




Developmental Malformations
Barth syndrome
May. 08, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
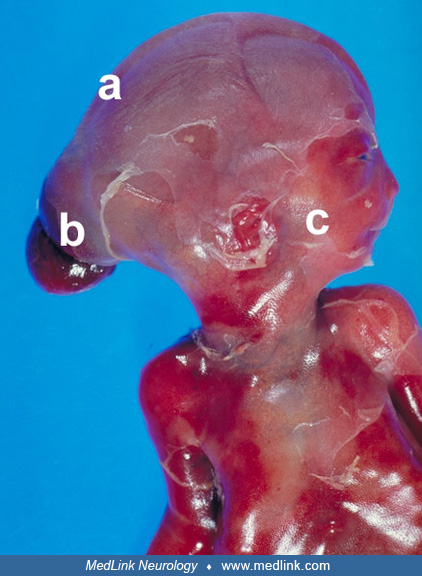
Meckel-Gruber syndrome is a lethal, inherited genetic condition with genetic heterogeneity and variable expression and is the most severe of the ciliopathies. It is generally characterized by three major findings: occipital encephalocele, postaxial polydactyly, and bilateral dysplastic cystic kidneys.
|
• Meckel-Gruber syndrome is the most common syndromic condition involving a neural tube defect. | |
|
• Phenotypic changes are highly variable. Major deficits of Meckel-Gruber syndrome include renal cystic dysplasia, hepatic fibrosis with ductal plate malformation, polydactyly, and occipital encephalocele (or less commonly, Dandy-Walker malformation, holoprosencephaly, or another CNS anomaly). | |
|
• Prenatal or perinatal demise is common. | |
|
• The disorder is genetically heterogeneous and is inherited on an autosomal recessive basis, with a recurrence risk of 25%. |
Thought to be originally described by Johann Friedrich Meckel in a pair of siblings in 1822, the syndrome was further defined by Grüber in 1934 and labeled "dysencephalia splanchnocystica" (68; 40). Evidence suggests, however, that Christopher Krahe described a similar syndrome in a child born in Denmark in 1684 (49). Much later reports described features of 13-15 trisomy syndrome with normal karyotype (63; 69) or a familial polydactyly with central nervous system dysplasia (102). The identity of this syndrome was established in 1969 by Opitz and Howe, who described one case and thoroughly reviewed 43 observations reported in the literature to date (73). They proposed the eponym of "Meckel syndrome."
A striking combination of clinical features characterizes the syndrome, which is lethal when fully expressed. “Formes frustes” are frequent, sometimes in association with other malformation syndromes, and they are difficult to differentiate from other disorders (67; 61). Helpful diagnostic criteria were suggested by Fraser and Lytwyn and further refined by Salonen, Blankenberg, and colleagues and Ahdab-Barmada and Claassen (34; 86; 14; 04). Frequently used eponyms include "Meckel syndrome," "dysencephalia splanchnocystica of Grüber," or "Meckel-Grüber's syndrome."
Typical clinical features of the Meckel-Gruber syndrome include posterior encephalocele, polydactyly, and polycystic kidneys.
In 1822, Meckel described the syndrome in two siblings, a boy and a girl, with microcephaly, sloping forehead, short neck, flat occiput, posterior exencephalocele, cleft palate, heptadactyly with syndactyly, large polycystic kidneys, small adrenal glands, short intestine with malrotated colon, and a small fibrotic liver. In addition, the boy had cryptorchidism (68). In 1854, Vrolik described two siblings with similar malformations, whereas in 1930, Cornelia DeLange described two female infants with cleft palate, fused fifth and sixth fingers, microcephaly with a sloping forehead and a large posterior encephalocele extruding from the posterior fontanelle, and polycystic abnormal kidneys with fibrosis and small adrenal glands. An absence of olfactory bulbs and hypoplasia of sella turcica, with variable dysplasia of the nervous system, was present in one case. The syndrome was better outlined in 1934 by Grüber, who described seven cases with various combinations of polydactyly, cystic kidneys and liver, cleft palate, "mild" arhinencephaly, abnormal eyes, sloping forehead with microcephaly, and posterior exencephalocele (40). Numerous authors, as listed by Opitz and Howe in 1969, described isolated case reports between 1854 and 1969 with similar features (73).
Postaxial polydactyly, cystic dysplastic kidneys, and hepatic bile duct proliferation with liver fibrosis were suggested as the most characteristic features (86; 14); a spurious morphologic heterogeneity of central nervous system anomalies has been described (43; 75; 64; 95). An attempt was made to delineate a constant pattern of central nervous system dysgenesis typical for this syndrome (04). Consistent features of central nervous system malformation include a combination of prosencephalic dysgenesis (arhinencephaly-holoprosencephaly and related midline anomalies) with occipital exencephalocele (an extrusion of the diencephalic-rhombencephalic dilated roof through the posterior fontanelle), expressing an associated rhombic roof dysgenesis.
Microcephaly, sloping forehead, eye anomalies, cleft or high-arched palate, and micrognathia reflect the underlying prosencephalic-rhombencephalic dysgenesis.
Malformations are lethal when fully expressed. Postnatal outcome in patients with encephalocele depends on the location and size of the cele and the presence of additional malformations (105). In a review of 36 fetuses with encephalocele, five were diagnosed with Meckel-Gruber syndrome; a total of six underwent surgery postnatally, but none were neurologically intact (23). Microcephaly with prosencephalic and rhombencephalic dysgenesis may be severe, and cystic dysplastic kidneys result in early renal failure. Associated liver fibrosis and pulmonary and cardiac malformations are also often lethal in the first hours of life. Rare reported patients with incomplete expression of the phenotype have survived months or even years (61; 72).
Typical case reports of newborn infants with Meckel-Gruber syndrome have been published (73; 04; 94). Infants with the fully expressed syndrome at birth do not survive, and no clinical symptoms are specific and diagnostic. A case in point is an infant delivered at 32 weeks’ gestation to a gravida 1, para 0, 14-year-old mother. Ultrasound diagnoses of oligohydramnios, microcephaly, and probable encephalocele had been made before delivery. Apgar scores were 1 at one minute and 1 at five minutes. Because of multiple congenital anomalies, no resuscitative measures were undertaken, and the infant died a few minutes after delivery.
Management is the only alternative to alleviate the clinical effects of the expressed malformation, such as respiratory distress, seizures, apnea, and severe metabolic acidosis–the result of pulmonary hypoplasia with respiratory insufficiency and hypoxia-anoxia. Shunting of hydrocephalus may result in a good early outcome (38).
Visualization of the surface anatomy of fetuses in utero at early gestational ages (7 to 13 weeks) may permit diagnosis of the malformation (80; 17; 89), allowing the choice of early termination of the pregnancy.
Meckel-Gruber syndrome is a type of ciliopathy. The etiology of Meckel-Gruber syndrome is heterogenous, with some 14 known disease genes, such as MKS1, MKS2/TMEM216, MKS3/TMEM67, TMEM231, CEP120, CEP290, RPGRIP1L, and CCD2A (104). Prevalences of the most common genes are C5orf42 (12% of cases), TMEM67 (10%), and AHl1 (8%) (50). One case of maternal uniparental disomy involving chromosome 8 has been reported (16). All of the encoded proteins are implicated in the function of primary “immotile” cilia (60). Therefore, Meckel-Gruber syndrome is placed in the expanding group of ciliopathies, one that also includes nephronophthisis, Joubert syndrome, and Bardet-Biedl syndrome. At least 89 loci have been implicated in the ciliopathies (66). Liu and colleagues state that cilia are found on almost all cells of the human body, and ciliopathies are characterized by the involvement of a core set of tissues--retinal, cerebral, and renal--that cause considerable dysfunction for the patient. (59).
These are some of the known genes related to the pathophysiology of Meckel-Gruber syndrome: MKS1, MKS2/TMEM216, MKS3/TMEM67, CEP290, RPGRIP1L, CCD2A, and TCTN2 (109). Novel variants in these genes continue to be identified (81). All of the encoded proteins are implicated in the function of primary “immotile” cilia (60). Meckel-Gruber syndrome is believed to be caused by dysfunction of primary cilia during early embryogenesis (01). Primary cilia are involved in signal transduction and play important roles in normal neurodevelopment. Mutations in genes responsible for Meckel-Gruber syndrome have been localized to the transition zone of the cilia. Srivastava and colleagues have seen a trend in which genes clustering in the same ciliary compartment tended to cause a similar phenotype (93).
Kyttala and colleagues identified the MKS1 gene (52). Their study involved Finnish families in whom the MKS1 locus was identified. In 70% of them, the patients were homozygous for the same haplotype on chromosome 17q23. DNA sequencing revealed a 29 bp deletion in intron 15. Analysis of 26 Finnish families with the common founder haplotype confirmed that all affected individuals were homozygous and parents were heterozygous for the deletion (now known as MKS1-Fin major mutation). Further work has shown that genes are highly conserved across species, including zebrafish, platypus, mouse, rat, chicken, wolf, and human (108). MKS1 is widely expressed in the fetal brain, liver, kidney, and digits (52). MKS1 has proved to be required for ciliogenesis in a kidney model (25). In 2025, Campobasso and colleagues identified a case report of a patient with a homozygous variant in the B9D1 that was previously known as the MKS1 gene (18). This gene has provided further insight into the pathophysiology of ciliary proteins.
MKS2 was mapped to chromosome 11q13. Mutations in the transmembrane protein TMEM 216 are identified as the causative MKS2 gene (100). TMEM216 localizes to the basal body, ciliary membrane, and microtubule structures and may have a role in signal transduction (100).
Smith and colleagues refined MKS3 mapping, suggesting a candidate gene TMEM67 (92). The gene is expressed in fetal brain, liver, and kidney. It encodes a transmembrane protein called “meckelin,” which localizes to the primary cilium and plasma membrane in ciliated cell lines (33). The description of this gene helps elucidate the role of meckelin in normal human development as well as in neural tube defects. Dawe and colleagues proposed that MKS1 and meckelin interact and are required for primary ciliary formation (25). Ahmed and colleagues found that TMEM67 is a component of the ciliary transition zone and works with ADAMTS9 for normal ciliogenesis (05).
MKS4 involves mutation of the protein CEP290 from chromosome 12q21.3. CEP290 localizes to the centrosomes of mitotic cells. CEP290 may have a role in primary cilium assembly (21). There are over 100 known CEP290 mutations. Mutations in CEP290 cause a broad range of ciliopathies, including Joubert syndrome, Senior-Loken syndrome, Leber congenital amaurosis, Bardet-Biedl syndrome, as well as Meckel-Gruber syndrome (60). There is extreme phenotypic variance of CEP290 mutations. Nine mutations in CEP120 have also been identified in patients with Joubert syndrome, Meckel-Gruber syndrome, and other ciliopathies (84).
RPGRIP1L (retinitis pigmentosa GTPase regulator-interacting protein-1 like) is a ciliary gene that is mutated in Meckel-Gruber and Joubert syndromes (48). This is MKS5 located on chromosome 16q12.2. RPGRIP1L may interact with CEP290 (19). Valentini and colleagues determined that mutations in this gene are associated with the most severe phenotype, which includes anencephaly and shortening and bowing of the long bones (101).
MKS6/CC2D2A on chromosome 4p15 was identified by Tallila and colleagues (97). CC2D2A is implicated in regulated intracellular calcium responses. Truncating mutations of CC2D2A cause Meckel-Gruber syndrome. Missense mutations cause Joubert syndrome (71).
TMEM17 was seen in a case of severe prenatal phenotype. This most severe phenotype was observed with loss of function in the homozygous state (76).
The ciliary transition zone continues to be elucidated. It is a diffusion barrier that organizes or compartmentalizes signaling proteins; mutations in transition zone proteins lead to Meckel-Gruber syndrome and other ciliopathies (26; 54; 56).
In Meckel-Gruber syndrome, the variety of defects described seems to represent scattered malformations, often noted in syndromes such as trisomy 13, triploidy, Smith-Lemli-Opitz syndrome, hydrolethalus syndrome (87), and C syndrome (08). However, a study of the pathogenetic characteristics of anomalies reported with Meckel-Gruber syndrome outlines a unifying trend in the pattern of somatic malformations: dysplastic cystic kidneys in Meckel syndrome are different from usual congenital polycystic kidneys in that dysplasia is a prominent feature in Meckel syndrome but is not usually found in polycystic kidney disease. Aberrant Wnt signaling, implicated in the pathogenesis of renal cysts, arises from mutations in ciliopathy genes (12). Exact mechanisms remain under study but include deficient metanephric differentiation (13), a lack of stromal-epithelial interaction (14), and altered cell proliferation or polarity (12). Skeletal anomalies arise from abnormal ciliary functioning in the Hedgehog pathway (44). With regard to CNS anomalies, the interaction between a perinotochordal matrix substance, prechordal mesoderm, and the overlying developing neuroectoderm may be defective (04). The major sites of somatic malformations in Meckel-Gruber syndrome are at the rostral and caudal ends of the notochord, where mesoderm and neuroectoderm are still closely apposed.
Incomplete induction of rostral prechordal mesoderm results in facial and brain anomalies, whereas more caudal impairment results in renal and genital anomalies. Such errors of morphogenesis within a defined embryologic developmental field have been considered downstream pattern-related events but were redefined as "syndromal pleiotropy" (65). Associated malformations reflect more diffuse impairment of such stromal-epithelial interaction. Similarly, involvement of homeobox genes coding for clusters of genes with specific anterior-to-posterior expression domains in the embryo is possible (15; 85). A close relation between cell adhesion molecules and Hox genes has been suggested (31). Early attempts at identifying the gene locus for Meckel syndrome using a genome-wide linkage study in five affected Finnish families pointed to markers assigned to chromosome 17, telomeric to the homeobox B region (74). It is interesting to note that many neuronal growth factors are located in this critical chromosomal region. Studies have outlined the role of transforming growth factor-beta3 in the abnormal lung development and cleft palate induction seen in mice lacking transforming growth factor-beta3, implicating this growth factor in epithelial-mesenchymal interaction (47; 78). Further implication of this growth factor and related proteins in dorsal differentiation and neuronal patterning of the roof of the neural plate has been reported (57). Bone morphogenetic proteins are required for mesoderm formation and patterning and are implicated in the induction of forebrain midline development (24; 36; 39) and notochord-mesodermal patterning (22). Primary cilia are implicated in the development of the corpus callosum in a mouse model of Meckel-Gruber syndrome (53).
Although the triad of polycystic dysplastic kidneys, occipital encephalocele, and postaxial polydactyly has traditionally been accepted as diagnostic of Meckel-Gruber syndrome (73), a working definition for the diagnosis of this syndrome was suggested by Fraser and Lytwyn in 1981. This included cystic kidney dysplasia plus at least two other defects, including "relevant" anomalies of the brain, face, eyes, lip, liver, genitalia, heart, and spleen (34). A study of 67 patients with Meckel-Gruber syndrome showed cystic dysplasia of the kidneys in all cases (86). Typical hepatic fibrosis with proliferation and dilatation of the bile ducts was also found in each of the 41 cases with liver available for study. Occipital encephalocele was observed in 57 of the 67 patients, usually with microcephaly. Polydactyly was present in 66 of the 67 patients. Additional anomalies include hypoplasia or aplasia of genitalia in boys; hypoplasia or atresia of the uterus and vagina in girls (02); hypoplasia or aplasia of the urinary tract, lungs, adrenal glands, or spleen; gastrointestinal malrotations; cystic or fibrotic changes of the pancreas; and heart defects in a smaller number of patients. In a comprehensive review by Ahdab-Barmada and Claassen in 1990 of all central nervous system anomalies reported in cases of Meckel-Gruber syndrome, a triad of major findings was outlined: (1) prosencephalic dysgenesis, including arhinencephaly, holoprosencephaly, agenesis of the corpus callosum, fused thalami, septo-optic dysplasia with small optic nerves, microphthalmia, small or absent pituitary gland, and cleft or high-arched palate; (2) rhombic roof dysgenesis, including mesodermal and neuroectodermal hypoplasia reminiscent of Chiari and Dandy-Walker malformations, absent brainstem tectum, cerebellar hypoplasia, agenesis-dysgenesis of cerebellar vermis, elongated flattened brainstem, aqueductal stenosis, and diverse glioneuronal heterotopia; and (3) occipital encephalocele, in association with mesodermal and rhombic roof dysgenesis, often with microcephaly and sloping forehead, sometimes to such a severe degree as to be labeled anencephaly in macerated fetuses (04). The association of a Dandy-Walker type of posterior fossa dysgenesis has been emphasized (103; 07; 107; 20), whereas the association of Chiari malformation was reported with an otopalatodigital syndrome (45). Syringomyelia has been reported in two fetuses (41).
Meckel-Gruber syndrome is a rare autosomal recessive disorder, with a prevalence estimated at one in 135,000 live births worldwide (42). However, the prevalence is much higher in some areas, especially those with increased consanguinity. In Finland, the birth prevalence is one in 9000, with a disease gene frequency of 0.01 (74). In some Belgian populations, the prevalence is estimated at one in 3000 (06).
No means of prevention are known. Prenatal diagnosis, particularly of CNS anomalies, begins with ultrasonography (89) and may be confirmed with fetal MRI (29).
Meckel-Gruber syndrome is an autosomal recessive disorder with variable expression of the mutant genes. Although the triad of cystic dysplastic kidneys, polydactyly, and occipital encephalocele is typical and diagnostic of this syndrome, many subtypes are recognized (see OMIM classification below). Such cases may mimic other genetic syndromes (67; 35).
Ciliopathies, or defects in the primary cilia, have been associated with a broad spectrum of more than 35 human genetic diseases. Liu and colleagues state that there is extreme heterogeneity and displays of allelism with other ciliopathies, such as Joubert syndrome, COACH syndrome, Bardet-Biedl syndrome, and oro-facial-digital syndrome (59).
COACH syndrome (oculo-encephalo-hepato-renal syndrome) exhibits both phenotypic and genotypic overlap with Joubert syndrome (96). In fact, Baala and colleagues discovered the MKS3 gene mutation in four patients with Joubert syndrome, suggesting that these are allelic disorders (09). Goldston syndrome represents a milder variant of Meckel-Gruber syndrome. In the condition, also known as cerebrorenal syndrome or Meckel syndrome type 7, one typically sees Dandy-Walker malformation and cystic renal dysplasia, which may appear as polycystic renal disease, and variable occurrence of hepatic fibrosis (03). Considerable overlap between phenotypes and genotypes in all of the foregoing syndromes obviously complicates diagnosis and awaits further delineation.
Turkyilmaz and colleagues have determined that many ciliopathies are allelic diseases, including Joubert syndrome, COACH syndrome, Bardet-Biedl syndrome, oro-facial-digital syndrome, and Meckel-Gruber syndrome (98). Lin and colleagues have even indicated that there is a proposed genotype-phenotype correlation; two null alleles of MKS1 result in Meckel-Gruber syndrome, one null allele and a non-truncating allele result in Joubert syndrome, and two non-truncating alleles result in Bardet-Biedl syndrome (58).
The other syndromes that may include features of ciliopathies are vast.
Microphthalmia, colobomas, and arhinencephaly in infants with polydactyly of the postaxial type and polycystic kidneys mimic the phenotype of trisomy 13. However, a meningoencephalocele is usually not seen with the trisomy 13 syndrome. Marshall and colleagues in 1964 and Miller and Selden in 1967 reported newborn infants with arhinencephaly, encephalocele, and 13-15 trisomy syndrome with normal karyotype (63; 69). Mecke and Passarge reviewed such cases in 1971, and they asserted that Meckel syndrome, as defined by Opitz and Howe, had a specific genetic identity and was not to be confused with similar phenotypes of other genetically different disorders (67). Familial occurrence of phenotypes simulating trisomy 18 (91) and Turner syndrome (55) are also reported.
Differential diagnosis with the Smith-Lemli-Opitz syndrome, which is a relatively common malformation syndrome, with a minimal incidence of one in 40,000 live births (61), relies on the presence of occipital encephalocele and cystic dysplastic kidneys in Meckel-Gruber syndrome. Microcephaly with holoprosencephaly, congenital cataracts, and hypoplastic kidneys with small cysts are more characteristic of the Smith-Lemli-Opitz syndrome, in association with postaxial polydactyly and syndactyly, more severe congenital heart defects, and common abnormalities of external genitalia. Similarly, the hydrolethalus syndrome (87) has been described as an occasional manifestation in the Smith-Lemli-Opitz and other syndromes and may be differentiated from Meckel-Gruber syndrome by the absence of occipital encephalocele or meningocele and the frequent lack of pulmonary lobation. Another closely associated congenital disorder is the C syndrome with trigonocephaly (08), but with an absence of occipital encephalocele and large cystic kidneys. Occipital encephalocele, polydactyly, or both have occasionally been reported in some patients with Joubert syndrome (09).
Prenatal diagnosis of Meckel-Gruber syndrome using ultrasonography is the norm and is possible as early as 10 weeks. The combination of occipital encephalocele, polydactyly, facial clefting, and increased renal size is typical of this syndrome (88; 82; 27; 30; 17). However, the diagnosis may be difficult when variability in the clinical expression of the phenotype is encountered (37). For example, congenital heart disease is present in a subset of patients (51), and unilateral renal agenesis is recognized, albeit infrequently (99). Single cases with megaurethra and female pseudohermaphroditism have been reported in affected fetuses (28). Diagnosis of Dandy-Walker malformation or variant may be particularly challenging (77). For this reason, a workup using both genotypic and phenotypic information is advisable (10). Transabdominal thin-gauge embryofetoscopy may offer excellent visualization of the surface anatomy of fetuses at early gestational age (seven to 13 weeks) (79). Meckel-Gruber syndrome can be diagnosed by first-trimester ultrasonography (89; 46; 83). With the discovery of causative gene mutations, gene testing is available and is evolving rapidly as technology is developed. Valentini and colleagues suggest that molecular diagnostic strategies include targeted multigene panel testing that can be expanded to exome if the panel is negative (101). Genetic counseling is imperative.
Genetic counseling of involved families is indicated. No surgical intrauterine treatment of cerebral or renal malformations has been attempted when prenatal diagnosis of Meckel-Gruber syndrome is made. In one large European review, therapeutic termination of pregnancy was chosen in over 75% of cases diagnosed prenatally (11).
Fontes and colleagues reviewed cases of patients with Meckel-Gruber syndrome who underwent neurosurgery. The longest patient survival was 11 months, and the median survival was 6 months of age (32).
Meckel-Gruber syndrome is an autosomal recessive congenital disorder. Responsible gene defects have been discovered. No teratogens have been implicated in the etiology of this syndrome. There is no known complication to the pregnant mother carrying a malformed fetus. Antenatal detection of the Meckel-Gruber syndrome in only one dizygotic twin fetus following in vitro fertilization and embryo transfer has attracted attention to the risks and difficulties associated with pregnancy termination in cases of multiple pregnancies with assisted reproduction (90). Xu and colleagues reported the first case of preimplantation genetic testing for a novel homozygous TXNDC15 variant (106).
It may be difficult to intubate a patient with Meckel-Gruber syndrome. Patients may have microcephaly, cleft lip or palate, mandibular hypoplasia, and other pertinent anatomic abnormalities. The challenges of airway management during general anesthesia have been described (70).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Robin Godshalk MS MHA
Dr. Godshalk of the Atlantic Health System in Morristown, New Jersey, has no relevant financial relationships to disclose.
See Profile
Harvey B Sarnat MD FRCPC MS
Dr. Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 16, 2026