Developmental Malformations
Barth syndrome
May. 08, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
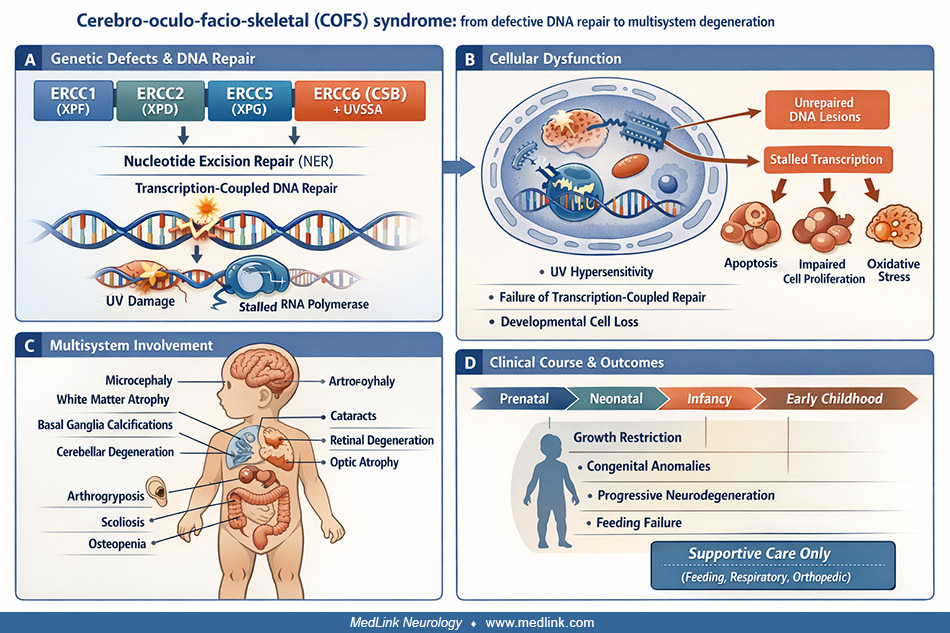
In this article, the author updates information on cerebro-oculo-facio-skeletal (COFS) syndrome. The condition is rare, with autosomal recessive inheritance, and manifests abnormal facies, ocular changes (eg, cataracts, retinal degeneration, microcornea, optic atrophy), in utero and postnatal growth retardation, severe psychomotor retardation, cerebral and cerebellar degeneration with calcification in basal ganglia and white matter, progressive joint contractures and wasting, and death in infancy or early childhood. In many instances, the disorder results from a mutation in the Cockayne syndrome group B (ERCC6/CSB) gene or xeroderma pigmentosum (ie, DNA repair) genes (ERCC2/XPD, ERCC5/XPG, ERCC1/XPF, or ERCC4), mirroring phenotypic and clinical similarities between these conditions.
|
• Cerebro-oculo-facio-skeletal syndrome is rare, with autosomal recessive inheritance, and manifests abnormal facies, ocular changes (eg, cataracts, retinal degeneration, microcornea, optic atrophy), in utero and postnatal growth retardation, severe psychomotor retardation, microcephaly with cerebral and cerebellar degeneration and calcification in basal ganglia and white matter, arthrogryposis with progressive joint contractures and wasting, and death in infancy or early childhood. | |
|
• The disorder results from a mutation in one of a number of genes, the Cockayne syndrome group B (ERCC6/CSB) gene or xeroderma pigmentosum (DNA repair) genes (ERCC2/XPD, ERCC5/XPG, ERCC1/XPF), reflecting phenotypic and clinical similarities between these conditions. |
Cerebro-oculo-facio-skeletal syndrome was first described by Pena and Shokeir in 1974. They identified 10 children, nine of whom were from two families of North American aboriginal background residing in or near the province of Manitoba, Canada, with a uniform constellation of congenital abnormalities. Based on the apparent heredity and affected systems, they titled their paper “Autosomal recessive cerebro-oculo-facio-skeletal syndrome” (45). This designation has been generally accepted. Since then, numerous case reports have dealt largely with clinical aspects of the disorder and its possible relationship to other disorders. A search for the molecular pathogenesis of the disorder advanced significantly in 1996 when XPF was cloned (17). The ERCC (excision repair cross-complementation) and XP (xeroderma pigmentosum) genes function as complexes that are essential to the repair of DNA (damaged by ultraviolet light, carcinogens, or mutagens) and, hence, are shown together in texts (29; 09).
Individuals with cerebro-oculo-facio-skeletal syndrome are usually identified at birth or shortly thereafter based on their physical appearance and severe psychomotor delay. However, some may present prenatally (with fetal features including thymus hyperplasia, splenomegaly, increased hematopoiesis, infratentorial anomalies like pontocerebellar hypoplasia, and supratentorial microcephaly with delayed gyral development) (30; 34). The appearance of those affected by the disorder is relatively characteristic and includes the major diagnostic criteria of (1) microcephaly; (2) ocular anomalies including cataracts, microphthalmia, optic atrophy, and blepharophimosis (18; 25; 15); (3) dysmorphic facies with a high and broad nasal bridge, large ears, overhanging upper lip, and micrognathia; and (4) musculo-skeletal abnormalities including flexion contractures of the limbs (arthrogryposis), scoliosis, hip dysplasia or dislocation, narrow pelvis, short stature, osteoporosis, dysplastic acetabula, and rocker-bottom feet with proximal displacement of the second metatarsals and longitudinal grooves in the soles along the second metatarsal. Infants may also have a short neck, hirsutism, widely spaced nipples, single palmar creases, axial hypotonia, peripheral hypertonia, and renal anomalies. Affected children are usually small at birth due to intrauterine growth retardation; growth progresses poorly in the postnatal period as well (45; 46; 31).
Affected infants may develop seizures, including infantile spasms (19). Motor delay is invariably severe. Few affected infants are capable of more than rolling over and, perhaps, sitting, smiling, and learning a few words. Nevertheless, variable progression has led some authors to suggest that there are two subtypes of cerebro-oculo-facio-skeletal syndrome (04). Vision and hearing are often impaired. The children suffer from feeding difficulties and failure to thrive. In the terminal stages, the children may lose weight despite adequate caloric intake by tube feeding. Repeated respiratory infections eventually lead to death in infancy or early childhood, usually by 6 years, although one well-documented patient died at 11 years of age. At that time, it had become apparent that he resembled children with Cockayne syndrome in appearance and in regard to hypersensitivity to sunlight.
Incorporate prenatal findings from recent fetal cases, including thymus hyperplasia, splenomegaly, increased hematopoiesis, and infratentorial anomalies like pontocerebellar hypoplasia, alongside supratentorial microcephaly with delayed gyri. Add hyporeflexia or areflexia, sensorineural hearing loss, syndactyly, camptodactyly, cleft or high-arched palate, small mouth, and clenched hands with abducted fingers, as consistently noted in updated reviews. Include modern genetic diagnostics (eg, NGS panels for ERCC1/2/5/6 mutations) and imaging details like cerebellar dysplasia, corpus callosum agenesis or degeneration, and progressive demyelination on MRI (34).
CT scan of the brain reveals enlarged ventricles and subarachnoid spaces with white matter hypointensity in the infantile period followed by progressive calcification in the basal ganglia, white matter, and sometimes the cortex (35; 06). Microencephaly with delayed gyral development, enlarged lateral ventricles, hypoplastic basal ganglia, callosal anomaly, and pontocerebellar hypoplasia have been reported in two studies comprising eight fetuses (08; 34).
Laboratory findings are nonspecific, and amino acid survey is normal. Patients may exhibit mild hepatic dysfunction, and liver biopsies may show fatty infiltration or mild cirrhosis (45). Some cases have elevated creatine kinase levels in infancy and muscle biopsies show slight sarcomeric disorganization and fibrosis (33; 13). Bone biopsies show necrotic chondrocytes and loss of osteocytes (24).
The neuropathological findings in cerebro-oculo-facio-skeletal syndrome include: (1) severe microcephaly and mild ventriculomegaly evident from birth; (2) possible delayed myelination in cerebral structures with otherwise normal myelin structure; (3) swollen, PAS-positive ubiquitinated granular glial cells in the white matter that appear shortly after the onset of myelination; (4) subsequent patchy or diffuse loss of myelin with atrophy and gliosis in the white matter; (5) progressive cortical atrophy; (6) parenchymal, pericapillary, and vascular mineralization in the globus pallidus and putamen, cerebral cortex at the depths of sulci, and white matter; (7) rare binucleate Purkinje cells; (8) severe degenerative changes in the internal granular and Purkinje cell layers of the cerebellum; and (9) retinal degeneration with optic nerve atrophy (06). At birth and in infancy, the brains have a normal gyral pattern, cortical architecture, and cortical thickness, suggesting that basic developmental processes are intact, although partial agenesis of the corpus callosum has been reported in two infants with cerebro-oculo-facio-skeletal syndrome (59; 53). Small brain size with ventriculomegaly at birth suggests that the degenerative process begins in utero.
Prognosis can be difficult to ascertain, given the diversity of findings in the syndrome (41). However, prognosis is uniformly poor for children who have been diagnosed positively with cerebro-oculo-facio-skeletal syndrome. Profound sensorineural hearing loss stems from accelerated cochlear nerve degeneration (12). The cause of this is unclear, but some phenotypic overlap is apparent in that cochlear changes can also be observed in Pena-Shokeir syndrome type I (28). Severe muscle weakness has been reported with end-stage muscle changes identified on biopsy (36). Ichthyosis has been noted in one male patient (61). Death ranged from the neonatal period to 6 years of age, although one child survived to 11 years of age. Most patients have severe cognitive and motor retardation and eventually die from respiratory illness (recurrent aspiration pneumonia and failure to thrive). Supportive management includes gastrostomy feeding; vision, hearing, or spasticity aids; orthopedic interventions for contractures; and respiratory support. Females exhibit ovarian dysgenesis precluding reproduction. Extreme hypersensitivity contraindicates chemotherapy or radiation. The cytotoxicity and DNA damage elicited by certain chemotherapeutic agents and radiation therapy trigger the nucleotide excision repair complex. Because ERCC1 is a major molecule in this pathway, mutations have a deleterious effect on the excision pathway (10). ERCC1 appears to play a prognostic or even carcinogenic role in some tumors (40; 49). Because the helicase family of enzymes is important to pathogenesis, future patients may respond to chemical rescue to restore enzymatic activity (57).
Cerebro-oculo-facio-skeletal syndrome, in most cases, is known or presumed to have a genetic basis. Most reported cases exhibit a familial tendency indicative of an autosomal recessive inheritance pattern (37). Parental consanguinity is common but not universal. A striking Finnish lineage has been identified, with a single common ancestor (ie, founder effect) traced to the 18th century (27). Chromosomal studies have generally been normal, although a case with 47XXX was reported (45; 44). A case of early-onset Cockayne syndrome, with phenotypic similarities to cerebro-oculo-facio-skeletal syndrome, was also reported as 47XXX (20). In both children, the chromosomal abnormality was considered to be coincidental. An Egyptian child with cerebro-oculo-facio-skeletal syndrome (and parents that were first cousins) was found to have a balanced translocation 46 XX t(1; 16) (q23; q13), inherited from the mother. It has been shown that some affected children related to the Manitoba Aboriginal population group within which cerebro-oculo-facio-skeletal syndrome was originally reported have a mutation in the CSB gene, located on chromosome 10q11-21. The mutation results in truncation of the Cockayne syndrome group B protein. Nucleotide deletions in the same gene at other sites result in Cockayne syndrome (05; 39). Other patients have been shown to have mutations in the xeroderma pigmentosum genes XPD and XPG, or in the ERCC1 (excision repair cross-complementing 1) gene (60). Reports include compound heterozygous ERCC2 variants with brainstem pilocytic astrocytoma (01) and Gly47Arg homozygous ERCC2 mutation with death at 21 months (52). Based on these findings, cerebro-oculo-facio-skeletal syndrome can be placed in a group of genetically and phenotypically related disorders, including Cockayne syndrome, xeroderma pigmentosum, and UV-sensitive syndrome (63). However, it should be emphasized that these conditions form a phenotypic spectrum without clear-cut diagnostic thresholds (30). The complexity of genotype-phenotype correlations depends on several genetic factors, among which are genetic variations due to parental consanguinity and differences in allelic expression or methylation status (54).
Affected individuals exhibit degeneration of brain and bone beginning before birth and generalized progressive wasting after birth. These features suggest that cerebro-oculo-facio-skeletal syndrome is a primary degenerative disorder affecting many cell types. The neuropathological changes and other phenotypic features are almost identical to those described in early-onset Cockayne syndrome (47; 07; 43; 06), and the two conditions are difficult to distinguish. Cerebro-oculo-facio-skeletal syndrome resides at the severe end of the spectrum, one whose heterogeneity is thought to be due to defects in DNA repair and transcription (30; 57). It has been known for some time that cell damage is important to pathogenesis. Cells from patients with cerebro-oculo-facio-skeletal and Cockayne syndrome exhibit defective growth in culture (44) and are abnormally sensitive to ultraviolet radiation (39). Cells with mutations in Cockayne syndrome group B are incapable of removing RNA polymerase II from sites of DNA damage (05). Neurodegeneration occurs in other disorders of DNA repair, including xeroderma pigmentosum (26). Because of these phenotypic similarities and the fact that patients with cerebro-oculo-facio-skeletal syndrome often have mutations in either the Cockayne syndrome group B gene (CSB) or the xeroderma pigmentosum genes (XPG, XPD, or XPF), it appears evident that these conditions (including trichothiodystrophy) should be considered part of the spectrum of nucleotide-excision repair syndromes (15; 60). All of these genes are involved in a variety of forms of DNA repair, including nucleotide excision repair, interstrand crosslink repair, and homologous recombination (56). Work has implicated DNA helicases, enzymes important to the separation of the DNA and RNA helix, and therefore, a host of genetic processes, including transcription, translation, replication, recombination, as well as repair (58). Thus, helicases play an important role in maintaining genomic stability (58). The ERCC1 and ERCC4 genes encode for the enzyme ERCC1-XPF nuclease. Deletions are incompatible with life, whereas mutations lead to a variety of disorders (38). Mutations in these genes are responsible not only for genetic diseases but also for some aging disorders and tumors. Because of the deleterious effect of ultraviolet light on cells, tumors may develop in areas of the body exposed to light, for example, carcinomas and melanomas of the skin and eyes (50). However, in another report, no increased risk of cutaneous malignancy was noted in the syndrome (64).
To date, 46 genes are known to be involved in excision repair; this large number explains the heterogeneity of disease and difficulty in diagnosis (11). It appears that nucleotide excision repair factors are also important to processes not associated with DNA damage; this may also help explain the clinical diversity of syndromes (22; 11). Perturbations in the balance between excision repair and nonexcision repair functions are thought to be responsible for this diversity (11). Interestingly, isoforms of XPD, derived from corresponding mutations, can result in genetic rescue and milder phenotypes (21). ERCC1 and ERCC2 play pivotal roles in the nucleotide excision repair pathway, which is essential for repairing bulky DNA lesions caused by ultraviolet light and other mutagens (41). Mutations in these genes result in defective repair of ultraviolet light-induced DNA damage, leading to the accumulation of unrepaired DNA lesions. This impairment is particularly detrimental during embryonic development, affecting rapidly dividing cells and resulting in widespread developmental abnormalities. ERCC5 encodes an endonuclease that excises damaged DNA, whereas ERCC6 is involved in transcription-coupled repair, a sub-pathway of nucleotide excision repair that specifically repairs genes actively being transcribed. Mutations in ERCC5 and ERCC6 disrupt these processes, leading to persistent DNA damage and triggering cell death (apoptosis) (32).
A study of 17 Iranian families identified two rare cases, including one with a UVSSA mutation and another with ERCC2-related cerebro-oculo-facio-skeletal syndrome (23). Findings align with global mutation patterns but also show population-specific differences. Another study described a pediatric patient with two unique compound heterozygous variants in the ERCC2 gene, linked to severe growth deficiency, microcephaly, facial dysmorphisms, and a brainstem pilocytic astrocytoma (01). The patient exhibited defective DNA repair following ultraviolet light exposure, indicative of impaired nucleotide excision repair due to these ERCC2 mutations. Tumor analysis revealed no cancer-related somatic mutations but identified a 2 Mb duplication in the 7q34 region, creating a KIAA1549 fusion protein, a common marker in pilocytic astrocytoma. These findings expand the clinical and genetic understanding of ERCC2-related disorders.
Cerebro-oculo-facio-skeletal syndrome is very rare (more than 1:1,000,000 live births; more than 100 cases now documented via genetic databases). The largest number of affected families has been among North American aboriginal populations of central Canada. Cases have also been reported in the United States (in both mixed-European and Afro-American backgrounds), Mexico, Finland, Italy, Egypt, Arabic countries, and Japan. Classification schemes continue to be developed (41). Autosomal recessive inheritance predominates; prenatal genetic testing via NGS panels (targeting ERCC1-8, XPA-G, etc.) enables early detection.
Genetic counseling is the best means of prevention. Protection from the sun should be pursued rigorously (14; 64).
Primary considerations are other syndromes with low birth weight, microcephaly, eye anomalies, and contractures. These include Neu-Laxova; Martsolf; Cataract, hypertrichosis, and intellectual disability; and MICRO syndromes. The latter condition is distinct from cerebro-oculo-facio-skeletal syndrome, differing in that neurologic degeneration is not as rapid and nucleotide excision repair studies in cultured fibroblasts are normal (16). Progressive microcephaly and growth failure/developmental delay are diagnostic criteria for Cockayne syndrome, especially if other findings are present, including photosensitivity, enamel hypoplasia, progressive sensorineural hearing loss, pigmentary retinopathy or cataracts, and enophthalmia (30). Syndromes with overlapping phenotypes (“overlap syndromes”) are recognized and include Fanconi anemia and Huntington-like disease (11). The molecular mechanisms responsible for these changes are under investigation and may well be heterogeneous or unexpected. For example, a mutation in ERCC6 (CSB) was found incidentally in an individual with Fanconi anemia and may have been responsible for the patient’s atypical skeletal defects (02). A study described a case of cerebro-oculo-facio-skeletal syndrome recurrence within a family, marked by prenatal findings of arthrogryposis and cataracts. Initially undiagnosed in the first case, COFS3 syndrome was confirmed in the second case through genetic testing, revealing compound heterozygous ERCC5 mutations (55).
The cerebro-oculo-facio-skeletal syndrome is diagnosed on the basis of an accurate phenotypic description with the addition of a skeletal survey and neuroradiologic work-up. MRI is valuable in identifying and following cerebral and cerebellar atrophy and loss of white matter (03). Magnetic resonance spectrometry has been used to demonstrate atrophy and hypo- or demyelination of white matter in affected patients (48). Cerebro-oculo-facio-skeletal syndrome has been suggested by ultrasound at 21 weeks (findings included microphthalmia, micrognathia, joint contractures, and rocker bottom feet) and subsequently confirmed at autopsy (42). Skin may show café au lait macules (62). Skin biopsy for fibroblast culture is successful in assessing nucleotide excision repair and ultraviolet sensitivity, whereas pertinent mutations can be identified by molecular testing (51).
Supportive care is indicated for feeding (gastrostomy), neurologic complications (poor vision and hearing, spasticity), and musculoskeletal (contractures) and respiratory complications.
Because of the poor prognosis, it is unclear if affected females will reach reproductive age. Moreover, ovarian dysgenesis in one 10-month-old female does not offer an optimistic outlook (03).
No specific information is available. Children with cerebro-oculo-facio-skeletal syndrome tolerate anesthetics for gastric, orthopedic, and ophthalmologic procedures.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Fardin Nabizadeh MD
Mr. Nabizadeh of Iran University of Medical Sciences has no relevant financial relationships to disclose.
See Profile
Harvey B Sarnat MD FRCPC MS
Dr. Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 16, 2026
Developmental Malformations
Apr. 16, 2026