Sleep Disorders
Sleep-related leg cramps
Jul. 03, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
The history of Wilson disease is a microcosm of the history of neurology, beginning with seminal clinical observations by the man whose name the disease bears, followed by pioneering work translating copper deficiencies in sheep to human treatment, and continuing on to identification of causative gene mutations. Amongst neurodegenerative diseases, it is unusual in that treatments can prevent and cure Wilson disease if they are given appropriately. Despite a long history, Wilson disease remains an important disease that is usually misdiagnosed. Misdiagnosis and delay in treatment are clinically relevant because if left untreated, Wilson disease progresses to hepatic failure or severe neurologic disability and death. Those adequately treated have a normal life span. In this article, the author discusses the neurologic features of Wilson disease, its diagnosis, and new treatment options.
• Wilson disease is caused by mutations in ATP7B that result in abnormal copper metabolism, and subsequent excess free copper is toxic. | |
• The most common clinical manifestations of neurologic Wilson disease include variable combinations of dysarthria, dystonia, tremor, and parkinsonism. | |
• Recognition of the disorder is important because effective treatment can prevent or improve disease features. | |
• The most useful screening procedure is a 24-hour urine copper test. In symptomatic Wilson disease, the 24-hour urine copper is always elevated to a value greater than 100 µg per 24 hours (normal is 50 µg or less). | |
• The aim of treatment is to reduce the amount of toxic-free copper. Agents available in the United States include zinc acetate and trientine. |
The historical landmark paper in this disease was written in 1912 by Samuel Alexander Kinnier-Wilson, an American neurologist working at The National Hospital at Queen Square in London (69; 15). He described a neurologic disorder associated with progressive lenticular degeneration and cirrhosis of the liver that came later to be known as Wilson disease, or hepatolenticular degeneration. The disease described by Wilson was one dominated by juvenile age at onset with dystonia and contrasted from the form of disease described by Westpahl in 1883 as pseudosclerotic, which had a young adult age of onset and symptomatically consisted principally of tremor and dysarthria. Only later was parkinsonism recognized as a major clinical feature of Wilson disease. Kayser and Fleischer described the corneal copper deposits associated with the disease, which later became known as Kayser-Fleischer rings. Several authors have described excess copper deposition, but Cumings was the first to postulate an etiologic role for copper and suggested that chelation therapy might be of benefit. A low biliary excretion of copper was later shown to be responsible for a failure to regulate copper balance, and the disease was found to be inherited as an autosomal-recessive disorder. Landmarks in treatment have included the development of penicillamine, trientine, and zinc therapies (64). The gene for Wilson disease has been identified, and an animal model of Wilson disease has provided insights into pathogenesis (11; 61; 70).
During early life, the patient is presymptomatic but accumulating copper, which invariably causes subclinical liver disease. Then, between early childhood and the fifth or sixth decade of life, but with a peak incidence of around 21 years, the patient presents with hepatic, neurologic, and psychiatric manifestations (10). It has been suggested that the hepatic, neurologic, and psychiatric presentations of Wilson disease occur in roughly equal proportions. An accurate estimate of the presenting proportions is challenging because many of the large case series have ascertainment bias based on clinical specialty and likely under-represent the psychiatric presentation.
The main clinical categories of neurologic Wilson disease have been traditionally divided variably into tremor, dysarthria, pseudosclerosis (tremor +/- dysarthria), parkinsonism, dystonia, or chorea (39; 21; 25; 56; 18; 65; 34; 60). Neurologic manifestations at initial presentation have been reported in approximately 18% to 68%. Mean age at onset of neurologic symptoms from large case series ranges from approximately 15 to 21 years of age (57; 10). The delay from disease onset to diagnosis is about 12 months (56; 65; 34). Initial misdiagnosis in Wilson disease includes over 100 different entities and spans a spectrum from “no diagnosis” to “neurologic problem” and includes many common and rare entities (43). The delay in diagnosis appears clinically significant as treatment outcomes have been reported to be better for those with a correct initial diagnosis.
Walshe, who introduced penicillamine, trientine, and tetrathiomolybdate therapy, stated that “no two patients are ever the same, even in a sibship, and there is no such thing as a typical picture of Wilson’s disease.” Because of the clinical heterogeneity, an understanding of the initial neurologic signs and symptoms of Wilson disease is diagnostically useful (see Table 1). In those studies that specify an initial manifestation, dysarthria is the most common, followed by dystonia, abnormalities of gait, tremor, parkinsonism, and chorea.
Tremor, dysarthria, and dystonia can be sole disease manifestations. More typically, combinations of neurologic features coexist, with a small number of features predominating. During the course of the disease, other neurologic features include chorea, athetosis, myoclonus, seizures, ataxia, pyramidal signs, drooling, and eye movement abnormalities (65).
|
(1) |
(2) |
(3) |
(4) |
(5) |
Total or (% average) | |
|
Number of patients |
31 |
27 |
136 |
119 |
48 |
361 |
(1) (56): initial signs of neurologic presentation
(2) (38): initial symptoms of neurologic presentation
(3) (65): initial signs of neurologic presentation
(4) (34): at time of diagnosis
(5) (54): first manifestations (%)
Table modified from (32)
Dysarthria is probably the most common neurologic manifestation of Wilson disease. Dysarthria in Wilson disease is most frequently of the mixed type with varying spastic, ataxic, hypokinetic, and dystonic components (56). Speech involvement is frequently concordant with the neurologic involvement in individual patients. In those with dystonia, speech frequently will have dystonic features with a strained or harsh quality. In those with parkinsonism, the speech quality may have hypokinetic properties (63). Ataxic dysarthria, with variation in word spacing and volume, is often found in association with other types of dysarthria and may be more common in those with tremor (56).
Dystonia can be focal, segmental, multifocal, or generalized and ranges in severity from mild to debilitating (58). A common focal dystonic manifestation of Wilson disease is the dystonic facial expression known as risus sardonicus. This type of dystonia inflicts the patient with a forced, often exaggerated, smile (58). Focal dystonia of the vocal cords, muscle of articulation, and swallowing frequently results in dysphonia, dysarthria, and dysphagia. Other focal dystonias include blepharospasm, cervical dystonia (torticollis), and writer’s cramp. As the disease progresses, focal dystonia can progress to segmental, multifocal, hemi- and generalized dystonia. Dystonia, like many neurologic features of Wilson disease, typically has a unilateral onset or predominance but can progress to bilateral or generalized involvement. Severe dystonia may lead to extreme posturing of the trunk, neck, or extremities. As in other forms of dystonia, sensory tricks can often lead to temporary improvement. The presence of dystonia has been demonstrated to correlate with MRI signal abnormalities in the putamen (56; 58).
Tremor in Wilson disease can occur at rest, on assuming a posture, with action, and its frequency and amplitude frequently change with differing activities. The tremor of Wilson disease can be identical to the tremor of essential tremor, with the arms most frequently involved but also involving the head and legs. The kinetic tremor is most frequently a distal, low-amplitude, medium-to-high-frequency tremor. Persistence of tremor asymmetry may differentiate this type of Wilsonian tremor from essential tremor, which is typically symmetric. The classic wing-beating tremor, thought to be associated with lesions of the dentatorubrothalamic pathway, is less frequently observed, proximally located primarily in the upper extremities, and typified by increasing amplitude with increased duration of posture holding. A unilateral isolated rest tremor is atypical in Wilson disease. When rest tremor is present, it is usually accompanied by postural and kinetic tremor, which may be more severe than the rest tremor (34).
Bradykinesia, imbalance, and cogwheel rigidity are the more common parkinsonian features. Parkinsonism is rarely an isolated clinical feature of Wilson disease, but like idiopathic Parkinson disease, parkinsonian features of Wilson disease are typically asymmetric.
Cognitive impairment in Wilson disease has been reported, but some have suggested that is primarily related to psychiatric and motor manifestations of Wilson disease (19; 01); others have considered it part of the neurologic manifestations of Wilson disease (29; 30). When present, cognitive impairment falls into two main, not mutually exclusive, categories: a frontal lobe syndrome or a subcortical dementia (29; 30). The frontal syndrome may manifest as impulsivity, promiscuity, impaired social judgment, apathy, decreased attention, executive dysfunction with poor planning and decision making, and emotional lability--at an extreme having pseudobulbar features. The subcortical dementia is characterized by slowness of thinking, memory loss, and executive dysfunction without cortical signs of aphasia, apraxia, or agnosia (29; 30).
Seizures are not an uncommon feature; they occur in about 6%, exceed the general population frequency by 10-fold, are rarely the presenting feature, and have been associated with the initiation of chelating therapy (20). Rarley, status epilepticus can occur (40). Ataxia has been reported to be a common feature by some (34; 60) and not felt to be prominent or present by others (56; 57; 65; 54). Cerebellar findings are rarely clinically relevant, and, on examination, frank ataxia is infrequent. Other cerebellar signs, such as overshoot dysmetria of the eyes and limbs, can occasionally be found.
Although symptoms are typically gradual in their progression, sudden worsening both with and without treatment has been described. It has been suggested that those with tremor-predominant symptoms may have a slower course than those with the predominantly dystonic form (21). Younger-onset patients are more likely to have chorea and dystonia and less likely to have a tremor-predominant manifestation, whereas those with an older onset tend to have tremor as the predominant sign (65).
In the absence of treatment, neurologic symptoms are progressive and result in a severely dystonic akinetic mute state with relative preservation of cognition (10). Before the treatment era, the median survival following the development of neurologic symptoms was approximately 5 years (20). The response to treatment is variable. Few studies have systematically investigated or reported the effect of treatment on individual neurologic signs or symptoms. It has been suggested that tremor may be more treatment-responsive than dystonia and dysarthria (21). Those who did not respond to treatment tended to have a more severe neurologic disease manifesting more frequently as dysarthria and dystonia (19). Following the initiation of chelation therapy, life expectancy has normalized (57).
Although psychiatric manifestations may be present as often as 30% to 50% of the time prior to a diagnosis of Wilson disease (19; 01), because psychiatric symptoms are often ill-defined and attributed to other causes, diagnosis of Wilson disease is rarely made during the period in which psychiatric symptoms are the sole manifestation. The lack of a diagnosis during this phase of the illness represents a lost opportunity to initiate treatment at a time when a favorable recovery may be more likely. At diagnosis, the most common psychiatric symptoms have been reported to be cognitive impairment, incongruous behavior, irritability, depression, and a personality change (19). During the course of disease, in addition to the persistence of the presenting symptoms, other features can develop and include impulsivity, disinhibition, irritability, reckless behavior, anxiety, substance abuse, catatonia, emotionality, and mania (50; 59). Depression has been estimated to occur in 20% to 30% of those infected by Wilson disease. It has been suggested that depression in Wilson may be reactive, but this does not explain those presenting with depression. Psychotic features do not appear to be a common manifestation of Wilson disease. Psychiatric manifestations of Wilson disease appear to be more common with neurologic involvement and are uncommon in the hepatic presentation (18; 19).
Psychiatric manifestations of Wilson disease also have a variable response to treatment. Incongruous behavior and cognitive impairment were reported to be more treatment-responsive than irritability or depression. Improvements tended to occur relatively early in the course of treatment, before 3.5 years, and then reach a plateau (19). Others have suggested that the most prominent improvement occurs between 6 and 18 months following initiation of therapy and then plateaus after 2 years of copper level normalization (01).
The hepatic presentation can vary in its onset from insidious, appearing much like chronic active hepatitis or alcoholic cirrhosis, to acute fulminant hepatic failure. Clinically, separating chronic active hepatitis due to Wilson disease from that due to other etiologies is impossible. Thus, such patients must be screened for the possibility of Wilson disease unless the diagnosis of viral etiology is clearly known. Similarly, patients with chronic cirrhosis from Wilson disease do not differ clinically from those with alcoholic and other kinds of cirrhosis. Again, screening for Wilson disease is important to identify the patients who can be treated. Patients who present with hepatic failure may have mild failure with jaundice, low blood albumin, and edema, but will not be in an acute, rapidly deteriorating, fulminant state. Other patients may present with acute fulminant hepatic failure. In these patients, hemolysis is present, because the acute hepatic necrosis releases so much copper that red blood cell damage occurs. The occurrence of hemolysis in hepatic disease should always trigger a search for Wilson disease because it is by far the most likely diagnosis. Patients with a neurologic presentation almost invariably have subclinical liver disease, usually mild-to-moderate cirrhosis (22).
In addition to the three major presentations, patients with Wilson disease may have a variety of other clinical manifestations (10). These include abnormalities of renal tubular function, including Fanconi syndrome. Renal stones and gallstones are not uncommon. Patients may have osteoporosis or osteomalacia, or they may have joint disorders such as arthritis or arthralgias. Female patients frequently have oligomenorrhea or amenorrhea. Abnormalities of the heart include interstitial fibrosis and myocarditis. Electrocardiographic abnormalities and orthostatic hypertension are not uncommon. Pancreatic disease, parathyroidism, and skin abnormalities may be present. In addition to Kayser-Fleischer rings, which do not produce clinical problems, the patient may have sunflower cataracts.
Untreated Wilson disease offers serious threats of progressive hepatic and neurologic deterioration and death. See the clinical manifestations section for details. Prompt therapy may prevent or ameliorate these complications. Overall neurologic outcome following treatment with anticopper therapy is favorable. Analysis of neurologic features at onset of treatment has identified tremor as a favorable prognostic sign, whereas dystonia is relatively refractory to treatment (12).
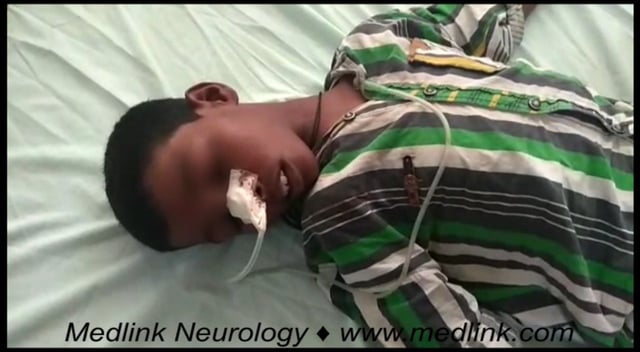
Patient 1: demonstration of early neurologic manifestations and favorable effect of penicillamine. A 14-year-old girl was seen in the university clinic with a 6-month history of increasing difficulty in her studies, nervousness, and tremor. Kayser-Fleischer rings were present, serum ceruloplasmin level reduced, and urinary copper increased. During the last few months, she had experienced swallowing difficulty. Speech was slow, and there was difficulty in articulation. There was tremor of extremities, hypersalivation, and diminished facial expression with stereotyped grin. Handwriting was impaired, verbal IQ was 90, performance IQ was 80, and her overall IQ was 83.
The patient had insight about her limitations and attempted to conceal the tremor. She was started on D-penicillamine therapy, at first 750 mg/day, later increased to 1500 mg/day, and this was continued for the next 9 years. At the age of 24, her handwriting was normal, as was her speech and behavior, and her overall IQ was 86.
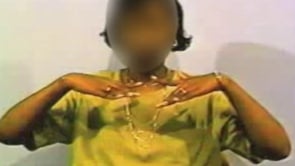
Patient 2: demonstration of neurologic worsening that may be associated with penicillamine therapy (42). A 9-year-old boy was evaluated because of hepatomegaly and elevated levels of transaminase. Wilson disease was diagnosed because of elevated levels of urinary copper at baseline (198 microgram/dl) and after penicillamine challenge (600 microgram/dl) and a high liver copper value (750 microgram/g dry weight). Baseline neurologic examination was normal, and slit-lamp examination failed to reveal a Kayser-Fleischer ring. Penicillamine therapy (20 mg/kg per day) in two divided doses by mouth and pyridoxine hydrochloride (25 mg per day) was started. After 10 weeks of treatment, a tremor in the upper limbs, which seriously impaired the patient's fine-motor activity, developed without any apparent cause other than the penicillamine therapy. The tremor was rhythmic with a variable oscillation--increased with movement, becoming accentuated as the limb approached its target--and disappeared with sleep. It became progressively more severe over a 2-week period, with a significant deterioration of the quality of life. Zinc sulfate therapy was started at a dosage of 100 mg three times per day, while the penicillamine dosage was gradually tapered over a 2-week period until complete discontinuation. Neurologic improvement was progressively observed within 2 weeks. Four weeks after the initiation of zinc therapy, fine motor activity was no longer impaired by tremor; 8 weeks later, the patient reached his [normal] neurologic baseline status. No other neurologic symptoms occurred during zinc therapy over a 30-month period.
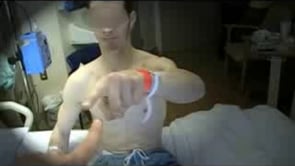
Patient 3: progressive neurologic symptoms that worsened after trientine therapy. A 25-year-old male patient developed tremors in his right hand 1 year before the filming of this video, which markedly worsened after initiation of valproic acid therapy for headaches.
The tremor was present in his right hand, primarily with action, and subsequently involved his left hand to a lesser extent. After he started treatment with trientine, 6 months before video filming, his tremors markedly worsened and were reported to have involved his whole body. Since that time, tremor has involved his whole body but has been worse in his legs and on the right side more so than the left. Lower extremity tremor developed 3 weeks before video filming. He has had progressive difficulty walking for at least 4 months, with marked worsening to the point where he could no longer stand unaided during the 3 weeks before video filming. Speech also has been involved for at least 6 months. It was mildly abnormal before treatment with Trientine; after treatment, it markedly worsened with the development of drooling and choking on food. Six months before video filming, he was found to have an elevated 24-hour urine copper of 171 µg, a ceruloplasmin of 6.6, and an ophthalmologic examination demonstrating Kayser-Fleischer rings.
Remarkably, the mechanism by which excess copper results in neuronal dysfunction and damage is unknown. Humans, including Wilson disease patients, take in about 1.0 mg of copper daily in their diet and require only about 0.75 mg. Thus, the extra 0.25 mg must be eliminated. The normal mechanism for eliminating excess copper is excretion in the bile for loss in the stool. Dietary copper is absorbed in the stomach and duodenum and transported via the portal vein to the liver, the main organ controlling copper regulation. Copper is absorbed into hepatocytes by copper transporter 1, and then ATOX1, a copper-specific chaperone protein, transports copper to ATP7B. The normal function of ATP7B appears to be the incorporation of copper into ceruloplasmin and the secretion of copper into bile. As a result of the mutation in the Wilson disease gene, the liver is incapable of excreting the excess copper into the bile, and a positive copper balance, averaging about 0.25 mg per day, is established. Copper accumulates over time, first in the liver and then in other body parts, such as in the brain. The damage from excessive copper appears to be oxidant in nature (10; 02; 06).
The Wilson disease gene was mapped to chromosome 13q14.3, and the causative gene was identified as ATP7B (11; 61; 70). ATB7B is a 1411 amino acid copper-transporting P-type transmembrane ATPase. The normal function of ATP7B appears to be the incorporation of copper into ceruloplasmin and the secretion of copper into bile. Abnormal ATB7B function leads to abnormal copper metabolism and subsequent copper deposition. Wilson disease is an autosomal recessive disease that occurs when a patient carries mutations in both copies of their ATP7B gene (24).
Mutational analysis has identified over 500 different Wilson disease mutations throughout the ATP7B gene, most of which are missense mutations (24). Wilson disease-causing mutations cause a wide spectrum of functional changes in ATP7B activity (33). Most mutations are inactivating or decrease protein stability, leading to either low or absent ATP7B activity, but up to 25% to 30% of mutations retain some function. Partial ATP7B activity may account for mild phenotypes. Most individual disease-causing mutations are rare, resulting in most patients being compound heterozygotes (33). The combination of a large number of low-frequency mutations occurring in variable combinations hinders phenotype-genotype correlations (24; 33).
Individual mutations have been associated with different ethnic populations. The H1069Q substitution is the most common mutation in white populations, representing 37% to 63% of identified mutations. The H1069Q is not prominent in Chinese or Indian populations, in whom other mutations occur at high frequency. The H1069Q mutation, particularly when homozygous, has been associated with a later-onset neurologic disease (55). Other genotype-phenotype correlations have been elusive. Few consistent genotype-phenotype correlations have been demonstrated. Within single families, concordance of clinical features has been demonstrated but the degree to which this is genetic or environmental is yet to be determined (13). It is unknown how the same mutation can cause a hepatic disease in some but a neurologic disease in others. A combination of environmental, genetic, and epigenetic factors is likely responsible (24; 33). A Finish study suggested that there may be reduced penetrance of ATP7B mutations (53).
Wilson disease appears to be typical of rare autosomal-recessive genes in that it is present at a low frequency in all populations. Estimates for the disease frequency in most populations are about 1 in approximately 30,000, which would lead to a carrier frequency of 1 in 90 (26). As with most autosomal-recessive diseases, there may be pockets of excess Wilson disease produced by a founder effect, particularly if consanguinity is common in the population (05). In some populations, the incidence has been reported to be much lower, such as 0.45 per 100,000 in Finland (53)
Preventive medicine is extremely important in this disease. First, early diagnosis is needed in symptomatic patients with either a hepatic, neurologic, or psychiatric presentation to lessen the likelihood of serious permanent residual disease manifestations. Thus, aggressive screening measures in clinic populations at risk are critical.
Secondly, an important target population is the siblings and parents of the newly diagnosed patient (31). Each sibling has a 25% risk of being in the presymptomatic disease stage. Because prophylactic therapy will prevent the onset of the disease, these patients should be aggressively examined to determine disease status. All full siblings should be screened for blood ceruloplasmin and 24-hour urine copper levels. The 24-hour urine copper is a much more reliable and sensitive procedure for diagnosing Wilson disease than the ceruloplasmin assay (10). In the case of the 24-hour urine copper in presymptomatic siblings, if the value is more than 100 it is diagnostic of Wilson disease. If it is less than 50 in an adult patient, it essentially excludes the diagnosis. If it is intermediate, it is compatible either with the carrier state because some of these patients have mild elevations of urine copper or the presymptomatic affected state because some of these patients do not have diagnostic levels of urine copper. The patients in this intermediate zone should have a liver biopsy to make the diagnosis. The liver copper level in such a patient is diagnostic. Because the risk of Wilson disease is significantly elevated in nieces, nephews (1/600), and cousins (1/800) compared to the general population, these relatives can be screened for ceruloplasmin and 24-hour urine copper levels.
Wilson disease must be distinguished from other common and rare neurologic diseases (45). Because Wilson disease is treatable, it should be considered as a possible diagnosis in every young person with a movement disorder.
The more common neurologic disorders that can mimic Wilson disease include essential tremor, young-onset Parkinson disease, and generalized dystonia. Rare juvenile genetic extrapyramidal disorders, including Huntington disease, neurodegeneration with brain iron accumulation disorders (formerly Hallervorden-Spatz disease), idiopathic torsion dystonia, chorea-acanthocytosis, and benign familial chorea can at times mimic Wilson disease. Neither chorea nor ataxia is typically found in isolation, and although Wilson disease can be considered, diagnostic consideration for causes of primarily choreic and ataxic diseases should be undertaken. The psychiatric abnormalities may be mistaken for a psychological abnormality, early schizophrenia, or drug abuse. The presence of Kayser-Fleischer rings and the coexistence of liver disease provide important clues.
The coexistence of chronic progressive extrapyramidal and liver disease with onset at school age to midlife suggests a diagnosis of Wilson disease. The hepatic manifestations cannot be distinguished from those of chronic active hepatitis, alcoholic cirrhosis, or acute hepatic failure. Acute hepatic failure is often associated with hemolysis, and its occurrence provides an important clue.
Other associated abnormalities: renal tubular dysfunction, including Fanconi syndrome; renal stones and gallstones; osteoporosis or osteomalacia; arthritis or arthralgias; oligomenorrhea or amenorrhea; interstitial cardiac fibrosis; myocarditis; Electrocardiographic abnormalities; orthostatic hypotension; pancreatic disease; parathyroidism; and skin abnormalities; Kayser-Fleischer rings; and sunflower cataracts.
The most common screening method for Wilson disease is a blood ceruloplasmin determination, although this is inadequate for either ruling in or ruling out Wilson disease. The ceruloplasmin value is usually low in Wilson disease, but in approximately 10% of patients, it may be normal or near normal. Further, about 10% of heterozygous carriers who will never have clinical problems have low ceruloplasmin values.
The most useful screening procedure is a 24-hour urine copper test (10; 05; 45). The 24-hour urine copper is always elevated to a value of 100 µg per 24 hours (normal is 50 or less) in symptomatic Wilson disease. The 24-hour urine sample must be collected in a container free of trace elements. The Leipzig score is recommended as a diagnostic algorithm, having shown high accuracy, particularly in pediatric cohorts (23).
Relative exchangeable copper determination, defined as the ratio of exchangeable copper to total serum copper, is a valuable tool with high sensitivity and specificity (REC > 18.5%). This assay is available for clinical use in France, Spain, Denmark, and India (23).
Another common screening procedure is a slit-lamp examination for Kayser-Fleischer rings. Visual inspection is not adequate. Kayser-Fleischer rings are invariably present in the psychiatric and neurologic presentations; however, they are present in only about 50% of patients who present with liver disease.
In a patient with classical clinical disease, Kayser-Fleischer rings, and elevated urine copper, the workup need not go further. If any question remains, the gold standard for diagnosis is a measure of quantitative copper of a percutaneous liver biopsy (45). The hepatic copper value in untreated Wilson disease is above 200 µg/g dry weight of tissue with the normal being 50 or less. Carriers of the Wilson disease gene may have mild elevations of hepatic copper but never to 200 µg/g. It is important not to rely on the stain for copper, for if the copper is still diffusely cytoplasmic, the copper stain may be negative in the face of great elevations of hepatic copper.
The gene for Wilson disease has been cloned (11; 61; 70), fostering hope for the development of a direct DNA test. This approach is not practical because more than 500 mutations that may cause Wilson disease have been identified (49; 02). However, if a mutation is identified in a proband, a search for this mutation in relatives can provide crucial information for the identification of as yet asymptomatic relatives and of heterozygotes and the opportunity for preventive therapy and genetic counseling. Mutational analysis appears to be improved by undertaking direct sequence analysis of the entire ATP7B coding region (14).
Various brain scans may be somewhat useful in the diagnosis. MRI scans are generally positive in patients who have neurologic or psychiatric symptoms but are often negative in patients with only liver disease.
In neurologically involved patients, the most common findings are T2 high-signal areas in the lentiform and caudate nuclei, thalamus, brain stem, and white matter. Involvement of white matter around the red nucleus can demonstrate the “face of the giant panda” sign (35). High-signal T1-signal, like those in portal-systemic encephalopathy, may also be observed (52). Signal abnormalities often improve with anti-copper therapy (16). Dopamine D2 receptor binding and regional cerebral glucose metabolism are reduced in the striatum and have been shown to return to almost normal levels after penicillamine therapy (48). Diffusion tensor imaging has been used to demonstrate widespread changes in both normal and abnormal appearing cerebral white matter (28).
The American Association for the Study of Liver Diseases, which outlines guidelines for diagnosing and managing Wilson disease, and the European Association for the Study of the Liver EASL-ERN Clinical Practice Guidelines on Wilson’s disease are excellent comprehensive references (47; 23).
Wilson disease is a condition that can be effectively treated. Treatment can be divided into initial therapy, with differentiation between the hepatic presentation and the neurologic/psychiatric presentation; maintenance therapy; treatment of the presymptomatic patient; and treatment of the pregnant patient (10; 45).
Pharmacologic agents include penicillamine, trientine, zinc acetate, and tetrathiomolybdate.
Zinc or trientine can be used for the initial treatment of patients presenting with neurologic or psychiatric disease, but like penicillamine, trientine can cause neurologic worsening in about 25% (08; 45). Zinc induces intestinal cell metallothionein, a protein that complexes intestinal food copper or endogenously secreted copper in saliva and gastric secretions. The complex cannot be absorbed into the blood, and as intestinal cells die, they, along with the complex, are sloughed into the stool, resulting in a negative copper balance. The net effect of zinc is an intestinal blockade of zinc absorption. Zinc has the additional effect of inducing hepatocyte metallothionein production, which reduces the toxic effect of free copper in these cells (05).
The de-coppering effect of zinc is slow; a period of 4 to 8 months of treatment is required to reduce copper to nontoxic levels. In symptomatic individuals, the natural course of disease continues during this period and can lead to permanent worsening. To avoid potential toxicities of trientine and penicillamine, some have successfully used zinc in the initial treatment of neurologic Wilson disease (27). An optimal treatment strategy may include a course of de-coppering with an agent such as tetrathiomolybdate followed by maintenance therapy with zinc. An advantage of zinc is its lack of serious significant side effects and safety in long-term use (04). Approximately 10% experience gastric discomfort or nausea on initiation of zinc therapy. The use of zinc acetate, compared to zinc sulfate, reduces gastric discomfort. Generally, gastric symptoms subside within days to weeks. For maintenance and prophylactic therapy in adults, 50 mg of zinc three times a day is recommended (10; 04; 27). It is important to separate the zinc from food by at least an hour. For maintenance therapy, zinc is favored over penicillamine or trientine because although all three drugs are equally effective, zinc has a superior side effect profile and does not appear to be responsible for neurologic worsening (10; 27). Because zinc therapy does not induce urinary excretion of copper like trientine or penicillamine, urine copper is an accurate reflection of body copper stores. Following a year of zinc therapy, a 24-hour copper of about 125 µg is a reflection of good copper control. Compliance with zinc can be assessed by monitoring 24-hour urine zinc excretion, which should be above 2 mg (05).
One note of caution for zinc therapy is that overtreatment leading to copper depletion has been associated with myelopathy and neuropathy (17). Fortunately, the symptoms and signs of copper treatment myelopathy may be reversible (62). Periodic monitoring of 24-hour urine copper and zinc levels can be used to avoid this rare potential complication of zinc therapy.
Trientine is a chelator that like penicillamine promotes urinary excretion of copper. Trientine is used in doses of 750 to 1500 mg for initial therapy and 750 to 1000 mg for maintenance therapy, divided into two or three doses per day. It should be given 1 hour before or 2 hours after meals. No well-designed clinical trials have determined the optimal initial trientine dose. Initial therapy with lower doses may reduce excess free copper and the risk of neurologic deterioration. A suggestion would be to initiate treatment with 250 mg twice daily and closely follow clinical signs and 24-hour urine copper levels. Although trientine has fewer side effects than penicillamine, it shares a common mechanism of action and can cause neurologic worsening. In a double–blind study comparing tetrathiomolybdate to trientine, 24% of those treated with trientine experienced neurologic worsening (08).
A study described a new formulation of trientine, trientine tetrahydrochloride, as noninferior to penicillamine (46). In spring 2023, trientine tetrahydrochloride (Cuvrior™) received FDA approval in the United States for treating adult patients with stable Wilson disease who are de-coppered and tolerant to penicillamine. A potential advantage of trientine tetrahydrochloride is that it does not require refrigeration.
While on trientine, urine copper is a reflection of enhanced urinary copper excretion and total body copper load. On initial treatment with trientine, 24-hour urinary copper excretion may be 1000 to 3000 µg (normal 20 to 50 µg/24 hours). After a few weeks, the urinary copper decreases to 500 to 1000 µg per 24 hours, and after approximately 1 year of therapy it should decrease to 200 to 500 µg per 24 hours. Nonceruloplasmin bound copper, or “free copper,” is a useful measure for monitoring efficacy of trientine and can be calculated by the formula: (Total Serum Copper in µg/ml) X 100-(Ceruloplasmin in mg/dl x 3)= Free Copper (Normal range is 5 to 15 µg/dl). Another way that free copper can be accurately estimated is by subtracting 3µg for every 1mg/dl of ceruloplasmin from the serum copper, expressed as µg/dl. During the maintenance phase of trientine therapy, a free copper below 25 µg/dl is the goal.
Penicillamine carries the risk of severe neurologic worsening in about 50% when used as initial treatment of neurologic Wilson disease, and 50% of those who deteriorate never recover to their prepenicillamine baseline (09). Because of initial neurologic worsening in those treated with penicillamine and availability of safer agents, its use is not recommended. If it is to be used, the standard starting dose is 250 mg four times daily or 500 mg twice a day. It has been suggested that starting with lower doses, such as 250 to 500 mg daily for a few weeks, may lessen side effects (45).
When initiated, the patient should be carefully monitored for various kinds of toxicities, including bone marrow and kidney toxicities. An acute hypersensitivity reaction occurs in about 25% of patients, which can be dealt with either by corticosteroid therapy or withdrawal of the drug and readministration in very low doses.
Tetrathiomolybdate has been studied for treatment of Wilson disease (08; 07). Tetrathiomolybdate acts by forming a tripartite complex with copper and protein. Given with food, tetrathiomolybdate binds food copper and endogenously secreted copper, preventing copper absorption (03). Given between meals, tetrathiomolybdate is absorbed into the blood and forms a complex with free copper and albumin. Free copper in the blood is in equilibrium with tissue copper so that binding of serum copper to tetrathiomolybdate shifts the equilibrium to mobilize copper from tissue stores into serum where it is bound by tetrathiomolybdate and excreted into bile. The result is a rapid removal of tissue copper with significantly less neurologic worsening compared to penicillamine or trientine. In a double-blind study comparing tetrathiomolybdate to trientine in the initial treatment of neurologic Wilson disease, both trientine and tetrathiomolybdate were found to improve neurologic function, but tetrathiomolybdate was significantly less likely to be associated with neurologic worsening than trientine (08; 07). Tetrathiomolybdate, 20 mg three times a day with meals and 20 mg three times a day between meals, or trientine, 500 mg two times daily, were given for 8 weeks with zinc, 50 mg twice daily, followed by zinc maintenance therapy. Tetrathiomolybdate was associated with reversible increases in transaminase levels and anemia in about 15%, necessitating close laboratory monitoring. In a proof-of-concept phase 2 trial, once-daily Bis-choline tetrathiomolybdate (WTX101) over 24 weeks rapidly lowered nonceruloplasmin-bound copper levels, and this was accompanied by improved neurologic status without apparent initial drug-induced paradoxical worsening, reduced disability, and stable liver function, with a favorable safety profile (67). A phase 3 trial of WTX101 is ongoing.
There was an ongoing effort to bring a tetrathiomolybdate formulation to market (ALXN1840), with promising initial results; however, the responsible pharmaceutical company has ended further development (68).
The combination of trientine followed by zinc maintenance may represent the best medical treatment. The trientine is given to obtain a rather robust negative copper balance. Zinc is given to induce hepatic metallothionein and allow that metallothionein to complex copper in the liver in a nontoxic form. This combination is used for 4 months and then the trientine is discontinued.
Where available, REC determination can be used to follow response to treatment.
In patients who present with liver disease, if the disease is fulminant, a hepatic transplant may be the only way to save the patient's life. However, in cases of mild hepatic failure, a picture of chronic cirrhosis, or a picture of mild hepatitis, medical treatment can be quite effective. The prognostic index of Nazer is useful in differentiating which patients will likely do well with medical therapy (37). Liver transplantation was given a weak recommendation on a case-to-case basis in patients with severe neurologic Wilson disease who do not respond to anti-copper (23).
Longitudinal studies on treatment outcomes in neurologic Wilson disease are surprisingly sparse and frequently anecdotal. In those studies reporting on the effect of treatment, the largest degree of improvement was observed between 6 months and 2 years of adequate copper control, with some benefit reported to occur for up to 3 years (19; 57; 44; 08; 36). No reliable predictors exist to differentiate those who will make an excellent recovery from those whose symptoms will stabilize but not improve.
Because pharmacologic treatment of dystonia can be less than optimal, deep brain stimulation has been considered, and at least in one case, bilateral globus pallidus deep brain stimulation improved dystonia and caregiver burden in Wilson disease (51).
Over the years, considerable attention has been paid to diet therapy for Wilson disease. Foods high in copper content include liver, shellfish, kale, sesame seeds, dark chocolate, mace, cashew nuts, chickpeas, prunes, mushrooms, sundried tomatoes, lean ham, soybeans, fermented soy (tempeh), goat cheese, avocadoes, and wheat bran. During the initial decoppering phase liver and shellfish should be avoided and other foods high in copper content should be avoided or restricted to small quantities (10). In the maintenance phase, only liver and shellfish are high enough in copper to be of concern and should be limited to single servings per week, or less. Other foods are not restricted because the content of copper is not high enough to be of concern. Occasionally, drinking water samples will be found to be high in copper. If the level is higher than about 0.1 ppm, the patient should use an alternative source of drinking water.
With all Wilson disease copper reduction treatments, it is important to monitor copper levels to ensure copper deficiency does not develop, as copper deficiency can cause irreversible myeloneuropathy (66).
As long as they do not have severe liver disease, patients with Wilson disease can tolerate pregnancy well. A study of pregnancy in Wilson disease provided valuable information regarding the risk of spontaneous abortion in Wilson disease, the risk of maternal decompensation, and the safety of chelating agents during pregnancy. The number of patients in whom some form of maternal demise occurred was low. Treatment of Wilson disease did not increase the risk of spontaneous abortion.
Undiagnosed and untreated Wilson disease resulted in higher rates of spontaneous abortion. This study provided clear evidence that patients with Wilson disease should continue treatment with close monitoring during pregnancy (41). Restrictions on patients with severe liver disease from Wilson disease are no different than restrictions on patients with other kinds of severe liver disease. Otherwise, the patient can generally tolerate pregnancy well and should continue therapy with anti-copper treatment during the pregnancy. Treatment with penicillamine can produce a fetal penicillamine syndrome; trientine has not been reported to cause fetal abnormalities in humans, although it is a known teratogen in animals. Zinc appears to be safe from the standpoint of teratogenicity.
For the treatment of the pregnant patient, because of the teratogenic effects of both penicillamine and trientine, zinc generally reduced to the minimum dose of 25 mg three times daily is used. Zinc has been specifically studied for its teratogenic effects, and none have been found (10).
Anesthesia is well tolerated by patients with Wilson disease. There are no restrictions other than those that would be placed on those patients who might have severe liver disease or severe neurologic impairment. Zinc therapy can be continued during the surgical period, although it is taken orally and may need to be interrupted while the patient cannot ingest orally. Zinc therapy can be safely interrupted for a few days during this period. Likewise, penicillamine or trientine should be taken up until the time of surgery and then resumed as soon as the patient can take the medication orally. Penicillamine does have some potential to interfere with wound healing.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Matthew Lorincz MD PhD
Dr. Lorincz of the University of Michigan has no relevant financial relationships to disclose.
See Profile
AHM M Huq MD PhD
Dr. Huq of Wayne State University has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Sleep Disorders
Jul. 03, 2026
Neurobehavioral & Cognitive Disorders
Jun. 17, 2026
Neurogenetic Disorders
Jun. 01, 2026
Movement Disorders
May. 24, 2026
Neurobehavioral & Cognitive Disorders
May. 20, 2026
Movement Disorders
May. 18, 2026
Movement Disorders
May. 18, 2026
Movement Disorders
May. 18, 2026