


Developmental Malformations
Barth syndrome
May. 08, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
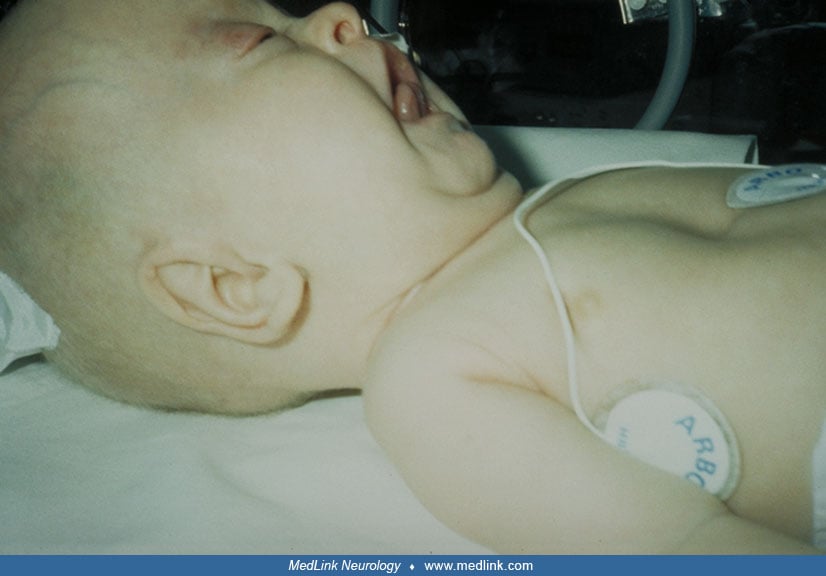
Yunis-Varon syndrome (“cleidocranial dysostosis”) is a rare, generally severe, and autosomal recessive condition manifested by numerous anomalies of the craniofacial complex (dolichocephaly, wide fontanelles, sparse hair, hypoplastic facial bones, thin lips, short philtrum, micrognathia, and variable changes of the CNS, including eyes), small/absent clavicles, small/absent thumbs and great toes, and other anomalies. Mutations in FIG4 have been identified in several unrelated or consanguineous families; a biallelic mutation in VAC14 has been identified in a single neonate with normal FIG4 sequencing. The syndrome continues to undergo phenotypic and molecular delineation.
|
• Major phenotypic changes in this rare congenital disorder involve the clavicles and head (cleidocranial dysostosis), mandible, and distal limbs. | |
|
• Craniocerebral anomalies include poor growth of cranial bones, macrocephaly, large fontanels, and cortical malformations (eg, polymicrogyria). | |
|
• Neurologic symptoms and craniofacial findings may include hypotonia and weakness, psychomotor delay, large, dysplastic ears, and cleft lip and palate. Seizures and psychiatric manifestations are recognized as well. | |
|
• Cardiorespiratory failure commonly leads to death in the neonatal period, although survival into the second decade has been reported. | |
|
• Familial recurrence, sometimes with a history of consanguinity, has suggested autosomal recessive inheritance in the past, and is supported by the finding of FIG4 mutations in several families. |
In 1980, Yunis and Varon described an infant boy and four infant girls from three Colombian families; one family had a single affected infant, and the other two families each had two affected infants. The patients had multiple malformations, including cleidocranial dysostosis, severe micrognathism, and hypoplasia or aplasia of metatarsal bones, phalanges, or thumbs. Changes in the cranium consisted mainly of “skull dysostosis,” characterized by wide fontanelles and separated sutures, craniofacial disproportion, macrocrania, hypoplastic facial bones, and micrognathia. Clavicular changes included uniform absence of the right clavicle and hypoplasia or absence of the left clavicle. All died before the age of 10 weeks. In two of the families, consanguinity was reported. The authors postulated this was a new recessively inherited syndrome (53). In 1983, Hughes and Partington reported a 4.25-year-old boy with the same pattern of malformations, shortness of stature, and mental retardation; they referred to the condition as “the syndrome of Yunis and Varon” (20).
Additional cases have been described with similar findings and some new features (35; 36; 19; 17; 01; 13; 38; 09; 51). Rabe and colleagues added a note that their patient had a sister with features of Yunis-Varon but did not describe her (38). The patient described by Partington had cardiomyopathy (35), and Ades and colleagues described a patient with congenital heart malformations (tetralogy of Fallot) (01). The patient reported by Dworzak and colleagues was a female with generalized lysosomal storage (13). A female baby of German origin had a severe hearing impairment between 1000 Hz and 8000 Hz, detected by auditory evoked potential studies (38). Christie and colleagues described a 28-year-old woman with multiple anomalies interpreted as Yunis-Varon syndrome (although the morphology of the clavicle was not described). By a variety of imaging techniques, the woman had atrophy of the left lobe of the liver and an anomalous dilated hepatic vessel at the junction of right and left lobes. Vascular compromise of the left lobe was suggested, but the patient was lost to follow-up before additional studies could be performed (09). Walch and colleagues reported a 15-week-old female infant with Yunis-Varon syndrome with microcephaly, hydrocephalus, and Dandy-Walker malformation; an additional case with associated Dandy-Walker malformation has been reported (41). Severe neurologic impairment associated with intraneuronal inclusions and vacuolar degeneration has been taken as evidence for a lysosomal storage disorder (51). However, the patient reported by Sumi and colleagues showed no evidence for such a disorder (45). Striatonigral degeneration has been associated with VAC14 mutations in a number of young patients (22; 30).
In the United States, there are estimated to be fewer than 1000 cases of Yunis-Varon syndrome (43). The most frequent congenital clinical features are severe hypotonia and weakness, wide fontanelles, and the following facial, hand, and foot abnormalities: sparse hair, micrognathia, anteverted nostrils, dysplastic ears, short upper lip, absence or hypoplasia of thumbs and halluces, hypoplasia or agenesis of nails, and short pointed fingers.
Cleft lip and cleft palate have been described in affected patients, although infrequently (37; 45; 41). Median pseudo-cleft of the upper lip has also been reported (24). Additional signs described by Garrett and colleagues included ectodermal abnormalities (sclerocornea and cataracts), cerebral malformations (arhinencephaly, corpus callosum agenesis with dilation of the occipital horns of the lateral ventricles), and hamartomatous lesion (17). Cortical anomalies are recognized. Pachygyria has been prominent in two affected sisters, as well as papillo-macular atrophic chorioretinopathy with salt-and-pepper appearing fundus (10). Polymicrogyria has also been reported (03). Cataract, but not sclerocornea, was also reported by Rabe and colleagues; they did not find major abnormalities in the differentiation of the white or gray matter or migration defects (38). Another group reported underdeveloped gyri, ischemic changes in the temporoparietal region, and bilateral lacunar infarcts in the middle cerebral artery region (24). In three cases, cardiomyopathy was found (35; 17; 13), and in one patient, acyanotic tetralogy of Fallot was described (01). The testes were enlarged in one patient (06). In several patients, radiological studies revealed that the dysmorphic abnormalities of face, hands, and feet are associated with absence or hypoplasia of clavicles, pelvic dysplasia, and hip dislocations. Narrow diaphysis and widened ends of the tibiae and femora have been described, with poorly understood “periosteal reaction” involving the midshafts of humeri and femora (06). The hypoplasia of digits varies considerably from mild to almost complete absence (02).
The clinical course of the disease is severe, with failure to thrive and marked psychomotor delay. Seizures and psychiatric difficulties complicate the course. Death usually occurs in the neonatal period due to cardiorespiratory failure. A few patients have survived longer. A patient with cleft lip and palate was doing well at 14 months (45). Another was alive at 4.25 years with short stature and mental retardation (20); one was reexamined at 11 years of age and had hypodontia, impacted permanent teeth, spinal defects, cardiomegaly, bilateral hearing loss, and metatarsus adductus (26); a 12-month-old boy with microcephalia and marked developmental delay was assessed as having the developmental level of a 3- to 4-month-old (01); a young woman was described with a hepatic vascular anomaly and atrophy of the left lobe of the liver, hitherto unreported findings (09). A 26-year-old man with dilated cardiomyopathy, short stature (treated with growth hormone), and renal artery stenosis with hypertension has been described (49).
The prognosis is severe, and patients usually die in the first few months due to cardiorespiratory failure. Pulmonary hypertension has been reported and may be more common than originally thought (39). The patient described by Rabe and colleagues presented feeding problems, lack of a swallowing reflex, and pyloric stenosis requiring myotomy. The child died at 3 months after progressive episodes of bradycardia (38). The age range of survivors is 12 months to 28 years.
Yunis-Varon syndrome is thought to be autosomal recessive, but exact genes or molecular mechanisms have not been identified until very recently. Campeau and colleagues identified mutations in FIG4, a gene that encodes phosphoinositide phosphatase and is responsible for PI(3,5)P(2) levels, endosomal trafficking, and autophagy (07). Biallelic null mutations have been postulated as the molecular mechanism responsible for the syndrome (34). Biallelic mutations have been found in VAC14, a scaffold protein bound to FIG4 (29; 31). Nearly one third of cases have presented with a history of consanguinity, with familial recurrence in some (24; 02; 10; 30). Complete loss of function of FIG4 causes Yunis-Varon syndrome (52).
Autopsies have been performed in at least 10 cases: an 18-week fetus (17), a 37-week stillborn baby (19), and seven patients ranging in age from 21 to 120 days (53; 17; 13; 38). Remarkable gross abnormalities were found in the 4-month-old boy described by Garrett and colleagues; these consisted of arhinencephaly, agenesis of the corpus callosum with dilation of the occipital horns of the lateral ventricles, and a hamartomatous lesion (17). Agenesis of the corpus callosum and dilation of the lateral ventricles were also found in a fetus examined by Garrett and colleagues (17). This same group identified hypoxic changes as well, including neuronal loss in the cerebral cortex, basal ganglia, cerebellar dentate nuclei, medullary olives, and anterior horn cells. In three other patients, deeper than normal brain fissures and normal histologic features were detected (53; 17). In two female siblings described by Garrett and colleagues, autopsy revealed a thick myocardium in one, whereas incomplete lung lobe fissures and incomplete separation of right and left liver lobes were found in the other (17).
In all but a single patient, chromosome analysis of lymphocytes, fibroblasts, or both has disclosed a normal karyotype. In the patient reported by Rabe and colleagues, chromosomal analysis was performed on cordocentesis material obtained at 32 gestational weeks because of fetal growth retardation and a mild ventriculomegaly. The analysis revealed a normal female karyotype with a large subcentromeric heterochromatic block in chromosome 1 (46, XX, 1qh++) (38). The block was also observed in the patient's mother, who was clinically normal. Routine laboratory investigations, including amino acid excretion, were normal with the exception of slightly high plasma creatine kinase (260 U-1 normal value up to 180 U-1) in the patient described by Dworzak and colleagues (13). In this patient, a muscle biopsy performed 18 days after birth revealed variability of fiber size with vacuoles in several fibers that were either apparently empty or filled with basophilic material; some vacuoles were positive for periodic acid Schiff and acid phosphatase.
Electron microscopy showed the vacuoles to be membrane-bound and to contain granular or membranous degenerated material.
Closely similar vacuolations were observed in the patient's fibroblasts as well as in the heart and cartilage at autopsy. In the brain, vacuolated neurons were observed in the cerebral cortex and in the dentate nuclei of the cerebellum, globus pallidus, thalamus, and subthalamic nuclei. Extensive biochemical studies on this patient only disclosed five abnormal urinary oligosaccharide bands that differed from those present in other lysosomal storage disorders. This finding led the authors to hypothesize that the disease was caused by a metabolic defect related to lysosome physiology. In the original paper of Yunis and Varon, urinary mucopolysaccharides were stated to be normal. Garrett and colleagues described similar vacuolation in their 4-month-old infant with cerebral abnormalities (17). The vacuoles were distributed in the basal ganglia, cerebellar dentate nucleus, medullary olives, anterior spinal horns, and layers 3 and 5 of the cerebral cortex. However, the authors suggested a hypoxic etiology for this alteration. In the muscle of the same patient, only moderate neurogenic atrophy was present, possibly related to loss of the spinal lower motor neurons. Walch and colleagues have identified neuronal inclusions with vacuolar degeneration, most often in the thalamus, cerebellar cortex and dentate nuclei, and inferior olivary nuclei (51). The findings were interpreted as arising from a lysosomal storage disorder. The role of FIG4 in oIigodendrocyte maturation is recognized. Inactivation of Fig4 in mice produces demyelination of Schwann cells and neuronal and axonal degradation in motor neurons (48); in fact, leukoencephalopathy has been observed in several patients with FIG4 variants (27). Knockdowns in the Drosophila homolog, dFIG4, result in aberrations in photoreceptor and motor neurons and other changes in neuronal function and eye development (25).
The syndrome is thought to be inherited in an autosomal recessive manner, as evidenced by consanguinity in at least five families (53; 36; 24; 41) and equally affected sibs of both sexes from five families (17; 38). Lesser evidence for this has been provided by Sumi and colleagues (45). Another report describes a newborn male with Yunis-Varon syndrome and previous sibling with similar features born to a nonconsanguineous couple (06). Consanguineous parents of patients are generally reported to be clinically normal. The only exception to this is the mother of the patient described by Hennekam and Vermeulen-Meiners, who showed asymmetrical face and shortened distal phalanges of all digits (19). Muscle biopsies performed in the healthy nonconsanguineous parents of the child reported by Dworzak and colleagues did not disclose any pathological features (13).
Yunis-Varon syndrome is related phenotypically to Charcot-Marie-Tooth type 4J, and although pathogenesis remains unclear, patients with FIG4 mutations can manifest findings common to the two syndromes (52). Some of these patients may manifest symptoms of parkinsonism (54). Clavicular anomalies are important in the differential diagnosis because other anomalies overlap considerably.
Beauregard-Lacroix and colleagues showed that osteopenia had not been seen in the past on further evaluation is seen in 30% of Yunis-Varon syndrome (04). This osteopenia is more likely secondary to slower bone formation rather than increased bone reabsorption.
Beauregard-Lacroix and colleagues also provided additional information about genotype-phenotype correlation as well as new information about osteopenia (04). They further delineated that the first third of the FIG4 gene is important in the interaction with the VAC14 gene. Complete loss of function of FIG4 (biallelic null variants) causes the most severe phenotype. When there is a compound heterozygote the phenotype seems to be dependent on the type of variant seen each allele. This is similar when there is VAC14 mutation. There is a wide clinical presentation when there is only one null variant.
The syndrome is recognized worldwide, but is very rare; it is estimated that there are fewer than 1000 cases in the United States (43). Both sexes are affected equally.
No prenatal testing is available, and familial recurrence can only be prevented by avoiding pregnancy.
The condition that most closely resembles Yunis-Varon syndrome is cleidocranial dysplasia syndrome. The characteristic features of this disorder are brachycephaly with bossing of parietal, frontal, and occipital bones; late closure of fontanelles; late ossification of cranial sutures; partial to complete aplasia of clavicles; and hand abnormalities. The hand abnormalities include asymmetric finger length with long second metacarpals, short phalanges of the second and fifth digits, and tapering distal phalanges. However, pelvic anomalies in cleidocranial dysplasia (delayed ossification of pubis, hypoplasia of iliac wings) are not present in Yunis-Varon syndrome, nor are the digital anomalies identical. Moreover, cleidocranial dysplasia syndrome is inherited in an autosomal dominant mode, with about one third of cases representing new mutations (21). Recessive inheritance, severe micrognathism, and distinctive facial and distal limb dysmorphisms are typical of Yunis-Varon syndrome, representing fundamental features for distinguishing the two disorders. Many of the individuals with cleidocranial dysplasia syndrome have a mutation in the RUNX2 gene (47).
Some features of Yunis-Varon syndrome are also found in Melnick-Needles syndrome (32). This is a severe congenital disorder that is usually inherited in an X-linked dominant or autosomal sex-limited dominant pattern, although an autosomal recessive form has been reported (46). It is characterized by exophthalmos, micrognathia, malalignment of teeth, flaring of the metaphyses of the long bones, S-like curvature of leg bones, irregular constrictions of the ribs, and sclerosis of the base of the skull. No patients have been reported with cranial dysplasia, distal aphalangia, and absent thumbs typical of Yunis-Varon syndrome.
Syndromes that share other features include Saint-Martin-Gardner-Morrisson syndrome, serpentine fibula syndrome, atelosteogenesis types I and III, fronto-otopalatodigital osteodysplasia, and boomerang dysplasia-lethal dwarfism (50). They can generally be distinguished by the following changes: Saint-Martin and colleagues described a lethal disorder in two brothers with dysmorphic facies, absent ossification of the cranium, curvature of long bones, and multiple fractures (40). Serpentine fibula syndrome manifests long curved fibulae, bowed radius, absent phalanges, and broad clavicles; it may be the same condition as Hajdu-Cheney syndrome (11; 15). Atelosteogenesis type I shows rhizomelic shortening of limbs, bowed, absent, or hypoplastic long bones, tarsals, carpals, and phalanges, but no clavicular anomalies; it has also been called spondylohumerofemoral hypoplasia (42) and may be related to boomerang syndrome (23). Atelosteogenesis type III has many of the same changes as type I, but long clavicles may be bowed or hooked (44). Boomerang syndrome is a bone dysplasia with an angulated (ie, boomerang-shaped) tibia, with possible absence of femur, humerus, ulna, and radius, as well as deficient ossification of the spine, possible absence of phalanges, and long clavicles; it may be identical to Piepkorn syndrome (37; 23).
The patient described by Dworzak and colleagues showed histopathologic features similar to lysosomal glycogen storage disease. However, patients with lysosomal glycogen storage survive longer than Yunis-Varon patients and do not have dysmorphic features; most reported cases of lysosomal glycogen storage have severe hypertrophic cardiomyopathy (12). An additional patient, a 15-year-old girl with ulnar ray oligodactyly and normal intelligence, has been described with a number of features resembling Yunis-Varon syndrome (16).
Diagnosis has been based on clinical features in the past, a fact altered by the discovery of FIG4 mutations in three families (07). In addition to genetic studies, physical examination and skeletal survey should be performed in a detailed manner. The latter continues to be refined (14). We recommend muscle biopsy and analysis of saccharides in urine to explore the possible association with lysosomal storage disease. Glaucoma has been reported in one patient (18).
Gavage feeding and oxygen administration may help these children to survive. Older patients will need a wide variety of treatments, which may be dental, medical, or surgical in scope (14). Experimental studies show that synthetic ligands of TRPML1 (transient receptor potential cation channel, mucolipin subfamily, member 1) are capable of rescuing abnormal lysosomal storage in FIG4 deficient cells and may, thus, be useful in treating patients with this deficiency in the future (55). Additional support for such an approach comes from the discovery that FIG4 regulates lysosomal membrane homeostasis (05) and that the inactive form of the FIG4 transgene provides partial rescue of neurodegeneration (28). In the adult mouse, Fig4 is important in the repair of white matter damage (33).
Loss of function mutations in FIG4 is thought to inhibit lysosomal function. Lysosomal dysfunction caused by FIG4 and VAC14 can be modified by CLCN7 in cultured cells, which may lead to a novel therapeutic approach (08).
Fetal ultrasonography is recommended. Polyhydramnios and fetal hydrops have been reported (06; 02). No histories of successful reproduction have appeared.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Robin Godshalk MS MHA
Dr. Godshalk of the Atlantic Health System in Morristown, New Jersey, has no relevant financial relationships to disclose.
See Profile
Ganeshwaran H Mochida MD PhD
Dr. Mochida of Boston Children's Hospital and Harvard Medical School has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 16, 2026