


Developmental Malformations
Barth syndrome
May. 08, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
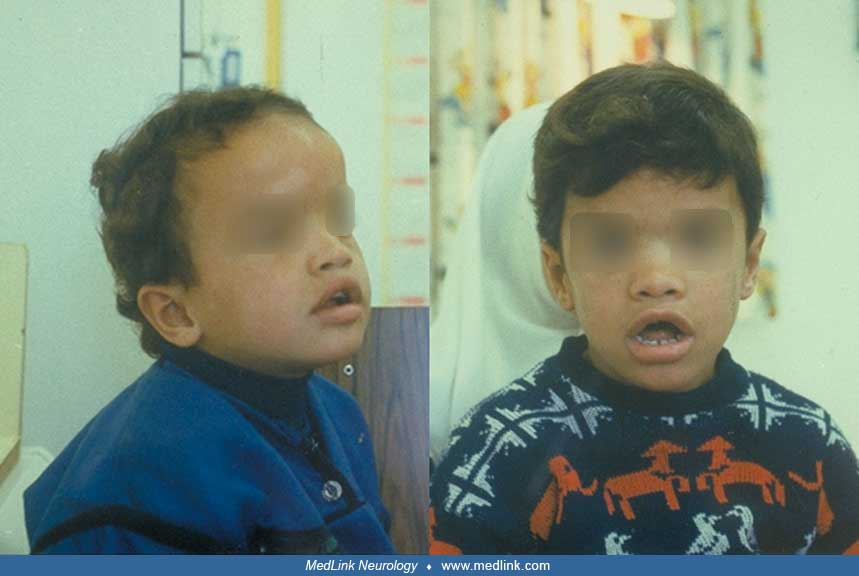
Acrocallosal syndrome (ACLS) is a multiple congenital abnormalities/intellectual disability syndrome. The clinical diagnosis is based on the presence of three of the four major criteria: (1) macrocephaly and facial dysmorphism, (2) partial or total agenesis of corpus callosum, (3) distal limb anomalies, and (4) intellectual disability. Biallelic mutations of the K1F7 gene would confirm the clinical diagnosis as it was implicated in typical acrocallosal syndrome. Further, a phenotype consistent with acrocallosal syndrome was identified in two patients harboring de novo/dominant mutations in GLI3. The phenotypic spectrum includes a severe form with in utero fetal death known as hydrolethalus-2 syndrome usually presenting with hydrocephalus or anencephaly. This clinical variability can be explained by the action of K1F7, as it is a regulator of the Sonic Hedgehog pathway by preventing an inappropriate activation of Gli2 and the processing of Gli3 into its repressor form. The clinical spectrum of acrocallosal syndrome and differential diagnoses are discussed.
|
• Acrocallosal syndrome is a ciliopathy disorder typically characterized by macrocephaly, agenesis/hypogenesis of corpus callosum, polydactyly, and intellectual disability. | |
|
• Hydrolethalus-2 is a more severe phenotype, including death in utero. | |
|
• Biallelic mutations in K1F7 gene are the most common mutations implicated in acrocallosal syndrome. | |
|
• De novo mutations in the GLI3 gene were identified in a few cases with acrocallosal syndrome (20). |
Acrocallosal syndrome (ACLS) was first recognized by Schinzel in two separate publications of two unrelated Swiss patients with macrocephaly, agenesis of the corpus callosum, hypertelorism, and polydactyly and was termed because of the unique association of corpus callosum and distal acral anomalies most commonly pre and/or postaxial polydactyly (18; 19). Biallelic mutations in KIF7 are delineated in the majority of patients, thus, autosomal recessive mode of inheritance. The phenotype is ranging from the lethal anencephaly to a previously unrecognized mild end of the spectrum. In contrast, de novo heterozygous GLI3 mutations were reported in two unrelated patients with acrocallosal syndrome. The exact frequency of acrocallosal syndrome is difficult to determine. The clinical spectrum has been broadened not only to include hypogenesis/agenesis of corpus callosum but also cystic malformation of the brain (25; 13). Midline defects such as cleft palate or congenital heart defects have been reported. Furthermore, anophthalmia, lack of nasal structures, and omphalocele have been described (06). Nevertheless, hypogenitalism was noted in few patients (23; 10; 16).
Nearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125