






Developmental Malformations
Barth syndrome
May. 08, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Focal cortical dysplasias are cytoarchitectural abnormalities most likely representing malformations of cortical development. Ranging from known genetic abnormalities to spontaneous mutations, these abnormalities most frequently present with intractable epilepsy. The author of this clinical article reviews the most recent proposed classification scheme for these disorders and looks at some of the recent genetic findings.
The term "cerebral cortical dysplasia" encompasses a spectrum of malformations of the cerebral cortex that arise during development and are associated with epilepsy in infants and children (165). Initially, cortical malformations were referred to largely by their gross characteristics (eg, lissencephaly, agyria or pachygyria, hemimegalencephaly, microgyria). As investigators discovered the range of microscopic cortical malformations that produce epilepsy but show no (or milder) gross abnormalities, additional terms such as "microdysgenesis" (162), "dysplastic cortical architecture" (206), "focal cortical dysplasia" (249), "generalized cortical dysplasia" (153), "synaptic dysgenesis" (19), and "cerebral dysgenesis" (213) were added to the literature. The term "focal cortical dysplasia" was first used to describe a specific malformation of the brain that consisted of disorganized cortex with enlarged irregular neurons and enlarged ballooned cells in some, but not all, cases (249), now corresponding to focal cortical dysplasia type 2. Dysplastic and megalocytic neurons were first identified by Crome and colleagues in 1957 (53).
Neuropathological classification scale. A classification scale to unify terminology of malformations caused by abnormalities of cortical development with emphasis on focal cortical dysplasias associated with epilepsy was proposed by a group of epileptologists, neuropathologists, and neuroradiologists (178). Various classification schemes of cortical dysplasias have been proposed (267; 165; 178; 122; 244). A resulting 2011 classification scheme was published under the auspices of the International League Against Epilepsy (ILAE); it represents a consensus statement with contributions by multiple European, North American, Japanese, and Australian neuropathologists who have special interest and long experience in epilepsy surgery (30), and has been validated by an international consortium of neuropathologists with experience in epilepsy (47). This scheme has a built-in flexibility for future revisions as data become available and interpretations evolve. It includes both focal isolated dysplasias of the cortex and also those associated with other lesions. It is expected that this scheme will become the template for uniform criteria in the neuropathological diagnosis of cortical dysplasias, analogous to the WHO (World Health Organization of the United Nations) classification of nervous system tumors, another consensus statement now accepted by neuropathologists throughout the world, who nearly always note the WHO grade when preparing reports describing primary cerebral neoplasms in surgical resections and autopsies. Another strength and modern approach of the ILAE classification is that although based primarily on neuropathological features, it also considers and incorporates aspects of neuroimaging and clinical aspects to be integrated with interdisciplinary correlations. Molecular, genetic, and immunocytochemical characterization of these cortical dysplasias will be added to the ILAE scheme. The temporal expression of neuronal proteins during cellular differentiation and the immunocytochemical demonstration of synaptogenesis are now better defined and often are altered in malformations, either as delayed, arrested, or precocious maturation (256; 214; 220). Developments in demonstrating tissue markers of lamina-specific gene expression and transcription products in specific layers of the cerebral cortex as well as previously unrecognized patterns of focal cortical dysplasia than those described in 2011, such as geographical microlesions of the cortex (18; 56; 172), require consideration for updating the neuropathological classification. The ILAE Commission on Neuropathology addressed these issues in 20108, and a revised classification scheme is proposed that also includes newly recognized entities such as minimal focal cortical dysplasias and oligodendrocyte proliferations (MOGHE) (169). Even more integrative proposals to incorporate genetic, imaging, and clinical data have been suggested (20; 174; 218). The neuropathological classification shown in Table 1 should not be confused, however, with other ILAE clinical and electroencephalographic classifications of epilepsies, including ICD coding for epilepsy and EEG criteria (103).
Table 1 summarizes the neuropathological criteria of focal cortical dysplasias. The principal difference between types 1 and 2 is that individual neurons are normal in morphology and size in type 1 and are dysplastic and megalocytic (enlarged) in type 2; both types exhibit abnormal cortical architecture with displaced and disoriented neurons and abnormal lamination. In type 2, subtype 2b includes balloon cells as well, large globoid cells of mixed cellular lineage that often express both neuronal and glial proteins, in addition to primitive proteins of early stages of differentiation, such as vimentin and nestin, also found in progenitor “stem” cells. Type 3 is not a distinctive or unique focal dysgenesis as are types 1 and 2 but represents type 1 associated with other lesions.
Focal cortical dysplasia type 1 (isolated) | |
• 1a: Focal cortical dysplasia with abnormal radial cortical lamination | |
Focal cortical dysplasia type 2 (isolated) | |
• 2a: Focal cortical dysplasia with dysmorphic neurons | |
Focal cortical dysplasia type 3 (type 1 associated with principal lesions) | |
• 3a: Cortical lamination abnormalities in the temporal lobe associated with hippocampal sclerosis | |
Revision of 2011 ILAE classification. Since 2011, considerably more experience and data regarding the pathogenesis and the genetic basis of focal cortical dysplasias have been gained (see below); therefore, a proposal to revise the above original classification scheme has been published (169), as well as recommendations about the neuropathological examination of surgically resected dysplasias at the gross and microscopic levels. Special histochemical and immunocytochemical applications reveal metabolic and cell lineage features not disclosed by hematoxylin-eosin stain (25).
The principal changes recommended are to simplify the scheme by combining or eliminating some subtypes. In type I, subtype Ia consists of radial micro-columnar architecture with little horizontal lamination; this pattern is well documented, has a developmental basis, and is recommended to be retained. Subtype Ib, by contrast, abnormalities of horizontal lamination, is much less well documented, is rare, and may have many causes unrelated to primary developmental malformation, including even ischemic laminar necrosis of the cortex. Subtype Ic is a combination of types Ia and Ib, hence, is not really a distinctive category. Type I is, thus, simplified to Ia, and Ib and Ic are deleted. Type II focal cortical dysplasia has dysmorphic, megalocytic neurons unlike type I in which neurons are morphologically normal but disoriented and displaced architecturally. The distinction between subtypes IIa and IIb is the absence or presence of balloon cells and, until it is better established whether this difference is important in pathogenesis or clinical presentation and prognosis, the revised ILAE classification will retain these two subtypes.
Perhaps the most important revision of the ILAE classification scheme relates to type III and its various subtypes (169). Type III is not really a distinctive form, but rather the presence of focal cortical dysplasia adjacent to another principal lesion. Subtype IIIa is problematic because it is a focal cortical dysplasia in the temporal lobe adjacent to hippocampal sclerosis. However, hippocampal sclerosis is never congenital; it is always an acquired lesion, so it is difficult to conceptualize how a developmental focal cortical dysplasia presumably from fetal life can form postnatally after hippocampal sclerosis appears. Subtype IIIb is adjacent to a brain tumor, usually a cortical epileptogenic tumor such as ganglioglioma. If these tumors are regarded as developmental dysplasias (see below), it is conceivable that focal cortical dysplasia may form at the same time. Subtype IIIc is focal cortical dysplasia Ia that forms next to a vascular malformation of the brain. This also is a feasible development because chronic ischemia induced by the vascular lesion may inhibit cortical maturation with persistence of the fetal architecture of focal cortical dysplasia Ia (see below), as it often does in cortex adjacent to a porencephalic cyst acquired in fetal life (Sarnat and Flores-214a). Subtype IIId is adjacent to almost any other lesion; in the case of porencephaly or infarcts in fetal life, the same reasoning can be applied as in focal cortical dysplasia IIIc. Focal cortical dysplasia adjacent to inflammatory lesions may be understood if cerebral inflammation occurs in fetal life, as it does in tuberous sclerosis and other developmental lesions (186; 228). Focal cortical dysplasia adjacent to postnatal cerebral contusions or other traumatic lesions is dubious.
The revised ILAE classification of focal cortical dysplasia still relies mainly on microscopic histopathology, but the neuropathological study of resected brain tissue with focal cortical dysplasia also requires immunocytochemical markers of cellular lineage and maturation, and recommendations for the technical study of such tissue also is progress (25). Incorporation of genetic profiles is also under consideration, as has been done with the WHO classification of brain tumors since 2016. The interpretation and incorporation of these data will likely influence revised classification in future.
• Focal cortical dysplasias are congenital developmental malformations of grey matter limited to focal zones in any lobe of the cerebral cortex. | |
• Focal cortical dysplasias are the most frequent cause of focal epilepsy in infants and children; seizures are the presenting symptom. | |
• The 2011 ILAE Neuropathological Classification of focal cortical dysplasia underwent revision in 2018. | |
• Focal cortical dysplasia type I is a focal malformation with morphologically normal neurons but abnormal cortical lamination and architecture: predominant microcolumnar rather than horizontal lamination. | |
• Microcolumnar architecture in focal cortical dysplasia I is reminiscent of the normal architecture of the fetal cortical plate in the first half of gestation and suggests that focal cortical dysplasia I is a maturational arrest. | |
• Focal cortical dysplasia type II consists of dysplastic, megalocytic neurons, and also abnormal cortical lamination/architecture. | |
• Focal cortical dysplasia type I does not yet have a genetic mutation as a correlate. | |
• Focal cortical dysplasia II is a postzygotic somatic mutation; its extent depends on timing in the 33 mitotic cycles of the early fetal neuroepithelium, so that small lesions limited to one gyrus are expressed late in the mitotic cycles, and large, extensive lesions are from expression in an early cycle, so that focal cortical dysplasia II and hemimegalencephaly are the same disorder with difference in timing. | |
• Focal cortical dysplasia II and hemimegalencephaly are the same disorder, with timing of onset of genetic expression determining the anatomical extent of the focal dysplasia. | |
• Focal cortical dysplasia type III is not a distinctive type in itself; it is focal cortical dysplasia I or II adjacent to another primary lesion, such as hippocampal sclerosis, tumor, vascular malformation, fetal cerebral infarct or porencephalic cyst, and others. | |
• Grey matter developmental tumors (ganglioglioma and dysembryoplastic neuroepithelial tumors) are now regarded as primary dysplasias more than neoplasias or dysplasias undergoing neoplastic transformation; hence, they are another form of focal cortical dysplasia. | |
• Major molecular genetic advances in identifying genes in the mTOR, AKT, PIK3CA, and RAS signalling pathways provide the etiology of focal cortical dysplasia II, hemimegalencephaly isolated or associated with neurocutaneous syndromes, and tuberous sclerosis complex, but not focal cortical dysplasia I. | |
• Minimal focal cortical dysplasia with oligodendrocytosis (MOGHE) is another distinct entity with a genetic basis demonstrated in some cases. | |
• Excessive neuronal heterotopic neurons forming synaptic plexi occur in the U-fiber layer beneath focal cortical dysplasia types I and II. |
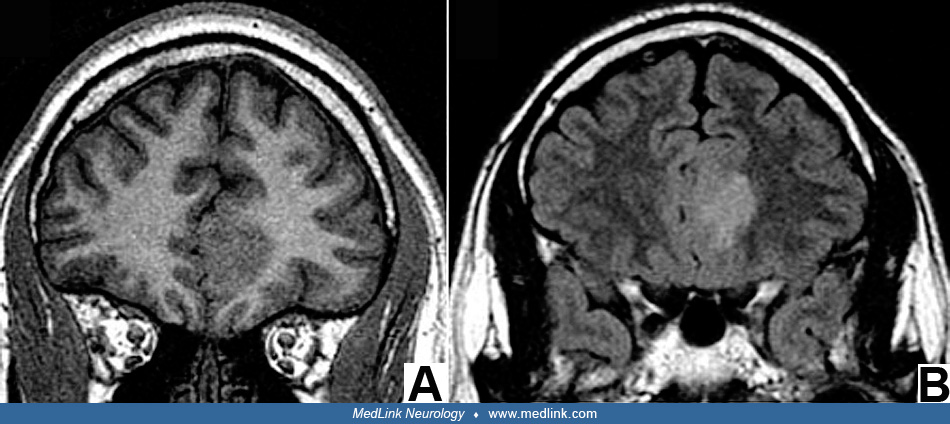
All focal cortical dysplasias are epileptogenic and the onset of seizures is usually the presenting symptom that leads to investigation by imaging and diagnosis. In some patients, seizures may begin as early as the neonatal period and may even begin by severe infantile epilepsies such as epileptic (infantile) spasms. Seizures also can begin in later infancy, childhood, or adolescence and often are progressive and refractory to medical control with antiepileptic and other drugs or the ketogenic diet. Early onset severe epilepsies are more often associated with tuberous sclerosis, hemimegalencephaly, or other type 2 dysplasias. As with other conditions that cause pediatric epilepsy, there is not a strict correspondence between the clinical phenomenology and the neuropathologic substrate. MRI often can distinguish foci of focal cortical dysplasia type 1 from type 2, but EEG patterns are less definitive in this regard (170). Even in MRI, histopathological type I lesions may not be detected because they consist of abnormal architecture and lamination of the cortex, but because the number and density of neurons is unchanged, microscopic cortical rearrangement or failure to mature beyond midgestational radial microcolumnar architecture does not necessarily cause a change in signal intensity or behave as a mass lesion; hence, the brain MRI may be designated as “nonlesional.” However, a frequent subtle MRI lesion in focal cortical dysplasia type I is the blurring of the grey/white junction at the site of the dysplasia, by contrast with sharp demarcation in other normal gyri of the same brain. The paucity of MRI findings in focal cortical dysplasia type I has even led some neuroradiologists to question the very existence of focal cortical dysplasia type I, but this would be a denial of histopathological abnormalities below the resolution of MRI. Functional MRI characteristics, such as hypometabolism, in regions of focal cortical dysplasia do not correlate with seizure onset (57).
The clinical form, age at onset of symptoms, and severity of epilepsy that a given patient with cortical dysplasia manifests appear to depend more on when during cerebral development the lesion occurred than on the specific type or topographic distribution of lesions (162; 99; 170). In general, the more severe the cortical dysplasia, the earlier the onset of symptoms and the more severe the epileptic syndrome (165). This may explain some of the heterogeneity of neuropathologic lesions seen in patients with pediatric epilepsy, because the clinical phenotype results not only from the lesion or putative "insult" to the developing CNS, but also from the subsequent developmental processes it affects. Infantile spasms, West syndrome, Lennox-Gastaut syndrome, complex partial seizures, partial motor seizures, and secondary generalized seizures have all been associated with cortical dysplasia (177). The most frequent pattern is that of focal epilepsy with onset in infancy or early childhood. The nature of the focal seizures depends on the site in the cortex. At times rare types of seizures, such as unilateral ictal blinking, may occur (263).
It was early recognized that in the focal cortical dysplasias of infancy, infantile spasms were generally easy to treat with vigabatrin or ACTH, but focal seizures were almost uniformly pharmacoresistant (148). Pharmacoresistance is due to both genetic and epigenetic factors, but no reliable biomarkers are yet available (155). Despite the histological appearance of more severe dysplasia, because neuronal cytology is involved in addition to disrupted tissue architecture, type 2 focal cortical dysplasias have a better prognosis for control of epilepsy than do type 1 focal cortical dysplasias, even with surgical resections. This difference may in part be related to more extensive lesions in type 1, so that the entire dysplasia is not resected. In one older adult case, epilepsia partialis continua occurred for 50 years before the correct diagnosis of a focal cortical dysplasia was finally made (92).
Developmental delays may be seen in patients with cortical dysplasia, but no clear relationship with the underlying pathology has been established (165), except in the context of specific diseases such as tuberous sclerosis complex or epidermal nevus syndrome. Focal neurologic deficits are rarely seen, and, when present, usually exist in association with an intrauterine destructive lesion or with extensive dysplastic lesions such as hemimegalencephaly or extensive polymicrogyria. The following are frequent in school-age children: global developmental delay in infancy and early childhood, cognitive impairment, attention deficits, and school learning disabilities (122; 90; 89; 48). A higher than control risk of autistic spectrum disorders occurs as well (74).
Genotype-phenotype correlations in focal cortical dysplasia (phenotype including clinical manifestations, imaging and electrophysiological findings, and neuropathological lesions) have been extensively studied and form a basis for integrated neuropathological as well as clinical diagnosis (24).
Prognosis depends on the extent of the cortical dysplasia, its location, and whether it is focal, multifocal, or diffuse (especially bihemispheric). Furthermore, the severity of the seizure disorder is associated with the histological grade (165). If the seizures are intractable, early surgical intervention leads to improved outcome. Focal lesions that can be surgically resected tend to have a better prognosis. The more complete the resection, the better the outcome in pediatric patients (121). Postoperatively, focal cortical dysplasia type 2 tends to do better in the long term than type 1 despite more severe cytological changes in individual neurons and glial cells (170). This apparent incongruity may be due to the often smaller lesions in focal cortical dysplasia type 2 and more extensive ones in focal cortical dysplasia type 1; hence, total surgical resection is more feasible in focal cortical dysplasia 2. In genetically determined generalized malformations of cortical development, biochemically associated abnormalities of the defective DNA may be important in both pathogenesis and prognosis. For example, a longer polyalanine expansion mutation in the ARX gene causes early infantile epileptic encephalopathy such as Ohtahara syndrome (110).
Tables 2 and 3 summarize the comprehensive features of the two principal types of focal cortical dysplasia.
|
• Neuropathology: micro-columnar cortical architecture; neurons not dysmorphic or enlarged. Synaptic plexi in U-fiber layer beneath focal dysplasias. | |
|
• Localization: posterior temporal, parietal, occipital. | |
|
• Clinical features: highly epileptogenic; onset of focal seizures in infancy or early childhood; high incidence of infantile spasms; developmental delay, cognitive deficits, school learning disabilities common; autistic spectrum disorder in some. | |
|
• Neuroimaging features: MRI shows blurring of grey/white junction in some, not all; no transmantle dysplasias; fMRI and PET may show hypometabolism at focus. | |
|
• EEG features: epileptiform focus not always well localized, even with intraoperative electrocorticography or depth electrodes. | |
|
• Genetics: SLC35A2 mutation in a minority of patients. | |
|
• Outcome of surgical resection: recurrent seizures more frequent than in focal cortical dysplasia type II, probably because of subtotal resection. |
|
• Neuropathology: neurons megalocytic and dysmorphic; transmantle dysplasias; cortical lamination disorganized with displaced and disoriented neurons, but micro-columnar architecture is not as prominent a feature as in FCD type 1a. Hemimegalencephaly may be isolated or associated with neurocutaneous syndromes, particularly epidermal nevus syndromes. Cortical tubers in tuberous sclerosis similar to other focal cortical dysplasia type 2 lesions and may have micro-calcifications in addition. | |
|
• Localization: any site of cortex, often temporal, frontal or insular. | |
|
Clinical features: highly epileptogenic; onset of focal seizures in infancy, childhood, or adolescence; developmental delay and cognitive deficits not characteristic except in tuberous sclerosis complex and epidermal nevus with hemimegalencephaly; autistic spectrum disorder infrequent. | |
|
• Neuroimaging features: MRI shows characteristic cortical lesions, usually at depths of sulci; transmantle dysplasias frequent; fMRI and PET may show hypometabolism at focus. | |
|
• EEG features: epileptiform focus often well localized even in scalp recordings but especially with intraoperative electrocorticography or depth electrodes. | |
|
• Genetics: postzygotic somatic mutation involving mTOR signaling pathway and pathways influencing mTOR AKT, GATOR, PIK3C, PTEN) and also TSC1 and TSC2 in tuberous sclerosis. | |
|
• Outcome of surgical resection: often complete abolition of epilepsy with total resection of lesion; in tuberous sclerosis, resection of most epileptogenic tubers improves frequency and severity of seizures. Hemispherectory or hemispheric disconnection for hemimegalencephaly may leave other focal neurologic deficits postoperatively, such as hemiparesis or hemianopia. |
A 37-weeks’-gestation male infant developed typical infantile spasms at the age of 5 days. Examination revealed increased head circumference (43 cm) and hypotonia. By 3 months of age, he manifested significant developmental delay. Seizures were unresponsive to phenobarbital and prednisone. MRI scan showed left hemimegalencephaly with lissencephaly of the involved left cerebral cortex; the right cerebral hemisphere appeared normal by MRI. An anatomical hemispherectomy was performed at 3.4 months of age; histopathology showed typical features of severe cortical dysplasia, and neuronal dysplasia and cytomegaly with cytoskeletal abnormalities but no neoplasia. Subsequently, the child showed modest improvement in developmental milestones and a decrease in seizure frequency with improved responsiveness to antiepileptic medications.
Though the etiology of focal cortical dysplasia type 1 remains somewhat elusive in most cases, a mutation of SLC35A2 gene is now demonstrated (128). Somatic pathogenic mutation of this gene also is associated with oligodendroglial hyperplasia in epilepsy (11). The mechanism of pathogenesis is now better understood in focal cortical dysplasia type I (see below) and suggests a focal event in the developing fetal brain that causes focal maturational arrest in organization of the cortex. Type 2 focal cortical dysplasia, by contrast, is now shown to be a postzygotic somatic mutation, which explains the cellular dysmorphism, and the extent of the lesion depends on the mitotic cycle of the periventricular neuroepithelium (see below). Specific genetic mutations and chromosomopathies are the underlying etiology of some cortical dysplasias, but these dysplasias usually are generalized and not focal. Genetic etiologies for cortical dysplasia have been proposed on both theoretical and empirical grounds (94; 230; 206). There are over 25 different syndromes of abnormal neuroblast migration, which are associated with a variety of inheritance patterns including autosomal dominant, autosomal recessive, and X-linked patterns, as well as familial cases with an unclear inheritance pattern (175). Familial cases with sibling involvement also are reported (136). Though distinct patterns of causation have not yet emerged for most types of cortical dysplasia, neurogenetic studies have demonstrated mutations within a number of genes (or chromosomal loci), which are associated with neuroblast migratory abnormalities, recognizing that cellular migration is only part of the pathogenetic mechanism.
Genomic DNA methylation is an increasingly recognized important factor in some focal cortical dysplasias and may distinguish histopathological subtypes (119; 272). Abnormal DNA methylation is already a known molecular mechanism in some other epigenetic disorders of cerebral development with clinical neurologic sequelae after birth, such as the “fetal alcohol spectrum disorder.” Dysregulation of the adenosine system is another pathological feature shared in both focal cortical dysplasia types I and II (81). Perilesional cortex adjacent to focal cortical dysplasias, often a highly epileptogenic zone, may be associated with disturbance in expression of Wingless (WNT) genes beyond the primary mTOR pathway of the focal cortical dysplasia itself (156).
Embryology of the developing cerebral cortex. Neocortical development can roughly be considered to be the result of a series of overlapping processes: (a) proliferation, (b) differentiation, (c) migration, (d) programmed cell death of neuronal precursors and neurons, (e) synapse formation and elimination, and (f) cortical remodeling (213; 206; 88). Abnormalities of these processes result in abnormalities of cortical architecture and resultant electrophysiological properties.
At approximately 4 weeks' gestational age, the neural tube forms and is covered by simple pseudostratified neuroepithelium, the component cells of which proliferate around the developing ventricular system. This zone is a mosaic of precursors giving rise to neurons, astrocytes, oligodendroglia, and ependyma of the developing brain (271).
The complex processes of cellular proliferation, determination of cell type, and differentiation are influenced both by cell lineage programs and by diffusible and soluble factors such as trophic factors, growth factors, and chemoattractant and chemorepellant molecules in the extracellular matrix. Cell surface interactions between cells and with the extracellular matrix also play a crucial role in these processes. Interactions are guided by cell adhesion and disadhesion molecules and extracellular matrix receptors. The influence of cell surface interactions and trophic factors may be critical in helping precursor cells determine their ultimate cellular fate. Cellular migration may use many of the same fundamental signals, as well as diffusible factors in initiating and maintaining appropriate migration.
Cells destined for the cerebral cortex migrate from their original location around the periventricular region to assume their appropriate location. About 80% of cortical neurons arrive at the cortical plate by radial migration along radial glial fibers of specialized cells for this purpose that really are progenitor cells themselves. Most of these neurons are glutaminergic. The other 20%, representing the population of GABAergic inhibitory interneurons, arrive not by radial but rather tangential migration along axons from the ganglionic eminence, a specialized part of the periventricular germinal matrix. Migration of neuroblasts to the cerebral cortex occurs in an "inside-out" fashion, with neuroblasts destined for the deepest cortical layers migrating initially and those destined for the more superficial cortical layers bypassing them (05; 240). Whereas cell adhesion molecules are needed to attach neuroepithelial cells to the radial glial fibers in the subventricular zone, disadhesion molecules are needed to detach these neuroblasts from the radial glia when they arrive at the cortical plate so that those behind them on the same radial glial fiber can bypass them to reach more superficial parts of the cortical plate. The bulk of neuroblast migration occurs between 7 and 16 weeks’ gestation. The initial architecture of the cortical plate during the first half of gestation is a radial microcolumnar arrangement or radial lamination. This histological organization changes to horizontal lamination, which is superimposed and gradually predominates, to remodel into a 6-layered cerebral cortex. Even after this time, however, some neuroblasts continue to migrate through the intermediate zone (future white matter), in a process that can continue up to a few months after birth (213). Synaptogenesis within the cortical plate is initiated at about 22 weeks’ gestation (225). Even during the physiological microcolumnar stage in the first half of gestation, and especially during the transition to tangential (horizontal) histological layering, there is already a distribution of specific types of neurons and primordial synaptic preparations forming horizontal lamination (212; 87). A complex combination of migratory patterns, including the radial glial scaffolding, allows for the development of a highly organized, 3-dimensional nervous system (192). Even in mature brains, a functional columnar organization continues to exist as functional columns or barrels (185; 91; 167; 193; 113; 111). In the occipital (visual) cortex, the on/off (ie, light/dark) inputs enable an invariant columnar architecture or barrel (132), and this is also the basis for the topographic visual sensory mapping in the cortex (120). Columnar functional and microscopic structural architecture is a general pattern in the developing forebrain, so that even the olfactory bulb exhibits such an arrangement (112; 276). Finally, some migratory neuroblasts appear not to use either radial glia or axons for guidance. The mechanisms guiding this type of migration are still unknown, although investigators have suggested that some neuronal precursors may migrate in closely associated chains (146).
Cajal-Retzius neurons of layer 1 of the neocortex and subplate neurons are crucial for normal cortical organization (17; 154; 219). The preplate, which consists of these neurons and a superficial plexus of corticopetal nerve fibers, is split by the newly arriving migrating neurons destined for the mature cortical plate. The reeler mouse is a mutant murine model in which there is failure of the normal “inside-out” pattern of neocortical formation (66; 58). A human genetic malformation in which inversion of the cortical layers often occurs is holoprosencephaly (222). The marginal zone before radial migration and the molecular zone (layer 1) after the cortical plate begins to form also provides another site of neurogenesis and gliogenesis in the early development of the cerebral cortex (49).
Throughout the second and early third trimesters, there is a transitory “subplate zone” beneath the developing cortical plate that contains large multipolar neurons. Subplate neurons mostly disappear in early postnatal life, but they are important in the fetus in organizing cortical connections in the developing cerebrum and in forming interconnections between the thalamus and cortical plate (219). Some subplate neurons are probably incorporated into layer 6, and others may contribute to the neuronal dispersion in the U-fiber later as the cortex matures. They may act as pioneer corticofugal axons. The U-fiber layer develops beginning at mid-gestation; its neurons, by contrast, are local short connections that do not project subcortically or in commissures (226). The subplate zone is structurally laminated as two layers at mid-gestation, which can be recognized postmortem in 3-Tesla MRI and confirmed neuropathologically; presumably the two layers are demonstrable prenatally by fetal MRI (184). Golgi impregnations and autoradiography also indicate lamination of the subplate zone, which blends with cortical layer 6 in the rat (260).
Programmed cell death or apoptosis is an essential mechanism of normal neural development. In the normal embryonic brain, there is an overproduction of neuroblasts, many of which will undergo programmed cell death (106). It is an active process involving gene transcription and protein synthesis. It includes, but is not confined to, apoptotic cell death. In vitro experiments using nerve growth factor provided the paradigm for programmed cell death in the nervous system (137). Competition of neurons for limited amounts of trophic factors at the time they are reaching their targets helps to create a competitive environment for trophic support, ensuring survival of the "best-connected neurons." However, the process of programmed cell death may play a far wider role in the developing nervous system than initially appreciated. This may serve as selection process for potential cortical neurons with the desired phenotype. In accord with this experimental finding, other investigators have found, using morphometric techniques to study human brain development, up to 70% loss of cortical neurons after the 28th week of intrauterine gestation; this suggests that programmed cell death within the cortex may play a larger developmental role than was initially appreciated (191).
The failure of normal programmed cell death in the genesis of some glioneuronal malformations has been investigated (280). The authors examined regions of poorly circumscribed cell clusters consisting of small oligodendroglial-like cells mixed with randomly oriented neurons. These clusters, which have been called “glioneuronal hamartias” (278), are present in some patients with cortical dysplasia and intractable epilepsy, although they are also seen in association with other epilepsy producing lesions. The small round cell component of these lesions expresses an embryonic form of the neural cell adhesion molecule and is negative for proliferation markers, suggesting a postmitotic state (278). Further, these cells strongly express the anti-apoptotic protooncogene Bcl2, which is not normally expressed in mature brain tissue (280), suggesting a role for an abnormality of normal programmed cell death in these epilepsy-associated lesions.
Supplemental to programmed cell death, a conspicuous elimination of synapses occurs during development and is essential to remodeling of the cortex. Synapse elimination is highly intertwined with the remodeling of cortical connections and is a highly dynamic process (189), demonstrated both in vivo and in vitro (261). The process of synapse formation, elimination, and remodeling continues beyond the period of development, and many of the molecules that are essential for these processes in development appear to continue to play a role in synaptic plasticity in the mature brain.
Continuing neuronogenesis. In addition to the fetal ventricular zone of the “germinal matrix,” the marginal/molecular zone of the early cerebral cortex and the external granular layer of the cerebellar cortex until 18 months of age, two sites of persistent resident progenitor stem cells are found in the fetal brain persist in the mature brain: olfactory bulb and hippocampus (229). The latter population of bipolar progenitor cells are located in the hilus of the dentate gyrus, just beneath the inner granule cell layer, and is known as the polymorphic zone. One of its polar processes extends through the dentate gyrus. These cells are capable of generating new dentate granule cells in particular and may involve a continuous turnover of granule cells during life, including new synaptogenesis, the proliferation increasing in epilepsy arising in the hippocampus (241; 239; 234). A role in the generation of new memory engrams throughout life is speculated, but the evidence remains inconclusive. Focal cortical dysplasia can be associated with hippocampal sclerosis as focal cortical dysplasia type 3a.
Pathogenesis of focal cortical dysplasias. Type 1 focal cortical dysplasia appears to be arrested maturation of the cortical plate. The radial microcolumnar architecture is normal in the first half of gestation but becomes pathological if tangential (horizontal layering) does not predominate in the second half or if microcolumnar architecture persists postnatally (223). Abnormal tangential lamination may result in some cases from selective lack of neurons from either radial or tangential migration. Totally disorganized cortex with no recognizable architecture might result from failure of disadhesion of neuroblasts from their radial glial fiber when they reach the cortical plate, so that neuroblasts behind cannot bypass them (220). Microcolumnar architecture in focal cortical dysplasia type 1 involves not only the arrangement of the neurons in the cortex, but also the synaptic layers that are vertical rather than horizontal, as demonstrated by synaptophysin immunocytochemistry (223). Radial synaptic lamination is not seen during the normal phase of microcolumnar architecture because there are no synapses or synaptophysin reactivity in the cortical plate until 22 weeks’ gestation (225; 223). Generalized cortical dysplasias type 1 may present neuropathologically as radial microcolumnar architecture involving all regions of cortex, exemplified in DiGeorge syndrome (22q11.2 deletions of maternal origin) and methylmalonic academia (223). These genetic diseases are germline mutations that occur at the time of fertilization; hence, every cell in the body carries the same genetic mutation.
The mechanism of type 2 focal cortical dysplasia is different than type 1. The cellular dysmorphism and dysregulation of growth is due to a postzygotic somatic mutation that affects some but not all premigratory neuroepithelial cells in the ventricular zone, destined for both radial and tangential migration, hence, a mixture of normal and dysplastic neurons in the cortical plate. This somatic mutation is similar to that of tuberous sclerosis complex and in hemimegalencephaly associated with mutations of the AKT3 gene in the isolated form or the AKT1 gene in Proteus syndrome (142; 183; 76).
The timing of the mitotic cycle in which the mutation occurs determines the extent of the resulting malformation and whether other tissues than the brain are involved (215; Sarnat and Flores-220). Of the estimated 33 mitotic cycles needed to produce all of the neurons of the human cerebral cortex (33), if the mutation occurs in a late cycle, a small focal cortical dysplasia lesion occurs; if the mutation is in an earlier cycle, the lesion is more extensive and may involve more than one gyrus; and even earlier mitotic cycle involvement produces even more extensive lesions as hemimegalencephaly or “total hemimegalencephaly,” which includes the ipsilateral cerebellum and brainstem because the neuroepithelium is continuous between the supra- and infratentorial space in late embryonic life (Sarnat and Flores-220; 215). Hemimegalencephaly is, thus, a spectrum of neuroanatomical extent (55). Earliest involvement may result not only in hemimegalencephaly but other organ involvement, hence, the close association of hemimegalencephaly with epidermal nevus syndrome (70). The neuroembryological demonstration that focal cortical dysplasia type 2 and hemimegalencephaly are merely different timings of onset of expression in the same genetic/metabolic disease has been confirmed by another approach of genetic studies (130; 60; 61).
Glutamate signaling is one of the strongest dysregulations in epilepsy, with layer-specific transcriptomic changes in multiple glutamate receptor genes (180). Axodendritic synapses of the molecular zone of the cerebral cortex are glutamatergic, but loss of dendritic spines can occur in focal cortical dysplasia type II and may be due to dysregulation of the complement system, which is important in the physiological removal of redundant microglia-mediated spines and is likely reactivated in focal dysplasia type II (207).
Astrocytes are another integral part of synaptic junctions. Astroglial vascular endothelial growth factor regulates glutamate transporter-1 expression via mTOR activation in experimental pilocarpine-induced status epilepticus in rodents and likely contributes to epileptogenesis of excitatory glutamatergic synapses in human cerebral cortex as well (101). Synaptic connectivity differs between focal cortical dysplasias types I and II, type I having a greater degree and wider area of cortical hyperexcitability than type II (236). Sleep-related epilepsy has a higher risk independent of seizure localization, even small lesions increasing the risk by 2.18 times in all focal cortical dysplasias (274). Maturational arrest of progenitor neuroepithelial cells also may contribute later to immaturity and compromise in the formation of neural networks in focal cortical dysplasias (06). Not only astrocytes, but oligodendrocytes, can also be affected and produce altered grey matter myelin, particularly in type IIb focal cortical dysplasia (209). Oligodendrocyte precursor cells show single-nucleus multiomics in focal dysplasia type IIIa associated with hippocampal sclerosis (144).
MRI provides a means not only of detecting focal cortical dysplasias but of providing clues to embryonic and fetal cerebral maldevelopment mechanisms (13; 02). Precise imaging of type 2 focal cortical dysplasias using a 7 Tesla MRI scanner are interpreted as defects in myelination at the grey/white junction where the normal sharp delineation is lost (292; 15; 273; 258), but this interpretation must consider patient age and the expected state of myelination of U-fibers that border gyri, serving to separate cortical grey matter from deeper white matter; except the amount of the perisylvian region and calcarine fissure, U-fibers are very late to myelinate relative to deep white matter, sometimes requiring years to even decades (226). Abnormal myelination in focal cortical dysplasia should not be confused with delayed normal myelination sequences of U-fibers. U-fibers are short axons that interconnect different parts of the same gyrus or immediately adjacent gyri. Because diffusion tensor imaging (DTI) is sensitive to axoplasm rather than myelin, it can show stronger contrast between grey and white matter, further improving presurgical delineation of focal cortical dysplasias and the boundary at the ventral surface of the cortex (129). Another advance in neuroimaging is functional MRI (fMRI) that suggests disrupted connectivity in large-scale neural networks including not only the cerebral cortex but also frontotemporal-cerebellar projections (143). Post-processing methods combined with positron emission tomography may detect focal cortical dysplasias previously diagnosed as MRI-negative (190).
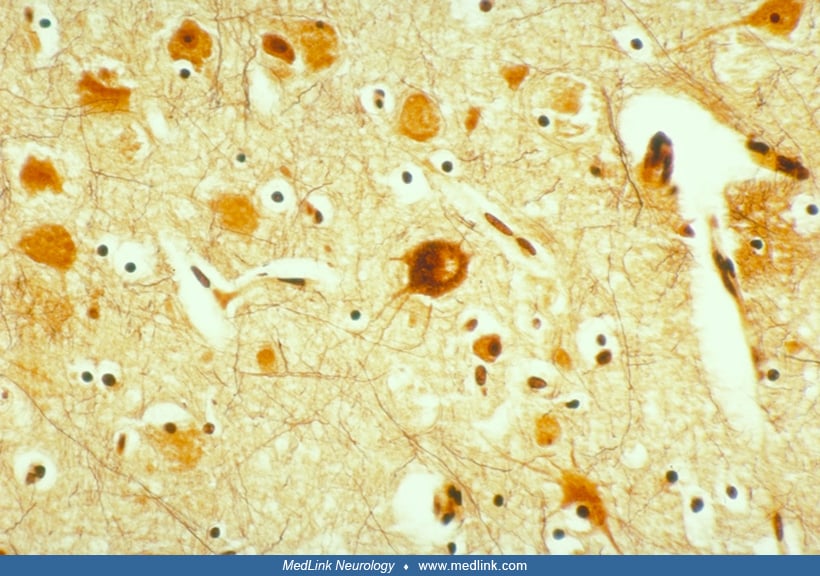
Additional factors that characterize focal cortical dysplasia type 2 are that they exhibit activation of the mTOR signalling pathway similar to tuberous sclerosis complex (50; 51; 210; 214; 255; 09; 140; 139; 116; 168; 248). Furthermore, mTOR disorders also exhibit upregulation of abnormally phosphorylated tau protein (214; 215). Tau is a microtubule-associated protein, as are many other proteins of genetic neuroblast migratory disorders, such as LIS1, doublecortin, and ARX. If tau overexpression occurs in the mature nervous system, late-onset adult tauopathies are characterized clinically by dementia but not epilepsy, and neuropathologically by neuronal degeneration and loss. Microtubules are essential cytoskeletal elements of developing neurons, and determine cellular lineage, orientation, polarity, growth, formation of neurites, and synaptogenesis. Abnormal phosphorylated tau in the immature neuroblast, thus, may cause cytological dysmorphism and growth abnormalities, as well as affect neuronal function. Such tau is upregulated in focal cortical dysplasia type 2 (but not types 1 or 3), in hemimegalencephaly, in tuberous sclerosis complex, and in a unique tumor; ganglioglioma in which the neurons are dysplastic and glial cells are neoplastic (214; 220; 215). This group of postzygotic somatic mutations has been called “infantile tauopathies”, though they lack some of the other associated abnormal proteins of adult dementia tauopathies, such as alpha-synuclein and beta-amyloid precursors (215). A murine model of autism improves with tau reduction (247), but whether this model might serve to study focal cortical dysplasia is not yet known.
Glial participation in focal cortical dysplasias is another factor that has not received the same attention as the neurons. Cytomegaly of glial cells in focal cortical dysplasia type 2 is well documented. Glial-neuronal interactions in focal cortical dysplasia type 2 are less well understood, as with dysplastic neurons and balloon cells, in terms of their epileptogenic potential (34). In focal cortical dysplasia type 2a, oligodendrocyte proliferation is impaired and maturation delayed, so myelination is also delayed (231; 63). Meningeal defects can be associated with focal cortical dysplasias (65). This may reflect a neural crest defect at the time that the cortical dysplasia was forming, but further data are needed.
Oligodendrocyte proliferation also might be a factor in the pathogenesis of subcortical white matter oligodendroglial hyperplasia in epilepsy (MOGHE) associated with some minimal focal cortical dysplasias. MOGHE occurs in young children with early onset and difficult-to-treat frontal lobe epilepsy. It is defined histopathologically by oligodendroglial hyperplasia and heterotopic neurons in the shallow subcortical white matter (233; 83; 266). Somatic variants of the gene SLC35A1 are identified in some MOGHE cases (243; 277; 10; 163). Some early cases were published in the literature as minimal malformations of cortical development (mMCD) or as variants of focal cortical dysplasia type 1. Somatic mutation of the SLC35A2 gene is demonstrated to be associated with both focal cortical dysplasia type I and also oligodendrocyte hyperplasia (128; 11). It is recommended that a revised ILAE neuropathological classification scheme recognize MOGHE as a specific disease entity preferentially affecting white matter (26).
Proteoglycans are intracellular matrix glycoprotein molecules in the CNS, secreted by astrocytes, that serve for axonal guidance because they repel glutamatergic (excitatory) axons and facilitate GABAergic (inhibitory) axons during fetal development. The most important proteoglycan in the nervous system of all vertebrates and most invertebrates is keratan sulfate (not keratin of fingernails). Keratan sulfate prevents aberrant decussation of ascending axons in the dorsal columns of the spinal cord and is also present in the forebrain (216). In the cerebral cortex, it first appears in the molecular zone at mid-gestation, then has a transitory patchy distribution within the cortical plate, and eventually is most concentrated in the deep cortical layers 5 and 6 and in the U-fiber layer, where it prevents axonal penetration from deep white matter heterotopia that mature late and project potentially epileptogenic axons after the keratan sulfate U-fiber barrier has formed. Though keratan sulfate is mostly in the intercellular matrix as granulofilamentous deposits, it binds to neuronal somatic membranes, except dendritic spines. Its transmitter specificity explains why axosomatic synapses are inhibitory and axodendritic synapses are mostly excitatory (216). Furthermore, keratan sulfate envelops axonal fascicles, including long tracts such as the corticospinal and short tracts such as the intrinsic axonal bundles within the globus pallidus and the pencil bundles of Wilson in the corpus striatum. This keratan “insulation” of fascicles and tracts ensures that axons cannot exit until they reach their destination and that axons from grey matter en route cannot enter the fascicle. Its presence in layer 6 of the cerebral cortex and in the underlying U-fiber layer serves to repel axons from deep white matter heterotopia, thus, preventing them from integrating into abnormal epileptic networks (226; 216). Keratan sulfate is also present in some other tissues, with the highest concentration in the cornea and in cartilage. Peripheral nerves do not penetrate cartilage.
It is recognized that inflammation may play an important role in the pathogenesis of cortical tubers in tuberous sclerosis complex, and many inflammatory markers are demonstrated in the brain tissue of fetuses with tuberous sclerosis complex (186). Inflammatory response and microglial activation is confirmed by other authors not only in tuberous sclerosis complex but in focal cortical dysplasia type 2, also an activation of the mTOR pathway (246).
Type 3 focal cortical dysplasia is usually type 1 microcolumnar cortical architecture, but at times can be type 2. It is associated with other types of lesions. If next to a porencephalic cyst, for example (type 3d) or next to a vascular malformation (type 3c), chronic ischemia during fetal life may be the cause of the arrested maturation of the cortical plate (223). Histopathological features are variable in the cortex adjacent to vascular malformations of cortex associated with epilepsy, with pseudolaminar necrosis, neuronal loss, and astrogliosis in addition to micro-columnar architecture (166). Focal cortical dysplasia type IIIa in the temporal neocortex near to hippocampal sclerosis is well recognized histopathologically (93).
Although focal cortical dysplasia types 1 or 2 can occur anywhere in the cerebral cortex, there are patterns of distribution for each. Classical focal cortical dysplasia type I of infants and young children typically occurs in the posterior hemisphere: posterior temporal, parietal, and occipital cortices (Table 2). The insula is increasingly recognized as a site of focal cortical dysplasia of both major types and an epileptogenic cortical region that produces complex clinical manifestations of autonomic regulation, olfaction, and mood and behavioral responses as a component of the “limbic system” (105). Because it is a deep structure, scalp EEG may be misleading, falsely negative, or mislocalizing, but intraoperative depth electrode recordings are more reliable (105).
Genetics. A number of genes have been identified, which are associated with normal ontogenesis and mutations or deletions of which result in brain pathology, many of which fall into the spectrum of cortical dysplasias. Type 1 lissencephaly and its association with mutations in the LIS1 gene, X-linked lissencephaly and its association with the doublecortin gene, tuberous sclerosis complex and its association with mutations in the TSC1 and TSC2 genes, and X-linked periventricular heterotopia and its association with the filamin-1 gene are well characterized examples. These genetically programmed malformations usually cause generalized rather than focal cortical dysplasia.
Many genetically determined disorders of cortical development and neuroblast migratory disorders exhibit abnormal cortical lamination and organization in a generalized rather than focal distribution, and subcortical heterotopia: lissencephalies 1 and 2 polymicrogyria, schizencephaly, subcortical laminar “band” heterotopia, and periventricular nodular heterotopia. Despite a known genetic basis for most of these and the fact that many are highly epileptogenic, none are focal cortical dysplasias. Though generalized lissencephaly and pachygyria are primarily due to genetic mutations not of the mTOR pathways, secondary mTOR defects may accompany them (286).
Focal cortical dysplasia is not usually familial, but familial cases are described that suggest a Mendelian inheritance, usually an autosomal recessive trait, in some families, but nearly all focal cortical dysplasia type 2 (136; 242). Tuberous sclerosis is a prototype form of multiple focal cortical dysplasias (ie, cortical tubers) of autosomal dominant transmission, and some familial simple cortical dysplasias type 2 are associated with the mTOR gene or its regulator, NPRL3 (173; 242), or with the PIK3CA/AKT pathway (141), as metabolic/genetic links to tuberous sclerosis. In tuberous sclerosis the defective mTOR (mTORC1) pathway begins in fetal life (186; 255), and it does so as well in focal cortical dysplasia II and hemimegalencephaly. Furthermore, variants of PIK3C initiate cerebral hyperactivity during gliomagenesis as in subependymal gliomas in tuberous sclerosis (285). Cortical tubers of tuberous sclerosis are essentially multiple focal cortical dysplasias. Abnormal RNA microarrays in focal cortical dysplasia II are useful in identifying defects in the Hippo signaling pathway as well, which is another genetic cascade (138). A unifying model for mTORC1-mediated regulation of mRNA translation is suggested with much supporting evidence (254). Pooled RNA sequences from multiple transcriptomes of patients with focal cortical dysplasia type II help characterize the single cell signatures in subtypes IIa and IIb (75). Tuberous sclerosis is an example of both germline mutation with Mendelian inheritance and also postzygotic somatic mutation related to a defective mTOR signaling pathway. An infant with a STXBP1 germline mutation and focal cortical dysplasia type 2 was described (238). In focal cortical dysplasia type II, potentially deleterious somatic variants in brain-expressed genes of the mTOR pathway were identified in six of 17 brain resections (289). The genetic defects in tuberous sclerosis also result in non-neuronal cells, especially glial cells, with distorted size and morphology (287).
In addition to the mTOR pathway being a common defective metabolism in tuberous sclerosis complex, focal cortical dysplasia II, hemimegalencephaly both isolated and associated with epidermal nevus and other neurocutaneous disorders, and ganglioglioma, other related integrated genetic metabolic pathways also are now demonstrated to also be defective and can contribute to dysmorphic neurons and altered neuronal function including epileptogenesis. The most important is the PIK3CA/AKT pathways (182; 97; 04). These pathways are known causes of hemimegalencephaly and epidermal nevus syndrome including variants such as the multi-organ overgrowth syndromes Proteus and CLOVES/MCAT (congenital lipomatous overgrowth, vascular malformations, epidermal nevi, scoliosis, skeletal, and spinal/megalencephaly-capillary malformation syndrome) (124; 70; 71; 114; 145; 157). Lymphatic and other vascular overgrowth syndromes, including Klippel-Trenaunay syndrome, are other somatic mutations due to PIK3CA mutations (150; 259). RAS pathways also are linked to these other genetic pathways and may be involved in these and other neurocutaneous syndromes, such as neurocutaneous melanocytosis (70). CCDC2 variants cause X-linked focal epilepsy and focal cortical dysplasia (86).
Though a genetic defect in focal cortical dysplasia type I remains unknown, a minority of patients (four of 48 cases = 8%) exhibit mutation of the SLC35A2 gene (128).
Mild focal cortical dysplasias with oligodendrocyte hyperplasia (MOGHE) show recurrent pathogenetic variants in the SLC3SA2 gene with mosaicism (181; 11). This mutation is present in a minority of cases of focal cortical dysplasia type I as well (128).
Destructive lesions associated with cortical dysplasia. Destructive lesions also appear to be capable of inducing cortical dysplasia, as evidenced by a number of animal models, and by clinicopathologic examples of cortical dysplasia in association with destructive lesions.
Cortical dysplasia has been noted in close proximity to small cystic infarcts (possibly occurring in utero) (165), adjacent to porencephalic cysts that formed due to middle cerebral artery occlusion at midgestation or earlier (223), other vascular lesions (200), and in association with intrauterine infections such as cytomegalovirus (84). These are designated in the ILAE classification as “type IIId”. In one autopsy series of patients with infantile spasms, 10% of the patients showed evidence for both malformative and destructive lesions (99). Another illuminating case report highlighted the role of discontinuities in the pial-glial barrier in association with massive neuroglial heterotopia into the subarachnoid space (41). Toxic and environmental insults may also cause cortical dysplasia, as they can affect the developing cortex during its time of greatest vulnerability. The presence of neuronal heterotopia in the survivors of the atomic bomb at Hiroshima (irradiation occurred between weeks 10 and 17 of development) clearly illustrates this point (192). Damage to radial glia and to the glia limitans has been suggested as a potential mechanism for generating the dysplasia.
Perhaps most convincingly, a variety of environmental insults ranging from chemicals, freezing injury, and gamma irradiation have all been used in animal models to produce lesions resembling cortical dysplasia. These data, which suggest that cortical dysplasia may be associated with both genetic and intrauterine destructive lesions, highlight the point that the pathology of cortical dysplasia is related to both the timing and the nature of the "disruption" of cortical development.
An initial report that papillomavirus was an acquired cause of focal cortical dysplasias (52) could not be confirmed by other investigators (237; 253; 28).
Animal models of cortical dysplasia. A number of important animal models have been developed in order to study the molecular basis of cortical dysplasia. These models range from naturally occurring genetic mutations to environmentally induced cortical dysplasia caused by exposure of the embryo or fetus to teratogenic toxins, including drugs and alcohol that the mother may consume early in gestation. Though good animal models of mTOR pathway disorders and focal cortical dysplasia type 2 have been developed, there presently are no animal models of focal cortical dysplasia type 1 (48). In a rat model of focal cortical dysplasia type II, electrographic epileptiform discharges appeared during the third postnatal week, and the first clinical seizures occurred a week later (107). In a murine model with mTOR mutation, not only was there a reduction in the number of GABAergic inhibitory interneurons in dysplastic cortex, but there were also postsynaptic alterations in GABA synaptic genes independent of interneuron paucity (290).
At least one murine model of focal cortical dysplasia type II not only shows histopathological features reminiscent of the human disease but generates a wide spectrum of high-frequency electrographic activity as an electrophysiological biomarker (43).
All animals use the mTOR signalling pathway, and it can be studied even in nematodes: upstream regulators that convey amino acid signals and mTOR complex I (mTORC1) controls cellular growth in response to amino acid levels (62). SAR1B senses leucine levels to regulate mTORC1 signaling by binding to leucine and targeting its activator GATOR2; silencing SAR1B results in lung cancer in mice but may also affect neuroepithelial cell growth and differentiation (38).
Spontaneous models. The tish rat is a spontaneously occurring autosomal recessive mutant, characterized by bilateral subcortical heterotopia beneath the frontal and parietal neocortices (134). These heterotopia, which project to subcortical targets, have bidirectional connections with the thalamus (232). They appear to be heterotopic nests of neurons intended for the neocortex and have the expected subcortical projections of neocortex, but they have a disorganized architecture and a “rim-to-core” orientation as opposed to the normal inside-out orientation of neocortex. They also bear electrophysiological properties similar to the seizures associated with human cortical dysplasia. Other work has shed light on the origin of these heterotopic neurons. As mentioned above, proliferation of cells destined for the cerebral cortex occurs in the ventricular or subventricular zones. However, in the tish rat, there appear to be two zones: (1) a ventricular or subventricular zone that gives rise to the normal neocortex and (2) a second misplaced proliferative zone within the intermediate zone that gives rise to both the normotopic and heterotopic neurons. These data show that this abnormality occurs early in development and is initiated before the migration of most cortical plate cells (131). Other genetic rodent models include the Reeler mutant rat, the Eker rat, and the ihari mutant rats (199; 171). Epilepsy in young Tsc+/- mice exhibits age-dependent expression that mimics that of human tuberous sclerosis complex (77).
Environmentally induced models. A number of environmentally induced models have been developed, highlighting the role of environmental factors in the development of cortical dysplasia, as well as providing an opportunity for studying the electrophysiology and molecular pathogenesis of these lesions. Methylazoxymethanol is a potent neurotoxin with anti-proliferative effects on dividing, but not quiescent, neuroepithelial cells. Intrauterine administration of methylazoxymethanol has been shown to result in epilepsy, producing lesions in the treated rats, which bear many pathologic features of cortical dysplasia, including abnormal cortical lamination, single heterotopic neurons in the neocortical molecular layer, marginal glioneuronal heterotopia, persistent subpial granular cells, and periventricular nodular heterotopia (40; 78; 45). Electrophysiologic, in vitro slice culture and connection tracing studies have shown the presence of abnormal electrophysiologic activity and kindling behavior (40; 79) with abnormal connections between heterotopic neurons in the CA1 region of the hippocampus and the neocortex (40; 79).
Other environmental models of cortical dysplasia include superficial cortical freeze lesions in which morphological analysis has been paired with electrophysiology to study the abnormal responses (149; 291). Redecker and colleagues have used the glutamatergic agonist ibotenate to induce cortical dysplasia-like lesions to study the electrophysiology and glucose metabolism of these lesions (197). Roper has utilized in utero irradiation followed by electrophysiology of slice cultures to study the electrophysiologic properties of these dysplastic areas (205; 204).
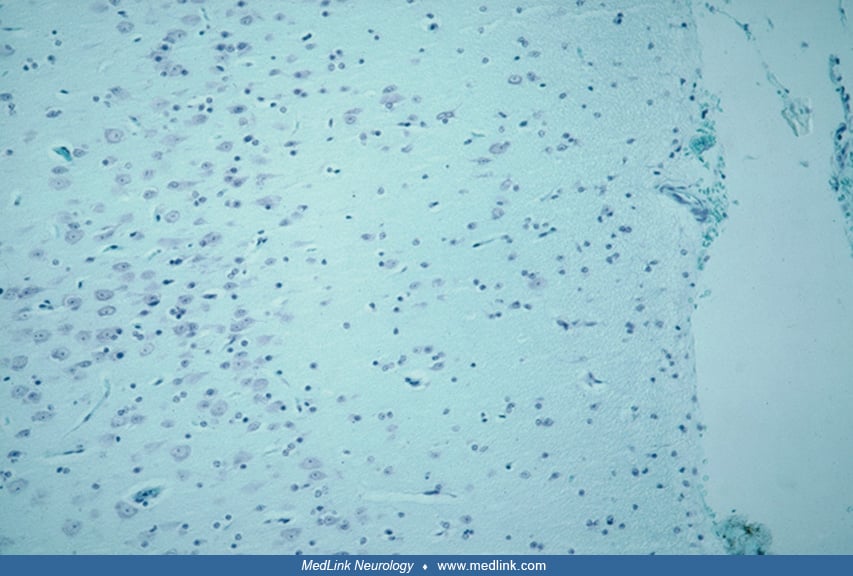
Neuropathology. Cortical dysplasia is best evaluated microscopically and can be characterized as easily identifiable abnormalities, summarized in Table 2. These microscopic features reflect disruption of normal developmental processes and can be used to stratify the lesions as resulting from early, intermediate, or late gestational events, which correspond, respectively, to severe, moderate, and mild cortical dysplasia (165). Other investigators have suggested that cortical dysplasia can be grouped into three main patterns: (1) cortical laminar disorganization, (2) neuronal cytomegaly with cytoskeletal abnormalities and balloon cells, and (3) hypercellular cortical molecular layer (188). The most important distinction in this regard is between (1) abnormal tissue architecture or distribution, arrangement and orientation of neurons and synaptic layers, and (2) cytological abnormalities with dysplastic and megalocytic neurons (27). Laminar-specific expression of gene products are demonstrated in some cases and can be shown in sections of resected brain tissue (56; 172). In a multicenter, international collaborative study involving neuropathologists in15 countries, it was demonstrated that the use of immunocytochemical reactivities brought greater diagnostic precision and agreement than simple hematoxylin-eosin histology alone, but that the integration of genetic profiles with neuropathology further enhanced this precision and agreement (26). Moreover, in situ hybridization in surgical specimens of focal cortical dysplasia type II demonstrates lamina-specific genetic expression in which the morphologically normal neurons show migration to correct layers of the cortex, unlike dysmorphic neurons and balloon cells that migrate abnormally and are displaced within the cortex (208).
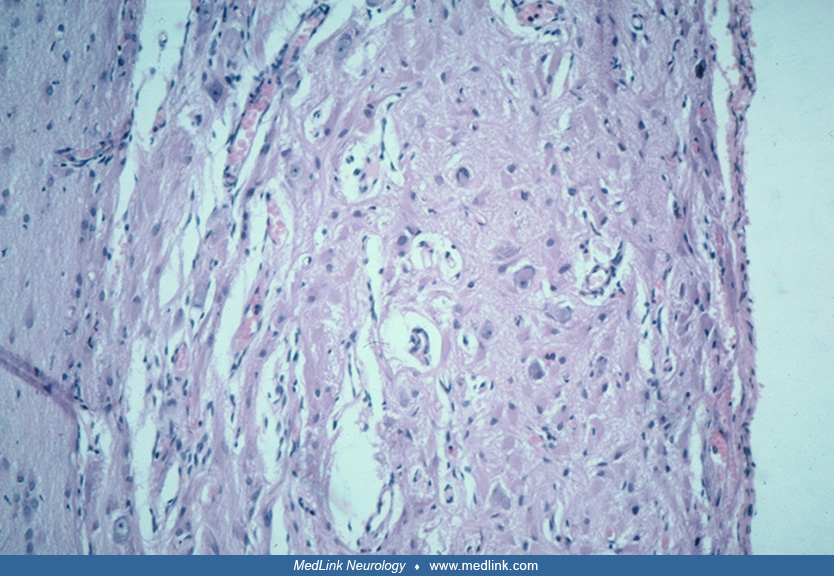
The unique “balloon cell” is characteristic of focal cortical dysplasia IIb, hemimegalencephaly, and tuberous sclerosis complex. Histologically, they are large globoid cells with eccentric nuclei and a homogenous eosinophilic cytoplasm. They have multiple short coarse processes that are not usually appreciated with hematoxylin-eosin stain but are well demonstrated by immunoreactivity for primitive proteins vimentin and nestin and also by heat-shock proteins such as alpha-B-crystallin. Balloon cells have mixed lineage, many expressing neuronal proteins including synaptophysin and also glial proteins, some showing a predominance of one and others of the other, as well as all constantly exhibiting immunoreactivity to filaments of progenitor “stem” cells (72; 282; 217). They are distributed throughout all layers of the cerebral cortex and also in subcortical white matter heterotopia.
|
• Cortical laminar disorganization or disorientation as radial rather than tangential (horizontal) orientation | ||
|
• Neurons in the neocortical molecular layer that are not Cajal-Retzius neurons| | ||
|
• Single heterotopic white matter neurons not forming a neuronal aggregate| | ||
|
• Persistent remnants of the subpial granule cell layer of Brun | ||
|
• Marginal glioneuronal heterotopia | ||
|
• Neuronal heterotopia in aggregate or sheets | ||
|
- Nodular | ||
|
• Abnormally formed convolutions as polymicrogyria with or without fused gyri, pachygyria, and lissencephaly | ||
|
• Neuronal and glial dysmorphism and cytomegaly with cytoskeletal abnormalities | ||
|
• Balloon cells, usually of mixed cellular lineage and expressing primitive, progenitor proteins, as well as both neuronal and glial proteins | ||
|
• Excessive neuronal heterotopia and synaptic plexi in the U-fiber layer just beneath focal cortical dysplasia types I or II. | ||
Though many neurons still reside in the intermediate zone or white matter in the last trimester of pregnancy and even into postnatal life (240), the phenomenon of single heterotopic neurons in the white matter is accentuated in cortical dysplasia (162). It was present in the vast majority of patients with cortical dysplasia and has been demonstrated using morphometric techniques (160). It is important to recognize that a certain number of subcortical white matter neurons are normal, particularly in the temporal lobe, so it is a semiquantitative distinction of how many such single displaced neurons cross the line from physiological to pathological. These white matter neurons were often called “isolated” single cells, but synaptophysin, a marker protein of the synaptic vesicle membrane, demonstrates that such cells are not isolated at all, but are in synaptic contact with others and with the overlying cortex; hence, they are capable of contributing to epileptic circuitry (225).
It was initially hypothesized that heterotopic white matter neurons result from injury to the radial glial fibers, resulting in becoming "stranded" or arrested in migration. However, more compelling data utilizing radioactive thymidine autoradiography and immunohistochemistry for microtubule-associated protein 2 and neuropeptides demonstrated that the subcortical neurons in the cat are remnants of subplate neurons (42). This suggests that the increased number of heterotopic neurons seen in cortical dysplasia may result from a failure of programmed cell death. The "normal" number of subcortical neurons has not been clearly established, and some regions of the brain, such as the temporal lobes, may contain a significantly higher population of heterotopic neurons than other lobes (203). Therefore, assessment of an increased number of neurons in the subcortical white matter may be difficult and may, unless obvious, require unbiased morphometric assessment.
Morphometric analysis has demonstrated a statistically significant increase in the number of neurons within the molecular layer of the cortex in epileptic patients versus controls (161), although the significance of this finding is unknown. Marginal glioneuronal heterotopia are excrescences of disorganized neuroglial tissue extending from the pial surface into the subarachnoid space. The persistence of the subpial granule cell layer has been seen in association with many cortical malformations (32). In some malformations, such as holoprosencephaly and some cases of lissencephaly, by contrast, the subpial granular glial layer of Brun may be deficient or absent, and glioneuronal heterotopia appear to result from overmigration beyond the pial barrier (213). Though this subpial granular layer of Brun may persist normally in the frontal sulci up to 2 to 4 months postnatally, its presence is considered a sensitive marker for cortical malformations. Marginal glioneuronal heterotopia may be associated with a failure of the glia limitans (41).
White matter neuronal heterotopia consist of disorganized masses of neurons in the white matter that usually occur in a periventricular position with a nodular morphology, although rare instances of laminar subcortical bands of heterotopic gray matter have been known to produce the appearance of a "double cortex" (73). It has been suggested that these are associated with injury to a group of radial glia leading to failure of a group of neuroblasts to migrate (213). Alternatively, a defect in genes controlling neuroglial interactions, neuroblast proliferation, and programmed cell death has been suggested as being causal (206).
Polymicrogyria describes aggregates of small gyri, often with bridging of the sulci by fusion of the molecular layers. Classic 4-layered polymicrogyria is most frequently considered to result from a destructive lesion that occurs at approximately 20 to 24 weeks' gestation, whereas an unlayered form is thought to result from an insult earlier in development (approximately 13 to 16 weeks’ gestation) (14). Whether polymicrogyria represents a destructive lesion with secondary malformation or a primary malformative lesion continues to be debated, but it is often seen as a feature in a variety of cerebral cortical malformations. Polymicrogyria often involves cortex lining both frontal and temporal lips of the lateral cerebral (Sylvian) fissure in schizencephaly and perisylvian polymicrogyria associated with severe epilepsy. One of the major defects that explain polymicrogyria is multiple discontinuities in the pial membrane that facilitates fusion of the molecular layer of adjacent gyri and overmigration of neuronal-glial heterotopia into the leptomeninges (245; 96; 217).
Neuronal cytomegaly describes the enlarged irregular neurons that are a distinctive feature of cortical dysplasia. Although not all cases of neuronal migration disorders contain these bizarre neurons, they were one of the characteristic features used in initially defining cortical dysplasia (249) and are seen in most cases of severe cortical dysplasia. These neurons, which are often present in hemimegalencephalic cortex, bear an extremely complex dendritic arborization as well as an abundance of perisomatic synapses and a paucity of axosomatic synapses (201; 59). Their increased neuronal size is associated with increased DNA and RNA content and increased nuclear and nucleolar volume suggestive of heteroploidy (21; 152). Many of these cytomegalic neurons contain cytoskeletal abnormalities. Argyrophilic neurofibrillary-like tangles and cytoplasmic vacuoles have been demonstrated within many of the cytomegalic neurons (268), as has the existence of paracrystallin intracytoplasmic structures visible on ultrastructural examination (59). These neurofibrillary-like cytoplasmic inclusions are, like the neurofibrillary tangles seen in Alzheimer disease, strongly immunoreactive with antibodies to high- and medium-molecular weight neurofilament, ubiquitin, and tau protein. However, they differ from the neurofibrillary tangles of Alzheimer disease in that they are not immunoreactive for paired helical filaments (64). Both phosphorylated and nonphosphorylated neurofilaments are found within the cell bodies of these cytomegalic neurons, suggesting abnormal phosphorylation of cytoskeletal proteins (64). A study showed that microtubule-associated protein 2 is strongly expressed in these enlarged neurons (281), particularly the embryonic isoform microtubule-associated protein 2c. Because microtubule-associated protein 2 (and especially its embryonic form microtubule-associated protein 2c) plays a role in growth and sprouting of neuronal processes, the authors suggest that this increased microtubule-associated protein 2 activity reflects elevated plasticity within the enlarged dysplastic neurons (281). Increased or aberrant expression of intermediate filaments may contribute to disruption of migration in the dysplastic neurons (250). Circulating micro-RNAs from serum exosomes may serve as a biomarker for diagnosis and even has possible therapeutic potential (39). Such micro-RNAs and also DNA methylation are two epigenetic mechanisms that have functional roles in focal cortical dysplasias (102). The actin cross-linking protein filamin-A is increased in resected cortical tissue in humans with epilepsy due to focal cortical dysplasia type 2 and also in murine model of this malformation, but its role in epileptogenesis remains poorly defined (288).
Immunocytochemical data analyzing the abnormal expression of glutamate receptor subunits in the dysplastic neurons of cortical dysplasia have generated a potentially interesting explanation for their abnormal electrophysiologic properties (07; 283). This group demonstrated that dysplastic neurons express NMDAR2A/B subunits and some differentially spliced forms of NMDAR1 proteins, which may result in a hyperexcitability of dysplastic neurons in cortical dysplasia (07; 283).
Calcium-binding proteins, such as calretinin and parvalbumin, are associated with GABAergic inhibitory interneurons that arrive at the cortical plate by tangential, rather than radial, migration, and also are strongly expressed in Cajal-Retzius neurons of the molecules later and neurons of the subplate zone in the fetal cortex; they are well demonstrated immunocytochemically (214; 215). These proteins exhibit differential reactivities in epileptogenic focal cortical dysplasias with selective parvalbumin impairment in dysplasias types 1 and 3, and abnormal but not reduced immunoreactivity in type 2 (159). In some cases in which GABAergic calcium-binding protein neurons are deficient in focal cortical dysplasia, this defect may be associated with type 2 potassium-chloride cotransporter immaturity (82).
Balloon cells have eccentric nuclei and ballooned opalescent eosinophilic cytoplasm. They often are binucleated and have dysmorphic nuclei. They tend to cluster at the cortex–white matter junction (268), sometimes extending into the subcortical white matter. Ultrastructurally, they are packed with filaments ranging in size from 400 to 600 nm in length and 30 nm in thickness, interspersed with nonmembrane-bound electron dense helical structures (67). Some of these balloon cells show dual staining with antibodies to both neuronal and glial markers (synaptophysin and glial fibrillary acidic protein, respectively) (268), implying either a failure of the cells to commit to a specific phenotype or a "dedifferentiation." Another interpretation is that balloon cells are of mixed cellular lineage (217). In any case, these findings suggest that an abnormality of development has occurred during the first trimester of intrauterine life. The resemblance of these balloon cells to cells found within the cortical tubers of tuberous sclerosis complex has suggested the possibility that cases of cortical dysplasia bearing balloon cell change may represent a forme fruste of tuberous sclerosis complex (177; 59; 67; 202). Balloon cells do not possess voltage-gated Na+ or K+ currents, hence, cannot initiate epileptic activity, but dysplastic neurons may do so (35; 01). They do, however, express the synaptic vesicle protein synaptophysin at their plasma membrane (217). Balloon cells do not show electrophysiological association with interictal spikes, fast gamma activity, and ripples that do dysmorphic neurons (194).
U-fibers surrounding cortical gyri often are selectively preserved in white matter edema, experimentally in cats, attributed to the richer capillary perfusion network in this region of white matter (36). Pericytes that surround intracerebral arterioles are involved in the pathogenesis of autosomal dominant arteriopathy that results in subcortical infarcts (80), but whether pericytes might play a role in the pathogenesis of subcortical lesions in focal cortical dysplasias is unknown. The U-fiber layer often exhibits “neuronal dispersion” or an excessive number of neurons displaced from layer 6 of the overlying cortex, particularly at the bases of gyri and depths of sulci, where most focal cortical dysplasia occur. These heterotopic single neurons form complex synaptic plexi that integrate into epileptic networks because many of their axons that do not form synapses with neighboring U-fiber neurons enter the grey matter of the adjacent cortex (226). Synaptic interactions initially form local circuits or plexi (179) and later extend these plexi to more distant sites in forming networks.
The percentage of neuropathological lesions of various types in cerebral resections for epilepsy in 9523 patients from 12 European countries over a period of 25 years in children and young adults revealed that hippocampal sclerosis was present in 36.4% of patients as the most common lesion, but 88.9% of these were adults. Tumors, mainly ganglioglioma, were found in 23.6%, and malformations of cortical development (focal cortical dysplasias) were present in almost 20% of total patients, mainly children. Focal cortical dysplasia II was more frequent than type I (29). More epidemiological data are needed, however, including differences that may exist between different countries and patients of different ethnic/genetic backgrounds.
No means of prevention are known.
Initial diagnosis is clinical, eg, an infant or child who develops severe or refractory focal seizures with a corresponding EEG focus of paroxysmal activity. MRI is usually reliable for focal cortical dysplasia type II with bottom-of-the sulcus lesions with or without a transmantle dysplasia. Preoperative and postoperative MRI are usually performed. Type I is not often easily diagnosed by MRI because there are no distinctive differences in tissue signal as there are in type II, but focal cortical thickening with blurring of the grey and white junction are clues. Ultrasonography may sometimes detect lesions in young infants but is useful intraoperatively in some cases and distinguishes focal dysplasia from other types of cerebral lesions (03).
One of the most important differential diagnoses is the distinction between isolated focal cortical dysplasia and association with another type of lesion. Type 1 focal cortical dysplasias are isolated; type 3 is the same as type 1 except that it is adjacent to or associated with hippocampal sclerosis (3a, always in the temporal neocortex), a tumor (3b), vascular malformation (3c), or another lesion, such as an infarct, porencephalic cyst, zone of chronic ischemia, zone of inflammation, etc. (3d). Type 2 focal cortical dysplasias may be isolated or exhibit more extensive brain tissue involvement (eg, hemimegalencephaly) or involvement of multiple organs (eg, skin, in the case of neurocutaneous syndromes; skin, heart, and kidney in tuberous sclerosis complex). Another important distinction is whether the cortical dysplasia is indeed focal or is more generalized throughout the cerebral cortex, as occurs in some chromosomopathies (eg, DiGeorge syndrome, trisomy-13) and in some inborn metabolic diseases beginning prenatally (eg, methylmalonic academia) (70).
The differential diagnosis for cortical dysplasia includes a variety of structural and nonstructural epilepsy-producing lesions. Nonstructural causes of seizures such as electrolyte imbalances, toxic causes, and febrile seizures always need to be excluded. Structural epilepsy-producing lesions, which are usually detectable with appropriate imaging, can be categorized into six broad groups: (1) malformative, (2) neoplastic (eg, ganglioglioma, dysembryoplastic neuroepithelial tumor), (3) familial or metabolic, (4) vascular or traumatic, (5) infectious or inflammatory (eg, Rasmussen encephalitis), and (6) hippocampal sclerosis (267). A combination of clinical, EEG, and imaging data can usually be used to differentiate these entities. One area of potential difficulty exists with the dysembryoplastic neuroepithelial tumor, which is an epileptogenic tumor of children and which has a multifocal pattern with adjacent cortical dysplasia. This diagnosis is confirmed by neuropathologic examination of biopsy or resected tissue, in conjunction with neuroimaging (46). Finally, it should be stressed that although cortical dysplasia (in the broad sense) is the most frequent malformative lesion associated with pediatric epilepsy, other malformations such as Sturge-Weber-Dimitri syndrome, vascular malformations, or meningioangiomatosis can all be causal.
Epileptogenic developmental cortical tumors. Two grey matter tumors of the cerebral cortex are regarded as a “borderland” between dysplasia and neoplasia: ganglioglioma and dysembryoplastic neuroepithelial tumor (DNET or DNT). The dysplastic part of the tumors is the neuronal elements, whereas the glial elements are regarded as the neoplastic component. These tumors do not metastasize, but they can enlarge disproportionate to brain growth in infants and children. They are almost certainly congenital lesions, but their clinical expression as epilepsy may be delayed for months or years though rarely until adult life. It is now questioned whether they are really malignant neoplasms at all, or another form of focal cortical dysplasia (252). Genetically, they share many mutations with focal cortical dysplasia II and hemimegalencephaly. Furthermore, ganglioglioma also shares an upregulation of abnormally phosphorylated tau protein with the others (215).
Focal cortical dysplasia-like histopathological features can be found incidentally in a subset of diffusely infiltrative gliomas of the brain, but the relationship is unclear (22).
In tuberous sclerosis, the cortical tubers are hamartomata that include both neuronal and glial elements, but periventricular nodules and subependymal giant cell astrocytomas are comprised of astrocytes without neurons that become benign tumors because of their growth and proximity to the foramen of Monro where they can cause obstructive hydrocephalus. Retinal astrocytic hamartomata also occur (195).
Focal cortical dysplasias may be causally associated with migraine and its pathogenesis in patients with both migraine headaches and epilepsy (69).
X-irradiation of the brain may induce neuronal hypertrophy and dysplasia, resembling focal cortical dysplasia type II (235).
The diagnostic workup for cortical dysplasia includes careful history (including family history), physical examination, EEG, and electrolyte, metabolic, and toxicological screens. Imaging (preferably MRI) is warranted to determine if a structural lesion is present and even to define the type of focal cortical dysplasia (108; 196).
Neuropsychologic testing may also be indicated. If a high likelihood of cortical dysplasia is established, the location and extent of resection can be determined by the localization of the structural lesion. The zones of epileptogenesis can be mapped using preoperative EEG and intraoperative electrocorticography. Stereoscopic EEG is a application for identifying epileptogenic foci in patients who do not exhibit an MRI lesion and may not even show a histopathological lesion at the EEG focus (127). There is an association between seizure-onset patterns in scalp and intracranial cortical surface EEG between histological types 1 and 2 focal cortical dysplasia (126; 95). In focal cortical dysplasia type IIIa, adjacent to hippocampal sclerosis, the type of dysplasia does not predict seizure onset, which can emanate from either the hippocampus or from temporal neocortex, based on depth electrode recordings (68).
The use of 7 or 9.4 Tesla MRI scanners, rather than the standard 1.5 Tesla or improved 3 Tesla apparatus, enables more precise diagnosis of type 2 focal cortical dysplasia and correlations with histopathological findings (198; 292; 251). Caution must be exercised in the interpretation of MRI intensity changes in lesions, however, because some changes may be transitory or reversible, probably due to epileptic discharge activity (125). Automated detection of focal cortical dysplasias using morphometric analysis in T1-weighted MRI may be useful in supplementing subjective visual identification of lesions (16). Automated detection of cortical dysplasias in high-resolution MRI and the application of artificial intelligence is an initial approach to supplement visual interpretation (104; 270; 269). Multimodal mapping of regional cerebral vulnerability was studied in 337 patients with focal cortical dysplasias from 13 sites worldwide and found to show promise as another tool of correlating cytoarchitectural alterations, postnatal synaptogenesis, and genetic mutations (133).
Diffusion tensor imaging reveals alterations in cortical tubers and perituberal cortex in tuberous sclerosis and is useful in detecting epileptogenic tubers from those that are less likely to produce seizures (284), but data are not yet sufficient to determine whether other types of focal cortical dysplasias display similar findings. Advanced diffusion MRI maps intracellular volume and diffusivity for multimodal imaging that includes functional MRI that show different changes across histopathological subtypes of focal cortical dysplasias (147). PET and SPECT increases the yield of identifying lesions that are not evident or ambiguous with standard MRI sequences and also help in predicting surgical outcome (98; 08). PET and electrocorticography abnormalities in cortex surrounding focal cortical dysplasia type II are likely secondary epileptogenic rather than the primary epileptic focus (151). Focal dysplasia may sometimes be demonstrated in “non-lesional” MRI by special techniques combined with PET (190).
Transcranial ultrasonography is usually limited to neonates and young infants with open fontanelles, but intraoperative ultrasonography of the exposed brain is another useful tool to help guide surgical resections (03; 187).
Because focal cortical dysplasia type II nearly always presents a lesion in MRI, usually at the base of a gyrus, depth of a sulcus, or as a “transmantle dysplasia” of grey matter heterotopia extending continuously from the ventricular wall to the base of gyrus, focal cortical dysplasia type I often shows no lesion evident on MRI because abnormal cortical lamination with normal neurons and with normal cellular density does not present contrast with surrounding normal cortex (257; 258). Blurring of the grey-white junction should be examined carefully because the neuronal dispersion and synaptic plexi in the U-fiber layer are adjacent to the junction (217). Furthermore, repeat brain MRI in children with “non-lesional” MRI may reveal a lesion not initially seen (100). More advanced myelination in infants may account for better visualization of some lesions.
Tissue adherent to explanted stereo-electroencephalographic electrodes in the presurgical evaluation of refractory epilepsy in patients with focal cortical dysplasia type II may assist in diagnosis and improve precision medicine by detecting somatic mutations (37; 118).
A web-based classification of neuropathological features of focal cortical dysplasia provides a unique algorithm guide as a novel educational approach for pathologists with limited experience in cortical developmental lesions (123). The use of artificial intelligence to detect focal cortical dysplasia in imaging and in histopathology is in initial stages and not yet clinically applicable but shows promise for a supplementary assessment in the future (269; 115).
Medical management, primarily with antiepileptic drugs, versus surgical resection depends on the extent, severity, and localization of the lesions. The extent of surgical resectability also needs to be considered, as well as location within or adjacent to the “eloquent” cortex, such as speech areas of the left hemisphere and primary motor and visual cortices. In the case of multiple cortical tubers in tuberous sclerosis, not all tubers are equally epileptogenic, and also in widespread hemispheric dysplasia in hemimegalencephaly, meticulous pre- and intra-operative electrophysiological studies are needed to identify those tubers or regions in which resection is likely to yield clinical benefit by seizure reduction.
The mTOR inhibitors, such as everolimus and sirolimus, have been used effectively for the subependymal nodules and giant cell astrocytomas in tuberous sclerosis. They do not cause as dramatic a reduction in size of cortical tubers as they do periventricular lesions, but they are often effective in treating the epilepsy associated with some tubers (211; 176; 54). A few non-tuberous sclerosis complex patients with mTORopathies, including hemimegalencephaly and focal cortical dysplasia II also have responded well (279; 109; 117). The toxicity and risk of these medications is low, but because there is insufficient documentation of evidence for effectiveness or clinically controlled trials, especially in children, it is difficult to secure permission to administer such drugs unless it is part of an approved protocol with the endorsement of an ethics committee. Thus, progress is slow in this promising treatment. The ketogenic diet may be effective in some cases because it inhibits the mTOR pathway (158).
Some cases of adolescents and adults with focal cortical dysplasia may tolerate surgery awake, without general anesthesia, so that intraoperative brain stimulation can induce functional responses (164). Anterior temporal lobectomy in type 3a (associated with hippocampal sclerosis) often provides effective seizure control, particularly if the lesion is in the right hemisphere (85).
Finally, antiseizure gene therapy shows promise with translational potential to treat the epileptic phenotype of mTOR-related focal cortical dysplasias type II, though still in the investigative phase (12).
Effective pharmacological seizure control is achieved in only a minority of patients with focal cortical dysplasia, but freedom from seizures is important for cognitive functioning and quality of life as well as the epilepsy itself. Focal cortical dysplasia is the cause of epilepsy in about half of all children who undergo surgical resection because of drug-resistant seizures (90).
The clinical outcome or prognosis of seizure control in surgically treated focal cortical dysplasia depends largely on the completeness of resection of the epileptogenic focus and of the dysplastic cerebral tissue. Complete resection is not always feasible, particularly if it involves eloquent regions of the cortex essential to motor or linguistic functions. The use of pre- and intraoperative EEG monitoring and specialized neuroimaging improves the outcome of seizure control and often results in improved postoperative cognitive and intellectual functioning, even in mild or anatomically small focal cortical dysplasia lesions (264; 265; 98). Intervention with surgery in early infancy in large focal cortical dysplasia type 2 causing pharmacologically resistant neonatal seizures is feasible and can improve long-term outcome (31). For extensive focal cortical dysplasias or hemimegalencephaly, hemispherectomy may be the most practical approach for abolishing refractory seizures, particularly if the involved hemisphere has little physiological function, such as for motor control, vision, or language. A valid scale of seizure recurrence risk based on 1237 patients across 32 centers with intractable epilepsy who underwent hemispherectomy was developed and showed 30% total cure of epilepsy and better control in many others (275). Functional hemispherectomy, consisting of cutting white matter connections without removing much tissue, is now used in some centers as an alternative to resection hemispherectomy, but results are not yet well established. Meta-analysis documents that the timing of surgical resection of epileptogenic dysplasias is a major factor in influencing long-term epileptic prognosis (262). Functional MRI mapping of cortical networks also may help predict postoperative seizure outcome (44). A meta-analysis documents that the timing of surgical resection of epileptogenic dysplasias is a major factor in influencing long-term epileptic prognosis (262). Functional MRI mapping of cortical networks also may help predict postoperative seizure outcome (44).
Despite the more severe cytological appearance in focal cortical dysplasia type II than in focal cortical dysplasia type I, surgical resection often is more effective for abolishing a seizure focus in focal cortical dysplasia II than I. The main reason is that the lesion of focal cortical dysplasia type I is often more extensive than in focal cortical dysplasia type II, and the margins are not as easily identified by neuroimaging or intraoperatively; therefore, focal cortical dysplasia type I resections are often subtotal, and epileptogenic tissue remains (135; 89). EEG studies also demonstrate that the epileptic zone extends beyond the apparent anatomical lesion (23; 89).
No known complications are related to pregnancy.
No interactions are known, except for possible interactions with antiepileptic medications.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Harvey B Sarnat MD FRCPC MS
Dr. Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 20, 2026