Epilepsy & Seizures
Driving and epilepsy
Jul. 08, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Benign adult familial myoclonic epilepsy (or familial adult myoclonic epilepsy) is an inherited epileptic syndrome characterized by cortical hand tremors, myoclonic jerks, and rare convulsive seizures. In most affected individuals, the disease takes a benign course; however, at an advanced age, worsening of the tremor and myoclonus is common, and slight intellectual disability is present in a subset of patients. Prevalence is unknown but is estimated to be less than 1 in 35,000. It is transmitted autosomal dominantly with high penetrance, and anticipation has been noted. Genetic studies of the families show heterogeneity, and different susceptible chromosomal loci have been identified. Diagnosis is based on clinical and electrophysiological findings. This is a well-delineated disease with remarkable features that clearly distinguish it from other forms of myoclonic epilepsy. Valproate, levetiracetam, and benzodiazepines are the most beneficial treatments. Precision therapeutic strategies, based on the underlying pathomechanisms, are on the way.
|
• Benign adult familial myoclonic epilepsy usually presents in the second decade of life with a mild hand tremor that is myoclonus-exacerbated by fatigue or emotional stress. Myoclonus usually appears around the same age and consists of erratic, arrhythmic, segmental jerks of small amplitude involving the distal upper limbs and heightened by posture and action. Rare or sporadic convulsive seizures are also a manifestation and are often precipitated by photic stimulation, emotional stress, and sleep deprivation. | |
|
• Diagnosis is based on clinical and electrophysiological findings. EEG findings include a photomyoclonic response along with abnormality of polyspikes and waves. Patients also display extremely enlarged cortical components of somatosensory-evoked potentials and an enhanced C-reflex. Jerk-locked average analysis reveals positive-negative, biphasic spikes preceding myoclonus. | |
|
• Benign adult familial myoclonic epilepsy is transmitted autosomal dominantly, and it must be differentiated from idiopathic epilepsy syndromes with prominent myoclonus features (eg, juvenile myoclonic epilepsy syndrome) and from progressive myoclonus epilepsies. | |
|
• Valproate, levetiracetam, and benzodiazepines are the most beneficial treatments on both tremor and myoclonus. Conversely, carbamazepine is not a drug of choice. |
The term “cortical tremor” was introduced by Ikeda and colleagues to indicate a postural and action-induced shivering movement of the hands mimicking essential tremor but showing the electrophysiological findings of cortical reflex myoclonus, that is: (1) brief EMG burst of about 50 msec duration; (2) no definite synchronization or reciprocity in antagonist’s muscles; (3) positive spikes preceding EMG bursts at the jerk-locked averaging; (4) enlarged cortical components of somatosensory-evoked potentials; (5) enhanced long-loop reflex I (or C-reflex) (24). Initially, it was likely conflated with other familial myoclonus epilepsies, especially progressive myoclonus epilepsies. As progressive myoclonus epilepsies became better understood clinically and genetically, this group began to stand out and was first recognized as such in Japan (03).
Uyama and colleagues first reported a patient with adult-onset fine finger tremulous movement, myoclonic jerks, and two generalized seizures coming from a family that was affected with the same condition through three generations with high penetrance (59). None of the patients showed other neurologic signs nor abnormal neuroradiological findings; the electrophysiological study indicated cortical reflex myoclonus. Subsequently, the same group reported four unrelated families, including 27 affected members spanning three generations, with high penetrance (58). In 1991, Yasuda used the term "benign adult familial myoclonic epilepsy" (BAFME) to describe two pedigrees in which affected members showed autosomal dominant hand tremor, myoclonus, and epileptic seizures with a nonprogressive course (65). Also, in these patients, electrophysiological studies showed evidence of cortical reflex myoclonus. In 1999, Plaster and colleagues and Mikami and colleagues mapped the disease on chromosome 8q24 (39; 43).
Although this condition was exclusively reported from Japan until the 1990s, several reports on pedigrees with similar clinical features but with different genetic identifiers appeared from different European countries and worldwide over the past decade with different names, such as autosomal dominant cortical myoclonus and epilepsy, familial adult myoclonic epilepsy, familial cortical myoclonic tremor, familial essential myoclonus and epilepsy, familial benign myoclonus epilepsy of adult-onset, and familial tremor and epilepsy. Despite phenotypic and genetic differences, the clinical and electrophysiological data point toward one syndrome (52; 53).
Genetic studies of families have revealed a large genetic heterogeneity with different identified loci, including 8q24 (FCMTE1), 2p11.1-q12.2 (FCMTE2), 5p15.31-p15.1 (FCMTE3), and 3q26.32-3q28 (FCMTE4), but a unique underlying causative mechanism, ie, pentameric intronic expansions (see section 4).
The main clinical features are persistent, continuous, fine, shivering-like hand myoclonus, resembling a tremor, often associated with occasional tonic-clonic seizures precipitated by sleep deprivation or photic stimulation. Age of onset is highly variable (usually 11 to 50 years of age). The initial sign, myoclonus, usually develops between the third and fifth decades and persists through life. After onset, myoclonus persists not only at rest but also on posturing or during the action and is increased by emotional stress, fatigue, lack of sleep, or intermittent light. Myoclonus is fragmentary or bilateral and is most marked in the distal portion of the upper limbs, and rarely, also the legs, head, trunk, proximal upper limbs, and facial muscles (ie, around the mouth and eyelids) can be involved (28). Exceptionally, patients may also present with drug-resistant focal seizures (20).
The clinical course is nonprogressive, allowing most patients a normal lifespan (27). Daily activities are not disturbed except for some difficulties in skillful finger movements, such as writing, buttoning, and picking up small objects. However, prolonged follow-up evaluation revealed walking impairment and a need for help in everyday life for individuals older than 80 years of age. Additionally, some extrapyramidal signs that are likely derived from valproate intake may also be aroused. Lastly, in some cases, slight impairment of the visuospatial processing, as well as of verbal and visual memory, could be observed. Whether these features may derive from the pathophysiological mechanism of the disease (ie, RNA toxicity) is also a matter of debate, although both EEG recordings and cognitive tests point toward some slow form of progression.
Intriguingly, a higher prevalence of mood disorders and a psychiatric burden has been observed in familial adult myoclonic epilepsy as compared to other forms of epilepsy and healthy controls, particularly depression and anxiety in association with pathological traits of personality, ie, paranoid, schizophrenia, and hypomania (06). Migraine is another symptom that is quite common in this population of patients (46; 10; 30; 01).
This condition usually has a benign course, although the myoclonic tremor is often refractory to treatment. A long-term (over 30 years) follow-up study of a 4-generation South African family confirms that this condition does not cause additional progressive neurologic deterioration, and the quality of life is mostly influenced by worsening of the cortical myoclonic tremor with age (60). However, in advanced age, a worsening of the myoclonus is possible as well as mild ataxia and difficulty walking (08). Some phenotypic differences across pedigrees of different ancestry may be found, possibly related to the underlying genetic variability. Indeed, even if Japanese patients show a worsening of both cortical tremor and amplitudes of giant somatosensory evoked potentials that are significantly increased over time (23), the disease is invariably “more progressive” in families of European descent (61).
A 68-year-old man had a family history of hand tremors and upper-limb jerks that began at 20 years of age and for which he had been treated for several years with propranolol (20 mg/day). Tremors consisted of continuous, rhythmic, distal fine twitches at the hands, enhanced by emotion or fatigue. Daily activities were not significantly disturbed except for some difficulties in skillful actions, such as writing, buttoning, and picking up small objects. Other than the tremor, the patient suffered from distal arrhythmic, mainly postural, myoclonic jerks, especially in the upper limbs.
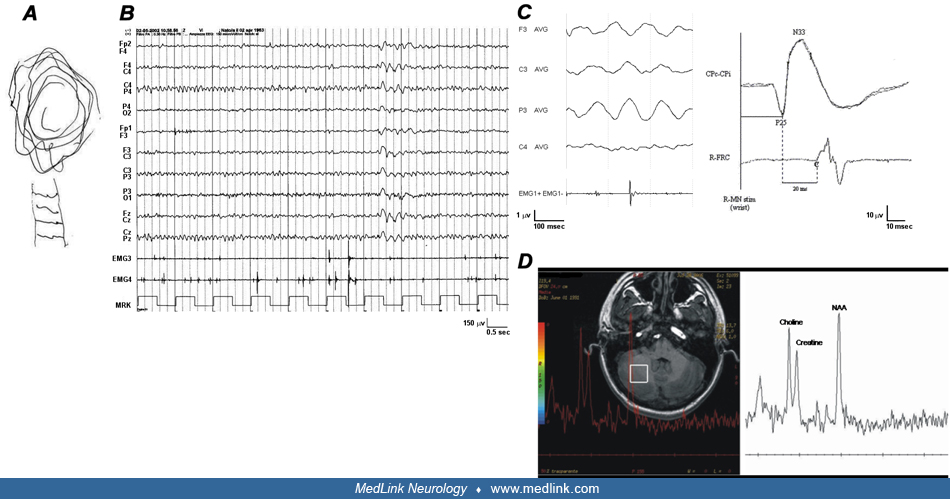
The patient experienced three tonic-clonic seizures in adult age, precipitated by sleep deprivation or emotional stress. Therefore, he was treated for many years with valproic acid (1000 mg/day) with some benefits. The patient had no signs of cognitive impairment and had a normal neurologic examination, except for hand myoclonus. His EEG revealed mild generalized paroxysmal abnormalities and photoparoxysmal response. Detailed electrophysiological investigations (jerk-locked averaging, giant somatosensory-evoked potentials, and long-loop reflex I) were consistent with the presence of cortical reflex myoclonus. Conventional brain MRI was unremarkable, but 1H-MR spectroscopy showed an abnormal spectral choline peak in the right cerebellar hemisphere. Add-on with levetiracetam (1000 mg/day) improved myoclonus and EEG abnormalities.
Genetics. Familial adult myoclonic epilepsy is caused by noncoding intronic repeat expansions and should be considered part of a broader phenotypic spectrum that overlaps with progressive myoclonic epilepsy rather than a strictly isolated syndrome (02).
The inheritance pattern is constantly compatible with an autosomal dominant trait. Five different chromosomal loci have been identified so far, but the underlying causative mechanism seems to be unique (ie, pentameric intronic expansion). Males and females are equally affected.
Firstly, by linkage analysis in one large Japanese kindred, Mikami and associates assigned the gene responsible for this disorder to the distal portion of 8q (39). A maximum 2-point LOD score of 4.31 with D8S555 was obtained at a recombination fraction of 0.0; the maximum multipoint LOD score was 5.42 for the interval between D8S555 and D8S1779. Mikami and associates also assigned a cytogenetic localization to 8q23.3-q24.11 (39). To date, one large pedigree and four small pedigrees originating from Japan were localized on this FCMTE1 locus (41). This FCMTE1 locus was also confirmed in a Chinese family, and the mapped genomic region was narrowed into an 18.4 Mb region at 8q22.3-q24.13, but the whole genome scan failed to identify the causative gene (05).
In non-Japanese benign adult familial myoclonic epilepsy patients, Labauge and colleagues reported in a pedigree in Spain absence of linkage to 8q24 (26). Subsequently, Guerrini and colleagues disclosed a linkage to 2p11.1-q12.2 in a large Italian family with autosomal dominant cortical myoclonus and epilepsy (20). However, the phenotype was not identical with benign adult familial myoclonic epilepsy as the affected patients showed complex partial seizures of frontotemporal origin along with generalized seizures. Linkage analysis in a Dutch family presenting with familial cortical tremor with epilepsy accompanying cognitive deterioration excluded linkage to chromosome 8q23.3-q24 (62). De Falco and coworkers described two benign adult familial myoclonic epilepsy families showing linkage to the same chromosomal region and suggested allelism with autosomal dominant cortical myoclonus and epilepsy, suggesting genetic heterogeneity (11). Locus heterogeneity in benign adult familial myoclonic epilepsy and the related disorder is present, although in most families with classical signs and symptoms, the disorders appear to result from a single gene on chromosome 8q (58).
In 2004, Striano and colleagues reported a family pedigree in Italy indicating an autosomal dominant inheritance characterized by cortical tremor, myoclonic jerks, and generalized seizures with a nonprogressive course (50). The family showed linkage to chromosome 2p11.1-2q12.2 (lod score value=1.55). This observation would confirm that benign adult familial myoclonic epilepsy is a genetically heterogeneous condition, with Japanese families linked to 8q23.3-8q24.13 and European families to 2p11.1-q12.2. In 2005, Striano and colleagues also reported a 5-generation family showing cortical tremor, myoclonus, and epilepsy from Italy (52). The electrophysiological study confirmed the presence of cortical reflex myoclonus in all affected members, and giant somatosensory-evoked potentials and enhanced long loop reflex I were found in three presymptomatic members who manifested hand tremor and myoclonus in the upper limbs after 1.5 years of follow-up. Genetic study revealed a significant linkage on chromosome 2p (maximum lod score=5.9).
Striano and colleagues also reviewed a familial case presenting autosomal cortical tremor, myoclonus, and epilepsy (54). They analyzed the phenotypic differences between the pedigrees according to the genetic acquisitions. They concluded that benign adult familial myoclonic epilepsy, familial adult myoclonic epilepsy, familial essential myoclonus and epilepsy, familial cortical tremor epilepsy, and autosomal dominant cortical myoclonus and epilepsy are the same clinical entity even if genetically heterogeneous. A founder effect may explain the high frequency of families coming from the same topographic area of Japan and Southern Italy (58; 34; 41). Over the years, genetic studies have narrowed the candidate FCMTE2 locus on chromosome 2 (34; 45; 10). In 2013, the critical interval was set to a 10.4 Mb segment, which included 59 genes, at 2p11.1-2q12.2 in a large Italian family (30). Later, an international effort on a familial adult myoclonic epilepsy cohort of 10 European families and one Australian/New Zealander family further narrowed the FAME2 locus to a 9.78 megabase interval within 2p11.2-q11.2, confirming the founder haplotype in four Italian families and allelic heterogeneity with at least four distinct founders responsible for familial adult myoclonic epilepsy at the FAME2 locus (21).
The FCMTE3 locus, linked to chromosome 5p15.31-p15, has been described in one French, two Chinese, and one Dutch family (13; 29; 31).
The FCMTE4 locus, mapped to chromosome 3q26.32-3q28, was identified in 2013 by a genome-wide linkage study (66).
A fifth locus (FCMTE5) was identified in a single consanguineous Egyptian kindred exhibiting adolescent-onset recessively inherited cortical tremor and epilepsy, and a single base-pair deletion was detected in CNTN2 (1q32.1) (47). However, the classification of this disorder as familial cortical myoclonic tremor and epilepsy remains controversial; the phenotype is consistent with temporal lobe epilepsy and carbamazepine-induced myoclonus (55).
Finally, mutations in some isolated families have been reported in different genes (ADRA2B, CTNND2, NOL3, ACMSD, PLA2G6) (44; 38; 12; 18; 67), but these findings have not yet been replicated and their pathogenicity is not proven.
Notably, a higher degree of clinical anticipation associated with a maternal transmission was reported in Japanese families, despite slight repeat expansions differences supporting the original hypothesis that the disease is associated with unstable expanding repeats (22; 23; 25; 69; 40), but additional factors could contribute to anticipation. Indeed, in April 2018, Japanese researchers uncovered expanded TTTCA repeats (between 440 and 3680 repeat units) in an intronic region of SAMD12 and at least two other genes (ie, TNRC6A and RAPGEF2) in 85 patients from 49 independent benign adult familial myoclonic epilepsy families (25). Common founder effects seem to play a role as individual loci expansions are expected in specific populations; however, Maroilley and colleagues identified the first family of confirmed European ancestry to carry repeat expansions of the SAMD12 locus, and yet, this is the shortest TTTCA expansion (about seven repeats) ever reported (36).
The familial adult myoclonic epilepsy international consortium showed that families linked to chromosome 2p11.1-q12.2 (FAME2) carry the same expansion of an ATTTC pentamer within the first intron of the gene STARD7. This expansion segregated familial adult myoclonic epilepsy in 154 affected individuals from 22 families (09). Also, families linked to chromosome 5p15 (FAME 3) have intronic TTTTA/TTTCA expansion in the gene MARCHF6 (formerly MARCH6) (16) and TTTCA and TTTTA pentanucleotide repeat insertions in intron 1 of YEATS2 (3q27.3) were reported as the causative mutation of FCMTE4 in the original family (68).
In summary, different genes may be involved, but this condition is homogeneously caused by intronic pentameric expansions occurring at a polymorphic short tandem repeat site initially composed of ATTTT repeats and containing an ATTTC insertion generally located at the 3’ end of the expansion (27; 14). A high somatic instability of repeat expansions, depending on DNA mismatch repair processes, is also present and may impact the progression of the disease (42; 14). Because the noncoding repeat expansions responsible for FAME are not detected by routine genetic tests used at this time, a clinical diagnosis accompanied by neurophysiological testing remains essential to guide the geneticist on the selection of the specific genetic technique (19). Studies have demonstrated that complex configurations of repeat expansions, particularly involving the MARCHF6 locus, contribute to phenotypic variability across familial adult myoclonic epilepsy and progressive myoclonic epilepsy (02).
Pathophysiology. The pathophysiological basis of this condition remains largely elusive. Both clinical and electrophysiological features of the syndrome suggest cortical hyperexcitability, which can be the result of a decreased cortical inhibition by the cerebellum via its cerebello-thalamo-cortical projections (52; 55a; 28; 17), encompassing a circuitry involving subcortical and pericentral cortical structures (ie, the sensorimotor cortex) (15). Accordingly, postmortem histological studies support the evidence of cerebellar pathology in these patients, and a few imaging investigations suggest the involvement of the cerebellum and its projection areas in these patients (62; 63; 58). MRI spectroscopy demonstrates elevated choline/creatine ratio in the cerebellum cortex of patients compared with controls (49; 32). A brain MR diffusion tensor imaging study compared cerebellar fiber density between patients with familial cortical myoclonic tremor with epilepsy and essential tremor, and it revealed significantly decreased mean fractional anisotropy (ie, microstructural damage of the cerebellar white matter) in familial cortical myoclonic tremor patients (04). Finally, resting-state functional magnetic resonance imaging revealed significant differences in the bilateral frontal lobe and fusiform gyrus correlated with tremor duration in familial cortical myoclonic tremor patients (64). The evidence that familial adult myoclonic epilepsy is caused by intronic pentameric expansions in distinct genes and that these expansions exhibit a high somatic instability, ultimately resulting in genomic rearrangements, suggests that a possible consequence of such repeat expansions is the loss of RNA and protein expression, eg, because of hypermethylation and silencing of the promoter.
An alternative mechanism is the formation of RNA structures filled in with AUUUU or AUUUC repeats that aggregate in the nucleus of cells (RNA foci), trapping numerous RNA-binding proteins that are unable to function properly (14). Notably, these RNA foci have been observed in postmortem brains of FCMTE patients carrying SAMD12 expansions from Japan (25), but they have not been observed in lymphoblasts and fibroblasts of individuals with MARCHF6 or STARD7 expansions (09; 16). This may indicate a tissue specificity with abnormal transcription and RNA toxicity mainly occurring in the neuronal cells, or yet abnormal transcription may be absent in other FCMTE subtypes, suggesting other disease mechanisms (14). Thus, a third disease mechanism could be the translation of the expanded repeat into a protein through repeat-associated non-AUG translation. This noncanonical protein synthesis, which begins at the site of the expanded repeats in the absence of an AUG codon, may lead to the synthesis of polybasic peptides and the formation of insoluble fibrillar aggregates through a toxic gain-of-function mechanism at the protein level (48; 14). Evidence supports a disease mechanism driven by RNA toxicity and dysfunction of the cerebellar-thalamo-cortical network, leading to cortical hyperexcitability and myoclonus (33).
Epidemiological features of benign adult familial myoclonic epilepsy are uncertain. Uyama and colleagues reported 54 patients in seven families, estimating a prevalence of approximately 1:35,000 based on their observation in Kumamoto Prefecture (58). A unique South Indian community has been described, including 241 patients with ADCME belonging to 48 families (35). The 48 families are domiciled in two southern districts of Tamilnadu, India, which belongs to a community called "Nadar" and whose nativity is confined to these southern districts, with reported unique genetic characteristics. This is the largest family reported worldwide.
Myoclonus is an important symptom of many neurologic disorders. When myoclonus is combined with generalized tonic-clonic seizures in adults, especially when a familial hereditary trait is present, progressive myoclonus epilepsies must be considered first (37). Progressive myoclonus epilepsies are characterized by myoclonus and progressive neurologic complications, such as dementia and cerebellar ataxia. In contrast, patients with benign adult familial myoclonic epilepsy demonstrate myoclonus and generalized tonic-clonic seizures without early signs of dementia and cerebellar ataxia.
Most myoclonus epilepsies are generally progressive, causing dementia, cerebellar ataxia, or other extrapyramidal symptoms in addition to myoclonus and seizures. Conversely, benign adult familial myoclonic epilepsy shows myoclonus as a primary symptom and is nonprogressive without accompanying cerebellar ataxia and mental deterioration. Benign adult familial myoclonic epilepsy is transmitted by an autosomal dominant trait with a high penetrance rate, whereas some progressive myoclonus epilepsies show autosomal recessive inheritance. There are a limited number of progressive myoclonus epilepsies transmitted by autosomal dominant inheritance, such as dentatorubral-pallidoluysian atrophy, in which cerebellar ataxia and choreoathetosis predominate, but myoclonus and dementia are rather mild.
Differentiation of benign adult familial myoclonic epilepsy from juvenile myoclonic epilepsy is not difficult because of their clinical features. In juvenile myoclonic epilepsy, myoclonic jerks occur frequently and often culminate in generalized tonic-clonic seizures. In benign adult familial myoclonic epilepsy, myoclonus persists for years, but generalized tonic-clonic seizures are rare, occurring only several times in a lifetime. In juvenile myoclonic epilepsy, the seizures are prone to occur on awakening. Such a circadian trait is not seen in benign adult familial myoclonic epilepsy. The onset age of benign adult familial myoclonic epilepsy is from the third to the fifth decade, whereas the peak age of onset of juvenile myoclonic epilepsy is in the second decade.
Accurate diagnosis of familial adult myoclonic epilepsy requires integration of clinical features, electrophysiological evidence of cortical myoclonus, and targeted genetic testing (33).
No hematological or biochemical findings have been reported. Electrophysiological investigations, including EEG, somatosensory-evoked potential, C-reflex, and jerk-locked back averaging, are essential to confirm the cortical origin of myoclonus. However, some of these electrophysiological features can be masked by antiepileptic treatments (54). The EEG background activity is usually normal or slightly slowed in the slower alpha band. Generalized paroxysmal abnormalities and photoparoxysmal response are frequently found in patients not receiving therapy (52; 58). Focal paroxysmal activity can occur in some patients in addition to generalized EEG abnormalities (20). Polymyographic recording helps to differentiate tremor and myoclonus. The EMG pattern is consistent with irregular, arrhythmic, or semi-rhythmic and high-frequency (around 10/s) myoclonic jerks. EMG bursts last about 50 ms and are usually synchronous between agonist and antagonist muscles, not showing the regular agonist/antagonist alternate as in essential tremor. Jerk-locked averaging analysis commonly discloses a positive-negative, biphasic, premyoclonic spike or a more complex pattern of wavelets related to myoclonus on the contralateral sensorimotor regions. The evaluation of somatosensory-evoked potentials and long-loop reflex I may show an enlargement of cortical components (P25-N33 amplitude larger than 8.5 to 15 µV) and enhanced long-latency (40 ms) C reflex response evoked by peripheral nerve stimulation. A reduction of the resting motor threshold intensity and the post-motor-evoked potential silent period has been reported in few patients evaluated by transcranial magnetic stimulation, indicating that central motor inhibitory mechanisms are impaired in these cases (20). MRI study is usually normal, even if minor, nonspecific abnormalities (eg, mild enlargement of the subarachnoid spaces of the lateral ventricles) are sometimes reported. An MRI spectroscopy study demonstrated elevated choline/creatine ratio in the cerebellum cortex of patients compared with controls (49; 32). The discovery that familial adult myoclonic epilepsy is caused by intronic pentanucleotide expansions in distinct genes now allows for confirmation of the affected status by targeted genetic analysis (highly sensitive RP-PCR assay), even in presymptomatic individuals. Long-read sequencing techniques, including nanopore-based approaches, have substantially improved detection of intronic repeat expansions and are increasingly regarded as the preferred genetic diagnostic method (57).
Treatment of familial adult myoclonic epilepsy remains largely symptomatic; however, evidence suggests that low-dose perampanel may reduce cortical myoclonus and tremor, with associated neurophysiological improvement in selected patients (07).
Cortical tremor is not responsive to alcohol, levodopa, or carbidopa but improves with antiepileptic drugs (24; 52; 58). Valproate, levetiracetam, and benzodiazepines produce the most benefit on cortical tremor and myoclonus as combining both antiepileptic and antimyoclonic activity. Perampanel could be useful as an add-on to first-line antiseizure medications. The effect of phenobarbital and primidone to control myoclonus is limited. Carbamazepine (CBZ) and gabapentin, commonly used in essential tremor, should be considered with caution in FAME as they can worsen myoclonus or lead to status epilepticus (51; 56). In these cases, a correct diagnosis and prompt discontinuation of the drug may reverse a potentially severe, life-threatening condition (51). In advanced age, worsening of the myoclonus is possible, as well as difficulty in walking and mild ataxia (08; 53). It is advisable for patients with photosensitivity to wear sunglasses in sunlight or to avoid places where light is reflected in the sea or snow. Identification of the gene and causative pentanucleotide expansions offers hope of preventive or curative options, such as RNA-targeting treatments, including antisense oligonucleotides, siRNA, and the CRISP-Cas9 technologies, which may help in reversing the pathological mechanism in the next few years.
The therapeutic landscape for benign adult familial myoclonic epilepsy continues to evolve, with management strategies primarily centered on symptom control. However, treatment response varies considerably among patients, reflecting the heterogeneity of disease expression and progression (58; 69; 63).
Seizures and myoclonic tremors are typically addressed with antiseizure medications, yet their efficacy differs on an individual basis. Levetiracetam, clonazepam, and valproate remain the most widely used pharmacological options, with levetiracetam often preferred for its favorable efficacy-to-tolerability ratio (58; 63). Despite pharmacological intervention, myoclonic tremors often persist, sometimes significantly impairing fine motor tasks, writing, and daily activities requiring precise hand control (69).
Although benign adult familial myoclonic epilepsy is not considered a progressive disorder, many individuals report a gradual worsening of tremor over time, particularly in tasks demanding fine motor precision (63). Although some patients maintain functional independence, others experience increasing difficulties with writing, object manipulation, or activities necessitating hand stability (58). Given these challenges, there is growing interest in nonpharmacological interventions, including transcranial magnetic stimulation (TMS) and deep brain stimulation (DBS), though evidence supporting their efficacy in benign adult familial myoclonic epilepsy remains limited (69).
Given the genetic and chronic nature of benign adult familial myoclonic epilepsy, particular care must be taken when addressing specific patient populations that may require unique treatment strategies and additional precautions (58; 63).
The U.S. Food and Drug Administration is advising healthcare professionals and women that the antiseizure medication, valproate sodium, and related products—valproic acid and divalproex sodium, are contraindicated and should not be taken by pregnant women due to the evidence that these medications can cause decreased IQ scores in children whose mothers took them while pregnant. More information can be accessed at www.fda.gov/drugs/postmarket-drug-safety-information-patients-and-providers/valproate-information. Women with benign adult familial myoclonic epilepsy who are planning pregnancy require careful treatment planning, as some antiepileptic drugs, particularly valproate, pose a high risk of teratogenicity and should be avoided whenever possible (69). The choice of alternative medications must balance seizure control with fetal safety, necessitating individualized treatment approaches. Furthermore, hormonal fluctuations during pregnancy can exacerbate myoclonus, necessitating close monitoring and potential dose adjustments (58). Preconception counseling and coordination with maternal-fetal medicine specialists are recommended to optimize outcomes (63).
Patients with benign adult familial myoclonic epilepsy undergoing surgical procedures require special anesthetic considerations to prevent exacerbation of symptoms (58). Certain anesthetic agents, such as ketamine and etomidate, have been associated with increased myoclonus, necessitating careful selection of alternative anesthesia protocols (63). Additionally, perioperative seizure control should be optimized, and patients on chronic antiepileptic drug therapy should have medication levels monitored to prevent breakthrough seizures (69). Multidisciplinary perioperative planning with neurologists, anesthesiologists, and surgeons is essential for minimizing complications.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Antonella Riva MD PhD
Dr. Riva of the University of Genoa received a travel grant from Biocodex and UCB Pharma as an attendant and from Jazz Pharmaceuticals as a guest speaker. She is also a member of the advisory board at Stroke Therapeutics.
See Profile
Pasquale Striano MD PhD
Dr. Striano of the University of Genova, Istituto Gaslini, has no relevant financial relationships to disclose.
See Profile
Jerome Engel Jr MD PhD
Dr. Engel of the David Geffen School of Medicine at the University of California, Los Angeles, has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Epilepsy & Seizures
Jul. 08, 2026
Epilepsy & Seizures
Jun. 02, 2026
Movement Disorders
May. 18, 2026
Neuropharmacology & Neurotherapeutics
May. 11, 2026
Epilepsy & Seizures
May. 08, 2026
Epilepsy & Seizures
May. 01, 2026
Epilepsy & Seizures
Apr. 30, 2026
Epilepsy & Seizures
Apr. 28, 2026