with flaccid quadriparesis does not respond to the correction of potassium or develops additional neurologic features along with a rise in sodium, osmotic demyelination syndrome should be suspected in the setting of SS-RTA.
Meningoencephalitis. Meningoencephalitis is a relatively common neurologic complication of Sjögren disease (23). It begins with headache, myalgias, confusion, and meningeal signs without fever; in some cases, sensorineural deafness may occur. Focal neurologic signs may be present; brain MRI shows hyperintense multifocal inflammatory changes in the cerebral white mater and cortex. The CSF profile is consistent with an aseptic lymphocytic meningitis, with up to 900 cells/mm3. Recurrences occur, and changes associated with vasculitis may be demonstrated by angiography.
Psychiatric and cognitive disorders. Central nervous system involvement precedes the symptoms of Sjögren disease in 52% to 80% of patients. In many patients, symptoms of depression and anxiety may antedate the diagnosis of Sjögren disease or become chronic accompaniments of the disease. Also common are cognitive changes with poor concentration and memory and abnormalities in neuropsychiatric testing, including executive dysfunction and frontal lobe deficits. The pathogenesis of these changes is not clearly understood, but it may be part of the so-called “vascular depression-vascular cognitive disorder,” resulting from ischemic interruption of prefrontal circuits important for mood and behavior. Sicca symptoms are more common in patients with cognitive disturbances and low inflammatory disease activity. A study comparing the risk of Alzheimer disease in patients with Sjögren disease and healthy controls revealed a 2.68-fold higher risk in the patients with Sjögren disease after 10 years (46). Seeliger and colleagues conducted a neuropsychological assessment using two standard tools: the extended German version of the Consortium to Establish a Registry for Alzheimer's Disease Neuropsychological Assessment Battery (CERAD-PLUS), and the test battery for attentional performance (TAP), widely recognized for evaluating attentional functions in German-speaking regions of Europe (73). They included 64 patients diagnosed with Neuro-Sjogren, who received treatment at a university hospital between December 2016 and January 2019. Their findings revealed cognitive impairment in 55% of neuro-Sjogren patients. The severity of impairment varied, ranging from mild (38%) to severe (17%). Notably, patients exhibited significant cognitive deficits in attentional and mnemonic domains. They assessed disease activity using the EULAR Sjogren's Syndrome Disease Activity Index (ESSDAI), revealing a significant correlation between disease activity and cognitive impairment (p = 0.0149). However, they found no correlation between disease duration and cognitive impairment (p = 0.3492). These findings suggest that a considerable proportion of Neuro-Sjogren patients experience cognitive impairment, possibly stemming from attentional deficits.
Optic neuropathy. Patients with Sjögren disease can present with bilateral visual loss secondary to retrobulbar optic neuropathy (23). In some cases, blindness secondary to bilateral optic neuropathy was the first manifestation of Sjögren disease. In about 12% to 15% of patients, the diagnosis was revealed by abnormal visual evoked potentials (23). The pathogenesis of optic nerve involvement in Sjögren disease is postulated to result from a combination of ischemic vasculitis and demyelination. The differential diagnosis should include multiple sclerosis and neuromyelitis optica.
Sjögren disease can be associated with neurologic and cognitive involvement, negatively affecting patients' quality of life. Goulabchand and colleagues conducted a nationwide retrospective study using the French Health insurance database to assess whether patients with Sjögren disease are at higher risk of hospitalization for neurologic diseases (33). The study selected patients hospitalized with new-onset Sjögren syndrome between 2011 and 2018 and compared the incidence of hospitalization for dementia, multiple sclerosis, encephalitis, and peripheral neuropathy with an age- and sex-matched (1:10) hospitalized control group. Adjusted hazard ratios (aHR) considered confounding factors, particularly socioeconomic status and cardiovascular diseases. The analysis included 25,661 patients hospitalized for Sjögren disease and 252,543 matched patients. The incidence of hospitalization for dementia, as well as the incidence of hospitalization for multiple sclerosis, encephalitis, and inflammatory polyneuropathies, were significantly higher in patients with Sjögren disease.
In a study, patients with Sjögren's disease and neurologic involvement had a different clinical phenotype than patients with Sjögren's disease without neurologic involvement (74). The study included 512 patients treated for Sjögren's disease without neurologic involvement (SjD) / neurologic manifestations of Sjögren disease (SjDN) between April 2018 and July 2022 (238 SjDN patients [46%] vs. 274 SjD patients [54%], cross-sectional design). The study found that male sex, older age at disease onset, hospitalization at first presentation, lower IgG levels, and higher eosinophil values were independent predictors of neurologic involvement in Sjögren's disease. Additionally, univariate regression showed older age at diagnosis, lower prevalence of rheumatoid factor, SSA(Ro)/SSB(La) antibodies, higher white blood cell count, and CK levels (treatment-naïve) in SjDN.
Regarding the role of peripheral nervous system involvement in Sjögren disease, a retrospective observational study by Quartuccio and colleagues demonstrated that the new onset or worsening of peripheral neuropathy during follow-up, along with activity in the biological and articular domains of the ESSDAI, was associated with poorer long-term outcomes (61). Specifically, these factors correlated with higher disease activity at the last assessment, after a mean disease duration of approximately 10 years (OR: 5.9 [2.4–14.5], p < 0.0001).
On the other hand, Fan and colleagues conducted a retrospective study at a hospital in China from January 2012 to December 2019 focused on central nervous system involvement in determining long-term prognosis in patients with Sjögren disease; 412 participants diagnosed with Sjögren disease were included (25). The study showed that the prevalence of CNS involvement in Sjögren disease patients was 10%, with 31% exhibiting neurologic manifestations as their initial symptom. Compared to Sjögren disease patients without CNS involvement, those with CNS involvement also exhibited kidney and lung involvement, hematologic abnormalities, positive antinuclear antibody (ANA), and anti-SSA antibody tests, as well as low C3 and C4 levels (all p < 0.05). Furthermore, the prevalence of lung involvement, immune thrombocytopenia, and high-titer ANA was significantly higher in pSS-CNS patients with disease activity compared to those in the moderately active group. Multivariate analysis identified lung involvement, positive anti-SSA antibody tests, and low C3 levels as prognostic factors for CNS involvement in primary Sjögren syndrome. Treatment with high-dose glucocorticoids and immunosuppressive therapy resulted in improvement in 60% of pSS-CNS patients, whereas 37% were unresponsive to treatment, and 3% died.
Therefore, these data support the hypothesis that neurologic involvement in Sjögren's disease has been underestimated until now. Physicians must be aware of these neurologic risks to choose the most appropriate diagnostic work-up.
A 40-year-old woman developed subacute persistent low-grade evening fever (up to 37.5°C), associated with sustained neutropenia (ranging from 1000 to 1200/μL) and intermittent lymphopenia (ranging from 700 to 1200/μL). An extensive evaluation for infections yielded negative results, and hematologic evaluation concluded for autoimmune neutropenia. Considering symptom persistence, she was referred for rheumatologic assessment. During rheumatologic evaluation, she reported a long-standing history of Raynaud phenomenon and a progressive onset of sicca symptoms over the preceding 4 to 5 years, including xerostomia and xerophthalmia, requiring regular use of artificial tears and frequent oral hydration. Objective testing confirmed glandular hypofunction, with pathological results on both the Schirmer test and unstimulated salivary flow rate. There was no clinical evidence of lymphadenopathy, salivary gland enlargement, cutaneous involvement, or other systemic extra-glandular manifestations. To further characterize the disease, a salivary gland ultrasound was performed, revealing marked parenchymal inhomogeneity. Serological studies showed antinuclear antibodies (ANA) at a titer of 1:640 with a fine speckled pattern, along with high-titer anti-SSA/Ro52, anti-SSA/Ro60, and anti-SSB antibodies. Additional laboratory investigations demonstrated polyclonal hypergammaglobulinemia (19 g/L), normal complement levels (C3: 130 mg/dL; C4: 25 mg/dL), and absence of rheumatoid factor and cryoglobulins. A diagnosis of primary Sjögren disease was established. Treatment was initiated with hydroxychloroquine (5 mg/kg/day) and a tapering course of oral prednisone (starting at 25 mg/day over 4 weeks), leading to resolution of febrile symptoms. Neutropenia, however, persisted. The patient remained clinically stable over the following 5 years, but hydroxychloroquine was subsequently discontinued due to suspected retinal toxicity.
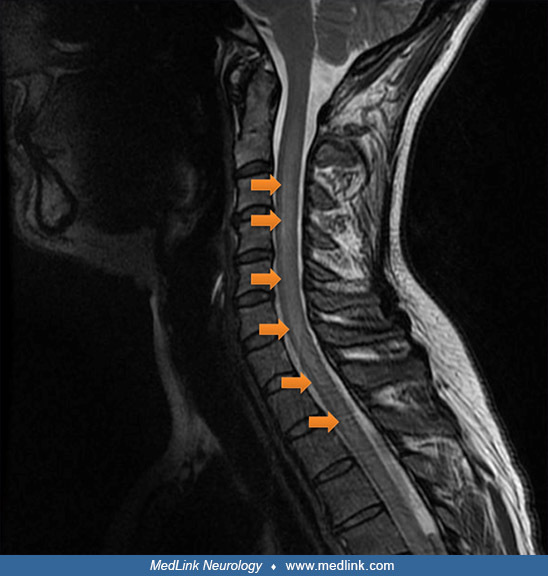
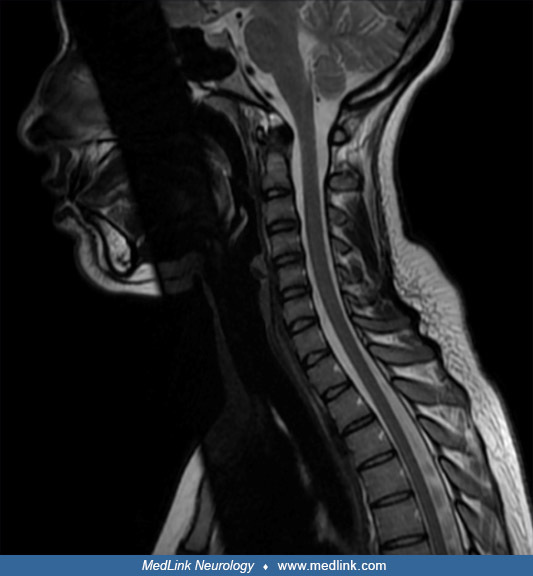
At the age of 45, the patient developed progressive dysesthesias and hypoesthesia affecting the lower limbs, accompanied by evolving spastic paraparesis. She subsequently experienced urinary retention complicated by overflow incontinence, necessitating regular intermittent catheterization, as well as severe constipation due to neurogenic bowel dysfunction, requiring frequent transanal irrigation to facilitate bowel evacuation. Neurologic examination revealed bilateral lower limb hyperreflexia, positive Babinski sign, and orthostatic hypotension suggestive of autonomic dysregulation. Cervical spinal cord MRI showed longitudinally extensive transverse myelitis, whereas brain MRI and visual evoked potentials were unremarkable.
Cerebrospinal fluid analysis revealed mild lymphocytic pleocytosis (73 cells/μL), elevated protein concentration (60 mg/dL), normoglycorrhachia (75 mg/dL), and absence of oligoclonal bands. Anti-MOG antibodies were undetectable in serum. Anti–aquaporin-4 IgG was identified at low titer in both serum and cerebrospinal fluid. A diagnosis of neuromyelitis optica spectrum disorder overlapping with Sjögren disease was made. Treatment with intravenous methylprednisolone (1 g/day for 5 days) was initiated, followed by a tapering course of oral prednisone (1 mg/kg/day over 12 months). Rituximab was administered with an induction regimen (375 mg/m² weekly for 4 weeks), followed by maintenance doses (1 g every 6 to 12 months) ensuring CD19+ B cell depletion. Under this regimen, she experienced marked improvement in motor and sensory function and partial resolution of bladder and bowel dysfunction. Cervical spinal cord MRI abnormalities resolved, and anti-aquaporin-4 antibodies became undetectable in serum at 6-month follow-up.
Two years later the patient developed progressive burning dysesthesias in both lower and upper extremities, associated with burning mouth syndrome despite continuing rituximab regimen. Neurologic examination at this stage was unremarkable, with preserved muscle strength, normal reflexes, and absent Babinski sign; however, the patient exhibited pronounced allodynia and hyperalgesia. MRI of the brain and spinal cord, as well as nerve conduction studies, yielded normal results, and anti-AQP4 antibodies remained negative. Subsequent rheumatological reassessment raised the suspicion of small fiber neuropathy associated with Sjögren disease. Quantitative sensory testing demonstrated psychobiological signs consistent with small fiber dysfunction, and skin biopsy of the right ankle confirmed reduced intraepidermal nerve fiber density, definitively establishing the diagnosis of small fiber neuropathy. Given prior intolerance to pregabalin and the potential sicca-related worsening with antidepressants, intravenous immunoglobulin was initiated at a dose of 0.4 g/kg/day for 5 consecutive days, repeated every 6 to 9 months. This approach, supported by some evidence from case series, led to a clinically meaningful improvement in burning pain, dysesthesias, and oral discomfort (30; 59). The patient continues regular rituximab infusions and IVIG therapy with stable neurologic status and sustained relief of neuropathic symptoms.
|
• Sjögren disease is an autoimmune disorder manifested by alterations of B-cell and T-lymphocytes occurring in individuals with a genetic predisposition. |
The pathogenesis of Sjögren disease has been reviewed by Fox (28). The exact mechanisms are still unknown, but the most prominent immunoregulatory alterations are B-cell hyperreactivity and enhanced levels of B-cell-activating factor/B-lymphocyte stimulator. The pathogenesis is multifactorial, whereby environmental factors activate glandular endothelial or epithelial cells, triggering inflammation in individuals with a genetic predisposition (HLA DR). The inappropriate B-cell activation can follow various stages of evolution, leading in extreme cases to malignant transformation of B cells. Patients with Sjögren disease have a 6- to 44-fold increased risk for developing non-Hodgkin lymphoma.
Polyclonal B-cell hyperreactivity in Sjögren disease accounts for the hypergammaglobulinemia, circulating immune complexes, and multiple autoantibodies directed against both organ- and nonorgan-specific autoantigens. Clinically, the most important and best characterized are the autoantibodies anti-Ro (SS-A) and anti-La (SS-B) directed against cellular heterogeneous ribonucleoprotein complexes consisting of antigenic proteins. The antibodies recognize autoantigens, which bind to ribonucleoprotein particles consisting of a 60kD SS-A/Ro RNA binding protein and hY1 RNAs and 48 KD RNA binding protein, which facilitates maturation of RNA polymerase III transcripts, such as precursors to tRNA and 5S-RNA. These antibodies are found in approximately 50% of the patients with Sjögren disease and tend to be associated more with severe glandular and extraglandular manifestations (28).
There are also alterations of cellular immunity in Sjögren disease. Mononuclear cells (primarily T-lymphocytes) infiltrate salivary and lachrymal glands with partial destruction of acinar and ductal structures. The T-lymphocytes and also the glandular cells cause the release of cytokines (especially interleukin-1, interleukin-6, and tumor necrosis factor alpha) (28). These cytokines, along with autoantibodies and metalloproteinases, cause a decreased release of neurotransmitters and a diminished response of the residual glandular cells to available neurotransmitters, resulting in the symptoms commonly seen in Sjögren disease (29). Interaction between constitutional factors (hormones and major histocompatibility complex) and environmental factors (most likely viruses) are thought to be important in the etiology of Sjögren disease. Females are affected in a ratio of 9:1 compared to males. Hormones such as estrogens, reactive hypothalamic and hypophyseal peptide hormones and dehydroepiandrosterone may play a role. Sjögren disease is associated with HLA-DR 3 and linked genes B8, DQ 2, and the C4 null gene in about 50% of the patients. Postulated infectious agents capable of triggering the immune process include herpes viruses (particularly Epstein Barr virus, cytomegalovirus, and human herpesvirus-6), H pylori and human retroviruses, in particular HTLV-1.
HTLV-1 and Sjögren disease. Transgenic mice expressing the HTLV-1 tax gene develop an exocrinopathy similar to that seen in patients with Sjögren disease. The expression of sequences homologous to the HTLV-1 tax gene has been found in labial salivary glands of patients with Sjögren disease. In Japan, Nakamura and colleagues investigated the presence of Sjögren disease in patients with HTLV-1-associated myelopathy, including a histological examination of labial salivary glands; definite Sjögren disease was found in 65% (13/20) of patients (55). More severe inflammatory cell infiltration in labial salivary glands was found in patients with HTLV-1-seropositive Sjögren disease than in seronegative controls. Pot and colleagues reported a spectacular radiological and clinical recovery of a patient with HTLV-1 myelitis and Sjögren disease who was treated with combined antiretroviral drugs (lamivudine and tenofovir) plus immunosuppressant therapy with prednisone and mycophenolate mofetil (60). Based on the aforementioned findings, it is advisable to test for HTLV-1 antibodies in all patients with Sjögren disease who develop a myelopathy.
|
• The incidence of Sjögren disease ranges between three and 11 cases per 100,000 individuals, whereas the prevalence ranges between 0.01% and 0.72%. |
The reported incidence and prevalence of Sjögren disease varies according to both the study design and the classification criteria used. The pooled prevalence rates in studies that used the 1993 European Classification Criteria was 12fold higher than the pooled prevalence rates in studies that used the 2002 American-European Consensus Group (AECG) criteria, whereas the pooled prevalence rates reported in populationbased epidemiological studies were slightly lower than that calculated in the total population. The incidence of Sjögren disease ranges between three and 11 cases per 100,000 individuals, whereas the prevalence ranges between 0.01% and 0.72%. It is also likely that there are asymptomatic cases that never were diagnosed. Only one study has evaluated the influence of race or ethnicity on the prevalence of Sjögren disease. This study found a significant two-fold higher prevalence among individuals with a nonEuropean background than in those with a European background in the general population of the Greater Paris area. At presentation of disease, the epidemiological profile of Sjögren disease is typical, which might aid early diagnosis.
Sjögren disease predominantly affects women. In fact, Sjögren disease has the most unbalanced gender ratio out of all systemic autoimmune diseases; nearly a 10:1 female:male ratio was reported in a big data study of more than 14,000 patients with Sjögren disease.
Although Sjögren disease can occur at all ages, it is mainly diagnosed between 30 and 50 years of age. Sjögren disease is rare in children, and the female:male ratio is less evident in children than in adults. Sjögren disease and other autoimmune diseases frequently coincide (associated Sjögren disease) in daily practice. In patients with systemic autoimmune diseases, the proportion of patients with concomitant Sjögren disease differs between conditions; 14% to 18% of patients with lupus, 7% to 17% with rheumatoid arthritis, and 12% with systemic sclerosis also have associated Sjögren disease. In clinical practice, the management of associated Sjögren disease should be the same as the management of Sjögren disease (14).
Neurologic manifestations have been reported traditionally in about 20% of patients with Sjögren disease (range 6% to 70%) (23). Goransson and colleagues performed a population-based study in Sweden and found that 27% of patients with Sjögren disease had neuropathy, including 31% with motor neuropathy, 13% with sensory neuropathy, and 11% with sensorimotor neuropathy (32). Central nervous system involvement ranges from 2% to 25% in hospitalized patients.
Ye and colleagues reviewed a large cohort of patients with primary and secondary Sjögren disease in a cross-sectional study and found that low complement (C3) levels, xerophthalmia, positive ANA, cardiac involvement, and labial salivary gland histologic results were useful in predicting the risk of neurologic complications (87).
Sjögren disease is a chronic disease that cannot be prevented. However, screening approaches that target specific subsets of patients with Sjögren disease might either prevent or ensure timely treatment of the main complications. The presence of autoantibodies (especially antiRo/SS-A antibodies) can be used for the early diagnosis of Sjögren disease in these patients. However, the frequency of antiRo/SS-A or antiLa/SS-B autoantibodies is often lower in patients with Sjögren disease who only have neurologic involvement than in patients with Sjögren disease who have no neurologic involvement, and these patients often require a salivary gland biopsy showing the presence of focal lymphocytic sialadenitis for the early diagnosis of Sjögren disease (14).
Depending on the form of neurologic presentation, the differential diagnosis of neurologic Sjögren disease include many processes that effect either the peripheral or central nervous system. As mentioned above, demyelinating diseases such as multiple sclerosis, neuromyelitis optica, and transverse myelitis are commonly on the differential for central nervous system Sjögren disease. Other multisystemic autoimmune disorders should be considered, as well as antineuronal antibody encephalitis and infectious processes (especially HLTV-1). The onset of transverse myelitis with thoracic back pain and rapid loss of motor function is reminiscent of nucleus pulposus herniation. Serum levels of vitamin B12 and methylmalonic acid are diagnostic for subacute combined degeneration.
Peripheral nervous system presentations vary widely and, therefore, the diagnostic work up should be focused on the patient’s symptoms, dependent on the clinical presentation.
The differential diagnosis of Sjögren disease includes a past history of head and neck radiation therapy, hepatitis C infection, AIDS, preexisting lymphoma, sarcoidosis, graft-versus-host disease, IgG4related disease (IgG4RD), and the recent use of anticholinergic drugs.
The diagnostic workup of Sjögren disease is outlined in Tables 1a and 1b. Sjögren disease often has a variable course and a wide spectrum of clinical manifestations, making the diagnosis difficult or delayed. Early recognition of Sjögren disease may prevent complications, and it allows for clinical surveillance of the development of serious extraglandular systemic manifestations. The diagnosis of Sjögren disease is strongly suggested in patients who present with signs and symptoms of oral and ocular dryness and who test positive for antibodies to the anti-SS-A or anti-SS-B antigen, or who have a positive salivary gland biopsy (14).
There is no gold standard test for the diagnosis of neurologic Sjögren disease. However, it is important to note that inflammatory CSF changes are rarely found and if so, a pathognomic pattern does not exist. CSF analysis is often used to exclude other autoimmune and infectious processes (56).
The therapeutic management of Sjögren disease has not changed substantially in recent decades: treatment decisions remain challenging in clinical practice, without a specific therapeutic target beyond the relief of symptoms as the most important goal. In the 2020 EULAR guidelines, the first line of therapy for ocular dryness should be volume replacement and lubrication using artificial tears and ocular gels, whereas severe or refractory keratoconjunctivitis sicca might require topical cyclosporine or autologous serum eye drops (16; 65). With respect to oral dryness, nonpharmacological glandular stimulation is the preferred first-line therapeutic approach in patients with mild glandular dysfunction, using gustatory or mechanical stimulants, whereas in those with moderate glandular dysfunction, pharmacological stimulation (pilocarpine and cevimeline) with muscarinic agonists may be considered (65).
With respect to extraglandular manifestations of Sjögren disease, only a few clinical trials have investigated the effect of systemic treatments (14). In addition, the lack of head-to-head studies comparing the efficacy and safety profile of immunosuppressive agents (leflunomide, methotrexate, azathioprine, mycophenolate, cyclophosphamide) does not permit a recommendation on the use of one agent over another, except when patient characteristics or comorbidities are considered with respect to the safety profile. Direct and indirect B cell blockade (rituximab, belimumab) seems to be the most promising approach in terms of biological therapies for the management of Sjögren disease, although other therapeutic targets are under investigation. However, the disappointing results of two large randomized controlled trials have opened a debate about rituximab use in Sjögren disease. These trials used a composite outcome based on the subjective evaluation of dryness, fatigue, and pain. The strong influence of personal and environmental factors on the intensity of this triad of symptoms could explain the lack of significant differences. In addition, inadequate patient selection, the influence of concomitant drugs, and the heterogeneity of diagnostic tests could have contributed to the lack of significant differences between placebo and therapeutic groups (14).
As a summary about the therapeutic management of systemic Sjögren disease, the 2020 EULAR guidelines recommend to limit the use of systemic therapies (glucocorticoids, antimalarials, immunosuppressive agents, intravenous immunoglobulins, and biologics) to patients with active systemic disease, always after a careful organ-by-organ evaluation of both severity and organ damage and following a schedule consisting of a 2-stage sequential regimen: first, an intensive immunosuppressive approach targeted to restore organ function as soon as possible (induction of remission), and a second therapeutic course aimed at maintaining the initial therapeutic response (maintenance of remission).
Isolated studies are exploring other drugs, such as fingolimod (an immunomodulatory drug) in patients with multiple sclerosislike CNS involvement and bortezomib (38; 78).
Vitamin B12 (cobalamin) deficiency. Andres and colleagues documented the common occurrence of vitamin B12 deficiency in 80 patients with primary Sjögren disease (05). Serum B12 levels below 200 pg/mL were found in 8.8% (7/80) of patients, and 56.2% of patients had B12 levels between 200 and 300 pg/mL, for an overall prevalence of B12 deficiency of 65%. In comparison, they found B12 deficiency in 5.3% of patients in an Internal Medicine clinic. In the general population, B12 deficiency occurs in 15% of people over 60 years of age. Cobalamin is first released from proteins in food by pepsin and stomach acid; then cobalamin is bound to the salivary vitamin B12 R-binder protein before it can be attached to intrinsic factor. Andres and colleagues postulated that B12 deficiency from food-cobalamin malabsorption is the result of a lack of saliva, which is typical of Sjögren disease (07).
Vitamin B12 deficiency causes a number of neurologic manifestations (68) that could worsen the neurologic complications of Sjögren disease. The most common manifestations of cobalamin deficiency include: peripheral sensory neuropathy; subacute combined degeneration of the spinal cord presenting with sensory ataxia and pyramidal tract involvement with bilateral Babinski sign (06); cerebellar syndromes; cranial nerves neuropathies, including optic neuritis and optic atrophy; urinary or fecal incontinence; stroke and atherosclerosis from hyperhomocysteinemia with cognitive decline or dementia; parkinsonian syndromes; and depression. In practice, it is advisable to exclude and treat B12 (cobalamin) deficiency in patients with Sjögren disease and psychiatric or neurologic symptoms, including the several types of neuropathies and myelopathy described.
The treatment of neurologic complications in Sjögren disease is dictated by the clinical symptomatology, the clinical course, and the implicated pathogenetic mechanism.
Peripheral nervous system. The common symmetric, distal axonal sensory and sensorimotor neuropathy is believed to be caused by perivascular cellular infiltration and necrotizing vasculitis. These axonal neuropathies usually follow a slowly progressive and insidious course that is often treated with the usual symptomatic therapy of Sjögren disease (salicylates, nonsteroidal agents, hydroxychloroquine, oral corticosteroids). IVIg has been shown to improve sensorimotor or nonataxic sensory neuropathies, but not in ataxic neuropathies. In cases with mononeuritis multiplex, multiple cranial neuropathies, pseudo-amyotrophic lateral sclerosis forms, or with lesions suggestive of severe vasculitis, first-line treatment appears to be intravenous corticosteroid therapy; however, when the patient’s course fails to improve or deteriorates, a nonsteroidal immunosuppressant agent should be considered. Patients with vasculitis- or cryoglobulinemia-related neuropathies have shown favorable responses to rituximab, which is currently considered the first-line therapy in this setting (82; 47). Plasma exchange remains a therapeutic option in severe cases of cryoglobulinemic neuropathy with peripheral nervous system involvement.
The sensory ataxic ganglionopathy with ganglion neuron antibodies and the demyelinating polyradiculoneuropathy of Sjögren disease respond poorly to triple treatment with prednisolone, cyclosporine, and cyclophosphamide. There is a case report of a patient with sensory neuronopathy treated with infliximab (18). There was marked improvement in clinical and neurophysiologic deficits associated with the neuronopathy using a dose of 3 mg/kg given at weeks 0, 2, 6, and every 12 weeks thereafter. Plasmapheresis and rituximab may produce improvement, and there is occasional response to treatment with IVIG (0.4 g/kg for 5 days). Yamada and colleagues reported excellent results with IFN-alpha treatment (3 MIU/day, three times weekly), resulting also in marked improvement of the clinical and laboratory manifestations of Sjögren disease (84). This treatment could potentially be useful also for predominantly autonomic neuropathies with elevated titers of ganglionic acetylcholine receptor autoantibody, trigeminal neuropathy, and for the painful neuropathy without sensory ataxia of Sjögren disease.
The EULAR-Sjögren Syndrome Task Force Group developed a therapeutic algorithm for first-, second-, and third-line treatments (65). They recommended general sequential use of the three main categories of immunosuppressive agents in Sjögren disease based on a similar approach to that reported for other systemic autoimmune disease, such as systemic lupus erythematosus or vasculitis, with no controlled studies supporting this approach in Sjögren disease. The immunosuppressive approaches traditionally used in multineuritis and axonal polyneuropathy (predominantly vasculitis related) are high dose of glucocorticoids as first-line treatment and oral immunosuppressive agents or rituximab as second-line. In refractory cases, cyclophosphamide or plasma exchanges are recommended as rescue therapy. Azathioprine and mycophenolate mofetil may be used in lieu of cyclophosphamide or following cyclophosphamide as “remission maintenance” agents, although neither medication has been evaluated for this purpose in formal clinical trials. In cases in which the axonal polyneuropathy is sensory, first-line treatment will be symptomatic, but in refractory cases, motor involvement, ganglionopathy, and or chronic inflammatory demyelinating polyneuropathy, EULAR Task Force Group recommend intravenous immunoglobulins 0.4 to 2 g/kg for 5 days as first-line treatment and methylprednisolone pulses as second-line or cyclophosphamide pulses 0.5 g/15 day (maximum six pulses) as rescue therapy.
The EULAR-Sjögren Syndrome Task Force Group recommended general sequential use of the three main categories of immunosuppressive agents in Sjögren disease based on a similar approach to that reported for other systemic autoimmu...
Skeletal muscle. Myalgias in patients with Sjögren disease may respond to hydroxychloroquine (6 to 8 mg/kg daily) (28). Additional treatments may include corticosteroids, azathioprine, and methotrexate.
Encephalic manifestations. Meningoencephalitis, focal manifestations, or seizures consistent with vasculitis should be treated with intravenous steroids. In severe cases, intravenous corticosteroids may be used in conjunction with cyclophosphamide, rituximab, azathioprine, cyclosporine, or methotrexate. Plasmapheresis, or treatments with IVIG, are probably indicated, but there are minimal reported data with these therapeutic modalities.
Spinal cord involvement. Rogers and colleagues have reviewed this topic (67), but these data are based on anecdotal reports. Nonetheless, it seems clear that treatment of Sjögren disease myelopathy with intravenous steroids alone is not adequate (67; 22). Combination therapy with agents such as cyclophosphamide, rituximab, azathioprine, cyclosporine, or methotrexate must be considered.
Cyclophosphamide is the historical agent of choice, given intravenously in pulse dosing (22). Current treatment regimens consist of a monthly intravenous bolus of drug for 6 to 12 months (22); then, if needed, every 3 to 6 months for the second year. Doses used are 0.75 to 1.0 gm/m2. Caution is required due to the high frequency of lymphoma in Sjögren disease. The most common severe adverse effects associated with cyclophosphamide are hemorrhagic cystitis and myelosuppression. Hemorrhagic cystitis can be prevented by adequate hydration and use of mercapto ethane sulfonate sodium, a sulfhydryl compound that binds to the toxic metabolite formed by cyclophosphamide, thus, preventing it from binding to the bladder wall and causing damage. Myelosuppression may be dose-limiting, with the white blood cell nadir occurring 7 to 10 days after treatment. Owing to the aforementioned side effects, the use of mycophenolate mofetil is being explored as an alternative to cyclophosphamide in the treatment of vasculitis. Rituximab is being used more often now because of the more favorable side effect profile and similar efficacy to cyclophosphamide.
Azathioprine has been used for relapses or intolerance to cyclophosphamide therapy. In uncomplicated Sjögren syndrome, azathioprine by itself was ineffective in a dose of 1 mg/kg/d for 6 months. The most serious side effects are dose-related myelosuppression, gastrointestinal disturbances, stomatitis, and hepatotoxicity.
Cyclosporine in combination with corticosteroids may be effective in the treatment of optic neuritis. Cyclosporine is a potent immunomodulator that decreases cytokines involved in T-cell activation and has direct effects on B-cells and macrophages. The onset of effect of cyclosporine is 1 to 3 months. It is only available in an oral dosage form. Serious adverse effects are hypertension, hyperglycemia, nephrotoxicity, tremor, gastrointestinal intolerance, hirsutism, and gingival hyperplasia.
Methotrexate at a dose of 0.2 mg/kg/week tends to improve subjective oral and ocular symptoms, without objective changes. It is an antineoplastic and antiinflammatory agent. Serious adverse effects include thrombocytopenia, gastrointestinal intolerance, stomatitis, hepatotoxicity, and pulmonary toxicity.
Other nonsteroidal immunosuppressant agents should be considered, especially when lack of efficacy or intolerance to cyclophosphamide appears. Other symptoms may benefit from concomitant immunosuppressant treatment. In addition, plasmapheresis appears to be useful when acute relief of symptomatology is needed, such as in a patient with rapid deterioration who is waiting for an immunosuppressant to work. IVIG has been successfully used in Sjögren CNS vasculitis. The dose used in the patient was 400 mg/kg/d for 5 days along with corticosteroids, with additional monthly doses of IVIG for 6 months and then every 2 to 5 months as indicated by the patient’s neurologic symptoms.
Rituximab, a chimeric monoclonal antibody directed against the B cell differentiation marker CD20, has been used with some success in the treatment of Sjögren disease (03). Most of the evidence consists of case reviews for treatment of extraglandular manifestations of Sjögren disease. To our knowledge, there are no randomized placebo-controlled clinical trials of rituximab in the treatment of neurologic manifestations associated with Sjögren disease.
The EULAR-Sjögren Syndrome Task Force Group therapeutic algorithm for central nervous system involvement is based on the type of neurologic manifestation (65). The authors suggested that corticosteroids plus cyclophosphamide could be the first- and second-line therapeutic approach in patients with CNS vasculitis and neuromyelitis optica spectrum disorder but that corticosteroids should be rapidly tapered with the aim of decreasing the risk of severe infection. Rituximab or/plus plasma exchanges may also be recommended for the treatment of this manifestation, especially in patients with severe or refractory disease, but its use is currently limited by the lack of US Food and Drug Administration or European Medicines Agency approval for these indications. When lymphocytic meningitis does not respond to symptomatic treatment, the same therapeutic algorithm should be followed.
The EULAR-Sjögren Syndrome Task Force Group therapeutic algorithm for central nervous system involvement is based on the type of neurologic manifestation. The authors suggested that corticosteroids plus cyclophosphamide could b...
Unfortunately, no controlled data were identified to support a differentiated organ-guided therapeutic approach for multiple sclerosis-like manifestations in Sjögren disease, and EULAR Task Force Group recommend specific multiple sclerosis therapies, with an individualized therapeutic approach being preferable.
Although today the true prevalence of CNS and PNS involvement in patients with Sjögren disease is still not known exactly, the analysis of the main studies carried out in the past 25 years suggests a figure between 5% and 10%. It is important to highlight the great variability of its neurologic symptoms and signs, which means that it can be confused with other neurologic diseases with a similar clinical spectrum. In clinical practice, the spectrum of neurologic manifestations in patients with Sjögren disease is broad, ranging from asymptomatic forms with the exclusive finding of demyelinating lesions in the white matter to forms of focal or diffuse cerebral involvement of spinal cord or epilepsy. The situations that most frequently induce greater complexity in the differential diagnosis of CNS involvement in patients with Sjögren disease are cerebrovascular disease and demyelinating lesions. There is no standardized treatment for CNS involvement in Sjögren disease. It will fundamentally depend on the type of neurologic involvement, the symptoms, and the possible coexistence of other associated processes. In severe forms of neurologic involvement, intravenous immunosuppressive treatment with pulses of methylprednisolone and cyclophosphamide should be used to reduce the number of neurologic sequelae; in refractory or life-threatening cases, the use of immunoglobulins, plasma exchanges, and, above all, the future role of biological therapies directed against B lymphocytes, such as rituximab, should be considered.
In a questionnaire-based study, Sjögren disease had no impact on pregnancy outcome before disease onset (34). The most important condition associated with Sjögren disease in anti-SS-A-positive mothers was congenital heart block in the offspring. Autoimmune congenital heart block is an immune-mediated acquired disease that is associated with the placental transference of maternal antibodies specific for Ro and La autoantigens. The disease develops in a fetal heart without anatomical abnormalities that could otherwise explain the block; it is usually diagnosed in utero but also at birth or within the neonatal period. Autoantibody-mediated damage of fetal conduction tissues causes inflammation and fibrosis and leads to blockage of signal conduction at the atrioventricular node. Irreversible complete atrioventricular block is the principal cardiac manifestation of congenital heart block, although some babies might develop other severe cardiac complications, such as endocardial fibroelastosis or valvular insufficiency, even in the absence of cardiac block (15). This condition is associated with high morbidity and mortality, and these patients should receive multidisciplinary care in a neonatal intensive care unit (58).
Pediatric onset of the disease is rarely reported, and there is no information on the frequency of pediatric presentation. In addition, a clear view of how the disease presents in children is difficult to obtain due to the scarce and heterogeneous available data. The main series that are currently published are characterized by a heterogeneous methodology using a variable definition of the pediatric age (from 14 to 18 years) and include up to 50% of patients who do not fulfill the current classification criteria as well as a mix of primary and associated forms of the disease (65).
Among the 12,083 patients included in the Sjögren Big Data Registry, 158 (1.3%) patients were diagnosed at an age younger than 19 years. There were 136 (86%) girls and 22 (14%) boys, with a mean age at first sign or symptom suggestive of the disease of 13.2 (SD 3.2) years and of 14.2 (SD 3.5) years at the time of diagnosis of primary Sjögren syndrome (63). With respect to systemic disease, the highest mean ESSDAI score was observed in patients with childhood onset, who also showed the highest frequencies of systemic disease in five (constitutional, lymphadenopathy, glandular, cutaneous, and hematological) out of the 12 ESSDAI domains and the lowest frequencies in four domains (articular, pulmonary, peripheral nerve, and central nervous system). A younger diagnosis was associated with an increased risk of presenting activity at diagnosis in some clinical domains (constitutional, lymphadenopathy, glandular, cutaneous, renal) but a decreased risk of presenting activity in others (pulmonary and neurologic domains). Although the nervous system was the organ-specific ESSDAI domain reported less frequently, some isolated cases of neurologic involvement have been reported in children with primary Sjögren disease (31). They mainly consisted of cerebellar ataxia and acute encephalitis, which are features that are not included in the neurologic ESSDAI domain. Some of these cases were even reported with other concomitant neurologic disorders (mental retardation, psychosis) (79; 49; 35).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Simone Longhino MD
Dr. Longhino of University Hospital “Santa Maria della Misericordia” has no relevant financial relationships to disclose.
See Profile
Maria Soledad Retamozo MD PhD
Dr. Retamozo of Hospital El Pilar Barcelona has no relevant financial disclosures.
See Profile
Francesc Graus MD PhD
Dr. Graus, Emeritus Professor, Laboratory Clinical and Experimental Neuroimmunology, Institut D’Investigacions Biomédiques August Pi I Sunyer, Hospital Clinic, Spain, has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Peripheral Neuropathies
Jul. 14, 2026
Neuroimmunology
Jul. 02, 2026
Neuromuscular Disorders
Jun. 16, 2026
Peripheral Neuropathies
Jun. 11, 2026
Peripheral Neuropathies
Jun. 11, 2026
Peripheral Neuropathies
Jun. 11, 2026
Neuromuscular Disorders
May. 27, 2026
Neuromuscular Disorders
May. 21, 2026