Neurobehavioral & Cognitive Disorders
Mental status examination
Jun. 17, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
This article reviews the clinical, biochemical, and molecular genetic characteristics of cerebrotendinous xanthomatosis. Cerebrotendinous xanthomatosis is an autosomal recessive disorder caused by mutations affecting the mitochondrial enzyme sterol 27-hydroxylase, with resulting cholestanol and cholesterol accumulation in virtually every tissue. There is variable clinical severity, with onset from infancy to adulthood. Typical clinical signs include early-onset cataract, tendon xanthomas, and neurologic signs that include progressive adult-onset dementia, psychiatric disturbances, and ataxia. Early treatment with chenodeoxycholic acid is beneficial.
|
• Cerebrotendinous xanthomatosis (MIM 213700) is caused by a deficiency of the mitochondrial enzyme sterol 27-hydroxylase. | |
|
• Sterol 27-hydroxylase (CYP27A1, NP_000775.1) is a key enzyme in the bile acid biosynthesis pathway. | |
|
• Cerebrotendinous xanthomatosis is a rare lipid-storage disease characterized by abnormal deposition of cholestanol and cholesterol in multiple tissues. | |
|
• Cerebrotendinous xanthomatosis is a treatable metabolic disease that requires early molecular diagnosis to prevent the dramatic, progressive, and unnecessary neurologic deterioration. | |
|
• Treatment with chenodeoxycholic acid may slow the progression of the disease and reverse symptoms in some patients. |
Cerebrotendinous xanthomatosis was first described in 1937 by Van Bogaert and colleagues, who reported two cousins with a slowly progressive neurologic syndrome with cognitive and motor impairment and cataracts (145). Tendon xanthomas were evident in only one patient at autopsy. The disease was described as a “cholestérinose généralisée.” Thirty years later, Menkes and colleagues identified the stored material as cholestanol, a metabolite of cholesterol present only in small amounts in controls (91). Since then, an increased serum cholestanol level has been a crucial diagnostic marker of the disease. In 1971, Salen reported that xanthomatosis was associated with defective bile acid pattern, with very low concentrations of chenodeoxycholic acid (124). In 1974, Setoguchi described a defect in bile acid biosynthesis, with incomplete oxidation of the C27-steroid side chain and with a greatly increased excretion of bile alcohols in bile, feces, and urine (131). In 1980, Oftebro and colleagues reported that a defect in the mitochondrial enzyme sterol 26-hydroxylase (currently designated 27-hydroxylase) is the cause of cerebrotendinous xanthomatosis (106). These authors detected absent sterol 26-hydroxylase activity in a liver biopsy from a subject with cerebrotendinous xanthomatosis. In 1984, Berginer and colleagues reported encouraging results of long-term chenodeoxycholic acid treatment on metabolic and clinical alterations.
The human CYP27A1 gene was localized on the long arm of chromosome 2 (26), and the first mutations were described (25). The structure of the CYP27A1 gene has been established by Leitersdorf and colleagues (84). To date, more than 50 different mutations within the CYP27A1 gene have been reported worldwide (57) (Human Gene Mutation Database at the Institute of Medical Genetics in Cardiff).
|
• Cerebrotendinous xanthomatosis usually manifests in the first decade of life with diarrhea, cataracts, and mild intellectual disability. | |
|
• Adolescent-to-young-adult-onset tendon xanthomas and adult-onset progressive neurologic dysfunction are typical manifestations of cerebrotendinous xanthomatosis. | |
|
• Tendon xanthomas, which are a hallmark of cerebrotendinous xanthomatosis, usually appear in the second or third decade of life. | |
|
• The most common neurologic abnormalities in patients with cerebrotendinous xanthomatosis are (in descending order) corticospinal tract abnormalities, ataxia, cognitive decline, and gait difficulty. |
Cerebrotendinous xanthomatosis (CTX) is an autosomal recessive lipid storage disorder that manifests with a wide range of systemic symptoms including neonatal jaundice or cholestasis, refractory diarrhea, juvenile cataracts, tendon xanthomas, osteoporosis, coronary heart disease, and multiple neurologic and neuropsychiatric manifestations (81).
Cerebrotendinous xanthomatosis usually manifests in the first decade of life with diarrhea, cataracts, and mild intellectual disability (81). Diarrhea develops within the first year of life, followed by motor and psychiatric symptoms, then cataracts and school difficulties between 5 and 15 years of age (38). Adolescent-to-young-adult-onset tendon xanthomas and adult-onset progressive neurologic dysfunction are typical but not universal manifestations. The clinical course of the disease is slowly progressive. Individual and intrafamilial variability is considerable (100; 149; 49; 96; 02). Considerable clinical heterogeneity can occur even in identical twins (163). Early treatment with chenodeoxycholic acid may stabilize or improve metabolic and clinical abnormalities (127).
In a review of the clinical features in 55 individuals with cerebrotendinous xanthomatosis by Mignarri and colleagues, cataracts were present in 49 (89%), tendon xanthomas in 43 (78%), osteoporosis in 20 of 30 assessed (67%), and diarrhea in 22 (40%). Neurologic involvement included peripheral neuropathy in 21 of 30 assessed (70%), paraparesis in 35 (64%), intellectual disability in 33 (60%), psychiatric disturbances in 24 (44%), ataxia in 20 (36%), seizures in 18 (33%), and parkinsonism in five (9%) (93).
A study of four Chinese pedigrees reported xanthomas and pyramidal signs in all identified cases, cerebellar ataxia in 67%, cognitive impairment in 67%, cataracts in 50%, peripheral neuropathy in 33%, and chronic diarrhea in only 6% (29). In a review of the clinical features in 43 individuals with cerebrotendinous xanthomatosis by Duell and colleagues, chronic diarrhea was present in 23 (53%), cognitive impairment in 32 (74%), premature cataracts in 30 (70%), tendon xanthomas in 33 (77%), neurologic disease in 35 (81%), and premature cardiovascular disease in three (7%) (47).
A multicenter, cross-sectional descriptive study documented the clinical characteristics of cerebrotendinous xanthomatosis patients of different ages, clinical presentations of early-diagnosed patients, and responses to short-term chenodeoxycholic acid treatment (76). Of 11 patients, three (27%) had neonatal cholestasis, seven (64%) had a history of frequent watery defecation that started in the infantile period, and eight (73%) had juvenile cataract. Seven (64%) were diagnosed in childhood. Four adults had pyramidal signs and parkinsonism symptoms.
In a retrospective, multicenter, descriptive study of16 clinically and genetically confirmed cerebrotendinous xanthomatosis cases, common clinical features included cognitive decline (75%), learning difficulties (69%), diarrhea (56%), cataracts (56%), gait issues (50%), and behavioral changes (44%) (02). Childhood diarrhea was strongly associated with earlier diagnosis. Initial misdiagnosis occurred in three patients, with an average diagnostic delay of 6.1 years, but shorter for children (2.7 years) compared to adults (11.6 years). Tendon xanthomas were observed in only one patient. Genetic testing identified seven CYP27A1 variants, highlighting genetic heterogeneity in this population.
The natural history varies for different phenotypes of cerebrotendinous xanthomatosis (ie, the classical, spinal, and “non-neurological” forms) (105); different symptoms and signs also have typical time windows of onset.
In general, those with adult onset have milder phenotypes with less or no evident neurologic involvement in many (167).
Natural history, disease progression, and symptom and sign timeline in each phenotype of cerebrotendinous xanthomatosis (classical, spinal, and “non-neurological” forms), according to age of symptom onset. (Source: Nobrega PR, ...
Diagnosis is often markedly delayed from symptom onset (167).
Xanthomas. Tendon xanthomas, which are a hallmark of the disease, usually appear in the second or third decade of life. First estimated to be almost invariably present (18; 29), they were found in 78% of patients in a study of 55 cases (93) and less than 50% of a series of Dutch patients (152). They are usually localized at the Achilles tendon; however, they have also been described on the extensor tendons of the elbow and hand, as well as the patellar and neck tendons. Xanthomas have been reported in the lung, bones, and central nervous system. Adult patients with cerebrotendinous xanthomatosis may present with tendon xanthomas as the sole or predominant feature, mimicking familial hypercholesterolemia (135). Diagnosis may be further delayed in patients with the cerebrotendinous xanthomatosis sans xanthomas (58).
Enterohepatic system. Chronic intractable diarrhea from infancy may be the earliest clinical manifestation of cerebrotendinous xanthomatosis (152; 31). Neonatal cholestasis has been reported (39). Gallstones have occasionally been reported.
Eye. In most affected individuals, cataracts are the first clinically documented finding, often appearing in the first decade of life (152; 143; 07; 52).
(Source: Fernandez-Eulate G, Martin GC, Dureau P, et al. Prospective cholestanol screening of cerebrotendinous xanthomatosis among patients with juvenile-onset unexplained bilateral cataracts. Orphanet J Rare Dis 2022;17[1]:434...
(Source: Fernandez-Eulate G, Martin GC, Dureau P, et al. Prospective cholestanol screening of cerebrotendinous xanthomatosis among patients with juvenile-onset unexplained bilateral cataracts. Orphanet J Rare Dis 2022;17[1]:434...
Cataracts may be visually significant opacities requiring lensectomy or visually insignificant cortical opacities. The appearance can include irregular cortical opacities, anterior polar cataracts, and dense posterior subcapsular cataracts (33). Other ophthalmological findings include palpebral xanthelasmas (113), optic nerve atrophy (128), and proptosis (99). In a series of patients reported by Dotti and colleagues (age range 32 to 54 years), ocular manifestations included cataracts in all cases, optic disc paleness in about 50%, and signs of premature retinal senescence with retinal vessel sclerosis in 30% (44). Cholesterol-like deposits along vascular arcades and myelinated nerve fibers were present in some patients.
Cardiovascular system. Premature atherosclerosis and coronary artery disease have been reported (144; 56). Lipomatous hypertrophy of the atrial septum has been described (43; 56).
Skeleton. Bone involvement is characterized by granulomatous lesions in the lumbar vertebrae and femur, osteopenia with increased risk of bone fractures, and impaired adsorption of calcium, which improves with chenodeoxycholic acid treatment (19; 50). Osteopenia is evident by total body densitometry in untreated individuals. Some patients may have marked thoracic kyphosis.
Endocrine abnormalities. Hypothyroidism has occasionally been reported (113; 22; 70).
Premature aging signs. Early-onset cataract, osteopenia with bone fractures and loss of teeth, atherosclerosis, and neurologic impairment with dementia or parkinsonism that are associated with the characteristic facies may suggest a generalized premature aging process (45).
Other systemic manifestations. In one series of 12 patients, a malar rash was present in 75%, but it resolved with chenodeoxycholic acid treatment (160).
The neurologic manifestations may include ataxia, dystonia, epilepsy, intellectual disability, dementia, peripheral neuropathy, parkinsonism, and intention or sometimes rest tremor (27; 51; 160; 167). In a systematic review of 91 cases of cerebrotendinous xanthomatosis, the most common neurologic abnormalities were corticospinal tract abnormalities (60%), ataxia (59%), cognitive decline (46%), and gait difficulty (38%); 68 (35.0%) had baseline cognitive problems (156). Ataxia, gait difficulties, and corticospinal tract abnormalities develop throughout life, whereas cognitive decline tend to begin later in life (156). Although less common, neurologic abnormalities, seizures, psychiatric changes, and speech changes develop throughout life, whereas parkinsonism and sensory changes generally begin later in life (156). The absence of neurologic symptoms at onset does not rule out the development of future neurologic symptoms (135).
Mental retardation and dementia. Mental retardation, or dementia following slow deterioration in intellectual abilities in the second decade of life, occurs in over 50% of individuals (152; 93; 29). Some individuals show mental impairment from early infancy, whereas the majority has normal or only slightly subnormal intellectual functions until puberty (61). In some cases, cognitive functions are nearly normal.
In a French multicenter cohort study of 27 patients, 44% (4 of 9) of children and 78% (14 of 18) of adults exhibited cognitive impairment, which could be severe (123). Subjects had significant impairment in executive, attentional, language, and visuo-spatial domains. Among adults, 16% (3 of 18) developed dementia after age 50; these patients had delayed chenodeoxycholic acid treatment and cerebral atrophy. Other than these three subjects, cognitive function was fairly stable, suggesting that cognitive impairment is usually due to a neurodevelopmental disorder that persists into adulthood. Cognitive impairment was less common in children, likely related to early chenodeoxycholic acid treatment in this cohort. The severity of magnetic resonance imaging abnormalities did not predict cognitive impairment.
Neuropsychiatric symptoms. Neuropsychiatric symptoms such as behavioral changes, personality disorders, agitation, aggression, depression, suicide attempts, hallucinations, and psychotic disorders may be prominent (53; 61).
Pyramidal and cerebellar signs. Pyramidal signs (ie, weakness and spasticity) and cerebellar signs (eg, ataxia) are almost invariably present starting at 20 or 30 years of age (73; 161; 29), although some studies reported lower frequencies (93).
A spinal form occurs in which spastic paraparesis is the main clinical symptom (150; 10; 158; 142; 06; 58; 64; 95). In the absence of xanthomas, the diagnosis of spinal cerebrotendinous xanthomatosis can be easily missed or delayed (06; 58).
Extrapyramidal manifestations. Extrapyramidal manifestations, including dystonia and atypical parkinsonism, corticobasal syndrome, postural tremor, ataxia, and myoclonus have occasionally been reported (41; 101; 62; 82; 121; 129; 93; 161; 138; 08). Parkinsonism is the most frequently reported movement disorder in patients with cerebrotendinous xanthomatosis, followed by dystonia, myoclonus, and postural tremor. Movement disorders are mixed in a quarter of the cerebrotendinous xanthomatosis patients with movement disorders and are usually part of a complex clinical picture (138). A movement disorder may be the presenting symptom in a significant proportion of cases (138).
Other neurologic and psychiatric manifestations. Other neurologic and psychiatric manifestations include epilepsy, dysphagia, palatal tremor, dysarthria, peripheral neuropathy, and hallucinatory psychosis resembling schizophrenia (75; 111; 141; 60; 93; 104; 29; 40; 165; 85; 72; 108).
If untreated, cerebrotendinous xanthomatosis is a slowly progressive lethal disease (145; 91; 124; 13; 133; 11). Long-term treatment may stop the deterioration and improve the neurologic functions (127; 28). Neurologic improvement is more likely when treatment is started in young patients (16; 14; 11; 146). Decrease of the size of tendon xanthomas was reported (102; 154). Complications of cerebrotendinous xanthomatosis include deterioration of vision, recurrent falls and bone fractures, urinary tract infections, pneumonia, cachexia, and heart attacks.
Case 1. A 40-year-old man had a 2-year history of progressive difficulty with walking and reduced balance (88). Physical examination demonstrated bilateral egg-sized, hard, smooth, and painless masses in the Achilles tendons. Neurologic examination showed mild cognitive and language impairment, gait ataxia, inability to perform tandem gait smoothly, mild muscle hypertonia, and pathologically brisk muscle stretch reflexes with extensor plantar responses and ankle clonus bilaterally.
MRI of the brain showed symmetric hyperintensities in the posterior limbs of internal capsules, paraventricular white matter, cerebral peduncles, and anterior pons on T2W and FLAIR images, with corresponding hypointensities on T1W images, which are along corticospinal tracts.
CT and MRI revealed cerebellar parenchymal abnormalities consistent with cerebrotendinous xanthomatosis.
MRI of an Achilles tendon xanthoma showed fusiform enlargement of the right Achilles tendon. The xanthoma was relatively isointense compared to muscle. An axial proton density image shows diffuse fat infiltration with low-intensity tendon bundles interspersed within.
An unspecified mutation was identified in the CYP27A1 gene.
He was started on chenodeoxycholic acid treatment (250 mg three times per day). His xanthomas of the Achilles tendons began to diminish with treatment, but no improvement was observed in his neurologic signs after more than 3 months of treatment.
Case 2. A 30-year-old Indian man, born to second-degree consanguineous parents, presented with progressive incoordination and gait ataxia since puberty (89). Examination revealed moderate intellectual disability, an immature cataract in the right eye, pseudophakia in the left eye, bilateral tendon xanthomas on the Achilles and patellar tendons, and cerebellar ataxia.
Arrows indicate bilateral Achilles tendon xanthomas. (Source: Mahadevan N, Thiruvadi V, Paranthakan C, Rejha A, Magesh A. Cerebrotendinous xanthomatosis: report of two siblings with the same mutation but variable presentation. ...
Laboratory investigations further revealed normal hematological parameters, a basic metabolic profile, and a normal lipid profile. Fine-needle aspiration cytology from an Achilles tendon xanthoma revealed foamy histiocytes in clusters and occasional inflammatory cells consistent with fibroxanthoma. T1-weighted MRI of the right ankle showed tendon enlargement with tendon xanthoma showing intermediate signal intensity. T2-weighted MRI of the brain showed bilateral dentate nucleus calcification and bilateral cerebellar atrophy.
Sagittal T1-weighted MRI. Arrow indicates Achilles tendon xanthoma. (Source: Mahadevan N, Thiruvadi V, Paranthakan C, Rejha A, Magesh A. Cerebrotendinous xanthomatosis: report of two siblings with the same mutation but variable...
T2-weighted MRI of the brain. (A) Sagittal section showing cerebellar atrophy (arrow) and (B) axial section showing bilateral symmetrical hypointensities involving dentate nuclei consistent with calcification (arrows). (Source:...
Genetic testing identified the pathogenic homozygous missense variant c.380G>A in exon 2 of the CYP27A1 gene, which results in the substitution of amino acid at codon position 127 (p.Arg127Gln). This patient was treated with atorvastatin 80 mg once daily and clopidogrel 75 mg once daily as chenodexycholic acid was unavailable in India.
Case 3. A 27-year-old man, the younger sibling of Case 2, presented with bilateral Achilles and patellar tendon xanthomas since childhood (89).
Arrows indicate bilateral Achilles tendon xanthomas. (Source: Mahadevan N, Thiruvadi V, Paranthakan C, Rejha A, Magesh A. Cerebrotendinous xanthomatosis: report of two siblings with the same mutation but variable presentation. ...
He also developed early-onset cataracts in both eyes, status post posterior chamber intraocular lens placement. Examination revealed moderate intellectual disability, an otherwise normal neurologic examination, and bilateral xanthomas. Fine-needle aspiration cytology of a xanthoma was consistent with fibroxanthoma. T1-weighted MRI of the left ankle revealed tendon enlargement with Achilles tendon xanthoma showing intermediate signal intensity. FLAIR images of the brain revealed normal bilateral dentate nucleus with no cerebellar atrophy.
Sagittal T1-weighted MRI showing left Achilles tendon xanthoma with intermediate signal intensity (arrow). (Source: Mahadevan N, Thiruvadi V, Paranthakan C, Rejha A, Magesh A. Cerebrotendinous xanthomatosis: report of two sibli...
Axial FLAIR image showing normal dentate nuclei with no cerebellar atrophy (arrow). (Source: Mahadevan N, Thiruvadi V, Paranthakan C, Rejha A, Magesh A. Cerebrotendinous xanthomatosis: report of two siblings with the same mutat...
Diagnosis of cerebrotendinous xanthomatosis was confirmed by genetic testing, which revealed the same homozygous missense pathogenic variant c.380G>A (p.Arg127Gln) in the CYP27A1 gene.
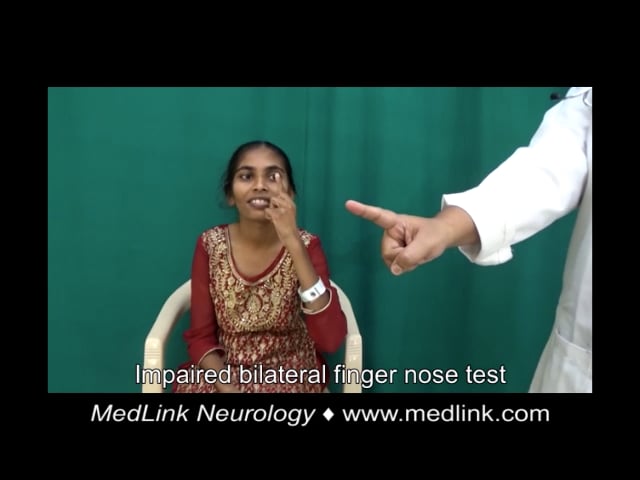
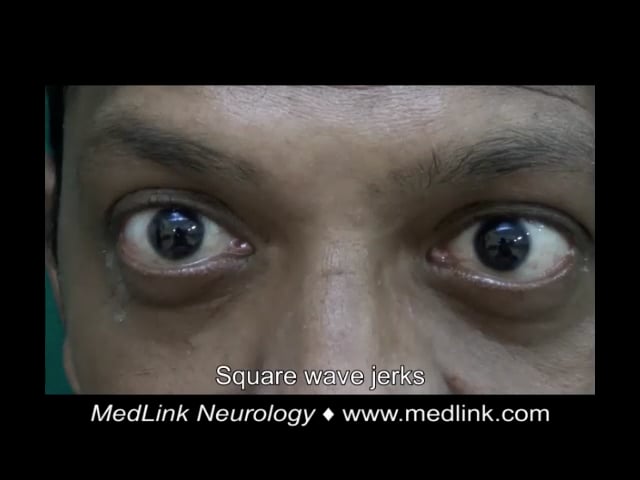
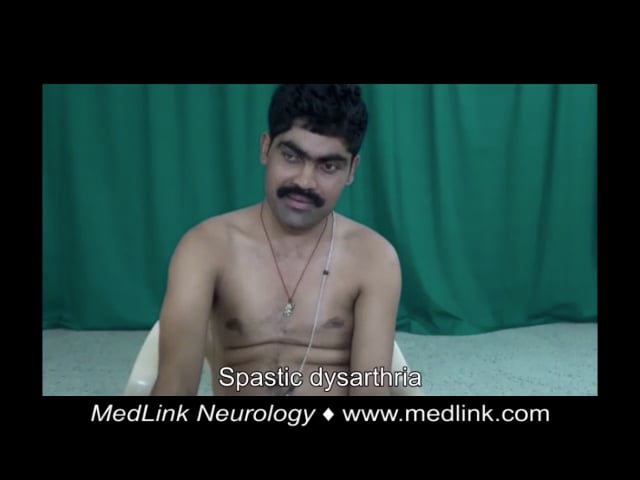
Case 4. A 22-year-old Indian woman presented with imbalance while walking, dysarthria, and hand tremulousness (74). There was no significant family history and no consanguinity. She had a history of intellectual disability and cataract surgery at the age of 17. Examination showed bilateral Achilles tendon xanthomas, ataxic speech, square wave jerks, saccadic intrusions, facial dystonia, palatal tremor, impaired bilateral finger-nose test and heel-shin test, and ataxic gait. MRI showed symmetric T2 hyperintense and FLAIR hypointense lesions along the dentate and cerebellar white matter bilaterally, T2 and FLAIR hyperintensity of the corticospinal tract along the periventricular white matter, posterior limb of internal capsule, cerebral peduncles, and pons, as well as hyperintensity of the inferior olives and middle cerebellar peduncles, and rarefaction of cerebellar white matter. Genetic testing was not done. Treatment with ursodeoxycholic acid (600 mg/d), atorvastatin (20 mg/d), baclofen (20 mg/d), and levodopa/carbidopa (300/75 mg/d) produced mild subjective improvement in gait after 3 months.
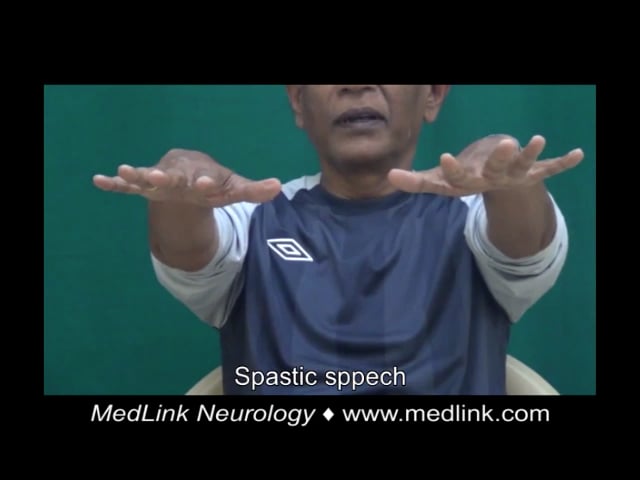
Case 5. A 44-year-old Indian man presented with walking difficulty, dysarthria, and pseudobulbar affect (74). There was no significant medical history. His parents were consanguineous. He had a history of seizures and cataract surgery at the age of 28. Examination showed bilateral Achilles tendon xanthomas, ataxic and spastic speech, square wave jerks, saccadic pursuit, impaired bilateral finger-nose test, dysdiadokokinesia, spastic gait requiring support to walk, brisk muscle stretch reflexes in all four limbs with ankle clonus, and bilateral Babinski signs. MRI showed symmetric hyperintensities on T2-weighted and FLAIR sequences in the periventricular white matter, corticospinal tracts, middle cerebellar peduncle, dentate and peri-dentate white matter, and inferior olives. The “hot cross bun” sign was evident within the pons. Long-segment hyperintensity of the dorsal and lateral columns was seen in T2 sequences of the spine MRI. Diagnosis of cerebrotendinous xanthomatosis was confirmed by genetic testing, which revealed a homozygous c.379C>G p.Arg127Gly missense was identified. Despite treatment with ursodeoxycholic acid (600 mg/d), atorvastatin (20 mg/d), baclofen (30 mg/d), amantadine (200 mg/d), and valproate (600 mg/d), he experienced relentless progression over the next 5 years to a bedbound state.
Case 6. A 66-year-old Indian man presented with slurred speech, dysphagia, pseudobulbar affect, walking difficulty, and forgetfulness. There was no consanguinity. He had a history of cataract surgery at the age of 56. Examination showed bilateral Achilles tendon xanthomas, spastic speech, mild spooning of fingers, mild ataxic gait, and postural instability. Fluid attenuated inversion recovery (FLAIR) images showed hyperintensities in the periventricular white matter and along the corticospinal tracts in the internal capsules, midbrain, and pons. The globus pallidi appeared hypointense on T1 and hyperintense on T2. The dentate nuclei appeared slightly hyperintense on coronal T2 images. Diagnosis of cerebrotendinous xanthomatosis was confirmed by genetic testing, which revealed compound heterozygous mutations (c.1184+1G> A/c.1537C> T splice site and NA/p.Arg513Cys missense). Despite treatment with ursodeoxycholic acid (600 mg/d), atorvastatin (20 mg/d), baclofen (20 mg/d), levodopa/carbidopa (400/100 mg/d), and escitalopram (10 mg/d), his deficits worsened after 6 months, and he died after 1 year.
Case 7. A 32-year-old Indian man presented with walking difficulty. There was no significant family history and no consanguinity. He had a history of developmental delay, intellectual disability, seizures, and cataracts with surgery at age of 12 years. Examination showed bilateral Achilles tendon xanthomas, spastic speech, gaze-evoked nystagmus, impaired bilateral finger-nose test, dysdiadokokinesia, impaired bilateral heel-shin test, spastic-ataxic gait, and ankle clonus. MRI showed mild diffuse cerebral and cerebellar atrophy with symmetric dentate nuclei hyperintensities on T2-weighted images. Diagnosis of cerebrotendinous xanthomatosis was confirmed by genetic testing, which revealed a homozygous c.526del p.Asp176MetfsTer6 frameshift truncation. He was treated with ursodeoxycholic acid (600 mg/d), atorvastatin (20 mg/d), and baclofen (30 mg/d). Follow-up results were not available.
Case 8. A 21-year-old Indian man presented with walking difficulty. There was no consanguinity. He had a history of cataract surgery at the age of 13. Examination showed bilateral Achilles tendon xanthomas, spastic speech, mild nonsustained gaze-evoked nystagmus, impaired bilateral finger-nose test, spastic-ataxic gait, brisk ankle jerks, and Babinski signs. MRI showed multiple chronic watershed infarcts in the internal carotid artery territory bilaterally and subtle periventricular white matter signal changes. Time-of-flight MR angiography showed complete occlusion of the intracranial segment of the left internal carotid artery. Genetic testing was not done. He was treated with ursodeoxycholic acid (600 mg/d), atorvastatin (40 mg/d), baclofen (20 mg/d), aspirin (150 mg/d), and clopidogrel (75 mg/d). Follow-up results were not available.
|
• Cerebrotendinous xanthomatosis is caused by a deficiency of the mitochondrial enzyme sterol 27-hydroxylase. | |
|
• As a result of sterol 27-hydroxylase deficiency, bile acid production is decreased. | |
|
• The absence of the negative feedback mechanism of chenodeoxycholic acid on 7alpha-hydroxylase, the rate-limiting enzyme in bile acid synthesis, leads to the accumulation of intermediates as precursors for cholestanol. |
Cholesterol chemical structure and metabolism. Cholesterol (5-cholesten-3 β-ol) is a C27 alicyclic compound with a fused 4-ring structure (labeled A-D), a single hydroxyl group at C-3, an unsaturated center between C-5 and C-6, an 8-carbon branched hydrocarbon chain attached to the D ring at C-17, and two methyl groups at positions 10 and 13 (numbered C-19 and C-18, respectively). The fused 4-ring structure with a hydroxyl group at C-3 is a sterol; there are three 6-carbon rings and a single 5-carbon ring. The letter identification of the rings is standardized, as is the numbering of the individual carbons, which applies to the sterol ring structure and to cholesterol itself.
Cholesterol is excreted in the bile, primarily as bile acids conjugated to glycine or taurine, the end products of cholesterol metabolism. Primary bile acids are synthesized in hepatocytes directly from cholesterol. The primary bile acids are 24-carbon compounds containing two or three hydroxyl groups, and a side chain ending in a carboxyl group. The carboxyl group is ionized at physiological pH, meaning bile acids are anions (even if the terms “bile acids” and “bile salts” are generally used interchangeably). The most abundant primary bile acids are cholic acid and chenodeoxycholic acid.
Hepatic production of primary bile acids is not sufficient to meet physiologic needs, so the enterohepatic circulation transports most of the excreted primary bile acids back to the liver. Bile acids that are not reabsorbed are converted to various secondary 24-carbon bile acids by gut bacteria. A portion of these secondary bile acids (particularly deoxycholic acid and lithocholic acid) is also passively reabsorbed and returned to the liver, where they are excreted into the gall bladder.
Bile acids serve several physiological functions: (1) the only significant means of cholesterol excretion; (2) solubilizing and transport agents (eg, preventing cholesterol from precipitating from solution in the gall bladder); and (3) a "biological detergent" (in the form of micelles) that facilitates hydrolysis of dietary fats.
Classic pathway of bile acid biosynthesis. Bile acid synthesis proceeds in the liver through a series of chemical steps beginning with cholesterol.
The classic (or neutral) pathway of bile acid biosynthesis produces about 90% of the bile acids.
The first several steps occur in the cytosol. The first and rate-limiting enzyme of the pathway is cholesterol 7α-hydroxylase (CYP7A1; cytochrome P450, family 7, subfamily A, polypeptide 1; also known as cholesterol 7α -monooxygenase), which catalyzes the hydroxylation of cholesterol to 7α-hydroxycholesterol. This step effectively determines the size of the bile acid pool. 7α-hydroxycholesterol then undergoes isomerization to form 7α-hydroxy-4-cholesten-3-one.
What follows is a complicated series of chemical reactions catalyzed by different enzymes. The net result, though, is synthesis of almost equal amounts of the two primary bile acids: cholic acid and chenodeoxycholic acid.
First branch. In one branch of the classic pathway, 7α-hydroxy-4-cholesten-3-one is converted into 7α,12α-dihydroxy-4-cholesten-3-one in the endoplasmic reticulum by the action of the enzyme CYP8B1 (cytochrome P450, family 8, subfamily B, polypeptide 1; also known as sterol 12-alpha-hydroxylase).
7α,12α-dihydroxy-4-cholesten-3-one is then converted into 5β-cholestan-3α,7α,12α-triol by reduction and dehydrogenation through the actions of 5β-reductase (AKR1D1, ie, aldo-keto reductase family 1 member D1) and 3α-hydroxysteroid dehydrogenase (3α-HSD; or AKR1C4, ie, aldo-keto reductase family 1 member C4).
5β-cholestan-3α,7α,12α-triol is converted into 3α,7α,12α-trihydroxy-5β-cholestanic acid (THCA) in the mitochondria through the action of CYP27A1 (cytochrome P450, family 27, subfamily A, polypeptide 1; commonly known as sterol 27-hydroxylase).
THCA then undergoes side-chain cleavage through peroxisomal β-oxidation followed by amino-conjugation (in the cytosol/peroxisome) to form cholic acid.
Second branch. In the other branch of the classic pathway, 7α-hydroxy-4-cholesten-3-one is converted into 5β-cholestan-3α,7α-diol. 5β-cholestan-3α,7α-diol is converted into 3α,7α-dihydroxy-5β-cholestanic acid (DHCA) in the mitochondria through the action of CYP27A1. Finally, DHCA is converted to chenodeoxycholic acid.
Alternate pathway of bile acid biosynthesis. An alternative pathway (also known as the acidic pathway due to the synthesis of acidic intermediates) generally produces less than 10% of available bile acids in normal adults but may be relatively more important than the classic pathway in patients with liver dysfunction and is the predominant pathway in neonates.
The alternative pathway is initiated by mitochondrial sterol 27-hydroxylase (CYP27A1).
The key regulatory steps of the alternative pathway are the first two steps: (1) 27-hydroxylation by mitochondrial sterol 27-hydroxylase (CYP27A1) to form 27-hydroxycholesterol, and (2) the subsequent 7α-hydroxylation by oxysterol 7α-hydroxylase (CYP7B1). CYP7B1 serves as the ultimate controller of cellular oxysterol levels.
Cerebrotendinous xanthomatosis, an inborn error of bile acid synthesis. Cerebrotendinous xanthomatosis, due to 27-hydroxylase deficiency, is a rare familial sterol storage disease with accumulation of cholestanol and cholesterol in most tissues, particularly in xanthomas, bile, and brain. Clinically, this disorder is characterized by dementia, spastic paresis, cerebellar ataxia, tendon xanthomas, early atherosclerosis, and cataracts.
Cerebrotendinous xanthomatosis is caused by autosomal recessive loss-of-function mutations in CYP27A1, a gene encoding a cytochrome p450 oxidase, a mitochondrial enzyme commonly known as sterol 27-hydroxylase. More than 20 different mutations have been defined in the sterol 27-hydroxylase gene (CYP27A1) of patients with cerebrotendinous xanthomatosis. A symptomatic heterozygote carrier with clinical disease and decreased enzyme function has also been reported; no second mutation was identified despite an extensive search (140).
The defect in sterol 27-hydroxylase leads to a block in bile acid biosynthesis, with a drop in levels of primary bile acids, accumulation of substrates for the mitochondrial 27-hydroxylase, and a pathologic and toxic increase in otherwise uncommon metabolites. In particular, cholesterol metabolism in cerebrotendinous xanthomatosis proceeds by otherwise minor metabolic pathways to form bile alcohols rather than bile acids. The absence of the negative feedback mechanism of chenodeoxycholic acid on 7α-hydroxylase, the rate-limiting enzyme in bile acid synthesis, augments the accumulation of intermediates as precursors for cholestanol. In cerebrotendinous xanthomatosis, these bile alcohols are excreted in gram amounts in bile and feces.
In cerebrotendinous xanthomatosis, the levels of cholestanol in serum are markedly increased, whereas the levels of cholesterol are generally normal. Metabolism of cholesterol in cerebrotendinous xanthomatosis includes, sequentially, the intermediaries cholest-4-en-3-one, 5α-cholestan-3-one, and, ultimately, 5α-cholestan-3β-ol (cholestanol) (126).
However, at least part of the excess cholestanol in patients with cerebrotendinous xanthomatosis is formed from the accumulated 7α-hydroxycholesterol and 7α-hydroxy-4-cholesten-3-one, the first two metabolic intermediates of the classic pathway of cholesterol metabolism (132). 7α,12α-Dihydroxy-4-cholesten-3-one, the product of CYP8B1 in the classic pathway of bile acid biosynthesis, is the most prominently elevated metabolite in serum and CSF of drug-naive patients with cerebrotendinous xanthomatosis (68).
In cerebrotendinous xanthomatosis, some intermediary metabolites from the classic pathway are metabolized into 5β-cholestane-3α,7α,12α,25-tetrol, 5β-cholestane-3α,7α,12α,23-tetrol, and 5β-cholestane-3α,7α,12α,24,25-pentol.
Cholestanol and 7α-hydroxycholesterol accumulate in many tissues (20), including the brain, leading to progressive neurologic dysfunction. The increased bile alcohol level may lead to disruption of the blood-brain barrier, which, in turn, may facilitate brain deposits of these substances (125). The storage of cholestanol in other tissues causes tendon xanthomas, atherosclerosis, lipomatous hypertrophy of the atrial septum, and cataracts (25). The reason for the accumulation of cholestanol and cholesterol in certain tissues in cerebrotendinous xanthomatosis is not understood.
An overview of cholesterol metabolism and the biochemical abnormality in cerebrotendinous xanthomatosis is shown in the following figure (24). The image shows the normal ("classical") metabolic pathway of cholesterol, which is mainly regulated by CYP27A1. The absence of this enzyme leads to a significant increase in activity in the cholestanol pathway, as well as an increase in upstream cholesterol metabolites. Chenodeoxycholic acid therapy inhibits CYP7A1, thereby halting utilization of the cholestanol pathway and accumulation of cholestanol metabolites in the CNS.
This image shows the normal ("classical") metabolic pathway of cholesterol, which is mainly regulated by CYP27A1. The absence of this enzyme leads to a significant increase in activity in the cholestanol pathway, as well as an ...
|
• The estimated prevalence of cerebrotendinous xanthomatosis in the general population is three to five per 100,000 but may be as high as 2% among patients with acquired juvenile-onset idiopathic bilateral cataracts. | |
|
• Some ethnic subgroups have a high cerebrotendinous xanthomatosis gene frequency. |
The estimated prevalence of cerebrotendinous xanthomatosis in the general population is three to five per 100,000 but may be 500-fold higher (1.6% to 1.8%) among patients with acquired juvenile-onset idiopathic bilateral cataracts (54; 07). Prevalence estimates are highest in Asians (1:44,407–93,084) and lowest in the Finnish population (1:3,388,767), whereas intermediate estimates are found in Europeans, Americans, and Africans/African Americans (1:70,795–233,597) (116). The prevalence of cerebrotendinous xanthomatosis related to the p.Arg395Cys mutation alone is approximately 1 per 50,000 among the Caucasian population (86). In a study population selected for early-onset idiopathic bilateral cataracts, cerebrotendinous xanthomatosis was present in 0.9% of enrolled patients and in 15% of patients who met criteria for genetic testing (55).
Some ethnic subgroups have a high cerebrotendinous xanthomatosis gene frequency. Cerebrotendinous xanthomatosis occurs at high frequency in both Sephardic and Ashkenazi Jews, and both groups may benefit from newborn and carrier screening for cerebrotendinous xanthomatosis (65). The prevalence in Sephardic Jews of Moroccan extraction was estimated to be 1 in 108 (12). In Jewish Moroccans, there is an estimated prevalence of 4 in 100,000, with cases resulting from the two mutant alleles in nonconsanguineous marriages (84). In a high-risk Israeli population, a 1 in 30 carriership frequency was determined for the c.355delC CYP27A1 gene variant newborns identifying as Druze (an Arabic-speaking ethnoreligious group originating in Western Asia), providing an estimated disease prevalence of 1 in 3600 in this population (35).
The prevalence of cerebrotendinous xanthomatosis is probably underestimated because of the wide spectrum of clinical phenotypes even without neurologic impairment, the wide range of age of onset, and frequent lack of clinician awareness of this disorder (167). In a retrospective study of 100 confirmed cases from 72 families, the mean age at diagnosis was 28.2 ± 14.3 years, and the diagnostic delay was 18.4 ± 13.7 years (167).
|
• Early diagnosis of at-risk family members allows initiation of treatment that may prevent or limit disease manifestations. |
Early diagnosis of at-risk family members using biochemical testing, or molecular genetic testing if the two disease-causing mutations in the proband are known, allows initiation of treatment that may prevent or limit disease manifestations. Prenatal diagnosis may be possible.
Different newborn screening biomarkers for cerebrotendinous xanthomatosis have been described. Both 5β-cholestane-3α,7α,12α,25-tetrol glucuronide (GlcA-tetrol) and the ratio of GlcA-tetrol to tauro-chenodeoxycholic acid (t-CDCA) (GlcA-tetrol/t-CDCA) are excellent cerebrotendinous xanthomatosis biomarkers suitable for newborn screening (69).
Sitosterolemia is an inherited sterol storage disease characterized by tendon xanthomas and by a strong predisposition to premature atherosclerosis. Serum concentration of plant sterols (sitosterol and campesterol) is increased. Primary neurologic signs and cataracts are not present. Spastic paraparesis may occur as a result of spinal cord compression by multiple intradural, extramedullary xanthomas (67).
Cerebrotendinous xanthomatosis is the second most common cause of early-onset cataract and neurologic disease (myotonic dystrophy type 1 is the most common cause). Unexplained juvenile-onset cataracts associated with infantile-onset chronic diarrhea and mental retardation or deterioration strongly suggest the possibility of cerebrotendinous xanthomatosis (32; 33; 152).
In hypercholesterolemia and hyperlipemia (especially type IIa) the plasma cholestanol level is normal. When xanthomas are not evident, the differential diagnosis includes all forms of progressive mental deterioration (59; 151).
When xanthomas are not evident, the differential diagnosis includes all forms of progressive mental deterioration or mental retardation (59; 164), and in one such case, the findings suggested mitochondrial disease and, specifically, myoclonic epilepsy with ragged-red fibers (MERRF) (78).
|
• Improved screening, clinical awareness, and prompt treatment are needed to prevent unnecessary and irreversible disease progression. | |
|
• Cerebrotendinous xanthomatosis should be suspected in individuals with the following findings: (1) infantile-onset diarrhea; (2) childhood-onset cataract, learning disability, or autism spectrum disorder; (3) tendon xanthomas with onset from adolescence to early adulthood despite relatively normal cholesterol levels; (4) adult-onset progressive neurologic dysfunction; (5) neuroimaging demonstrates cerebral, cerebellar, and spinal cord atrophy, and T2-weighted/FLAIR hyperintensities involving subcortical, periventricular, and cerebellar white matter and the brainstem and dentate nuclei; (5) multinodular thickening of the nerve roots and trunks of the lumbosacral plexus detected (without gadolinium enhancement) on MRI and MR neurography. | |
|
• Cerebrotendinous xanthomatosis is caused by a deficiency of the mitochondrial enzyme sterol 27-hydroxylase, with resulting cholestanol and cholesterol accumulation in virtually every tissue. | |
|
• The main laboratory abnormalities that distinguish cerebrotendinous xanthomatosis from other conditions with xanthomas include the following: (1) high plasma and tissue cholestanol concentration; (2) normal to low plasma cholesterol concentration; (3) markedly decreased formation of chenodeoxycholic acid; (4) increased concentration of bile alcohols and their glyconjugates in bile, urine, and plasma; and (5) increased concentration of cholestanol and apolipoprotein B in cerebrospinal fluid resulting from changes in the blood-brain barrier. | |
|
• A clinical prediction rule has been developed. |
Cerebrotendinous xanthomatosis is underrecognized (83; 63; 159). Improved screening, clinical awareness, and prompt treatment are needed to prevent unnecessary and irreversible disease progression (09; 104; 05; 38; 127; 143; 34). The average age at diagnosis is 32 to 35 years, with a mean delay in diagnosis of 16 to 17 years (93; 104; 47; 130). A 2-tiered newborn screening approach may be feasible (35). Forme frustes and the marked clinical heterogeneity can make diagnosis difficult (139); delays can result in worse clinical outcomes, so early diagnosis and treatment initiation are critical.
Cerebrotendinous xanthomatosis should be suspected in individuals with the following findings:
|
• Neonatal cholestatic jaundice | |
|
• Infantile-onset diarrhea | |
|
• Childhood-onset cataract (especially bilateral fleck-like opacities) (109; 74) | |
|
• Learning disability or autism spectrum disorder (47; 34). Metabolic screening for cerebrotendinous xanthomatosis should be performed in patients with autism spectrum disorder, particularly when it is accompanied by diarrhea, intellectual disability, juvenile cataract, or neurologic involvement (134). | |
|
• Tendon xanthomas with onset from adolescence to early adulthood despite relatively normal cholesterol levels. | |
|
• Adult-onset progressive neurologic dysfunction (dementia; psychiatric disturbances; pyramidal, extrapyramidal, or cerebellar signs; and seizures). It should be considered as a possible etiology of progressive ataxia with palatal tremor, even in the absence of tendon xanthomas and cataracts (120). | |
|
• Typical neuroimaging findings, particularly dentate nuclei abnormalities at MRI | |
|
• A sibling afflicted with cerebrotendinous xanthomatosis | |
|
• Neurologic symptoms in the family history (109) | |
|
• Consanguinity of the parents (109; 74) |
Clinical prediction rule (suspicion score; Mignarri score; Mignarri index). Mignarri and colleagues devised a clinical prediction tool or “suspicion index” that is simple, inexpensive, and yet highly effective as a diagnostic aid (93); this is now sometimes referred to as the Mignarri score or Mignarri index. Clinical features were classified as moderate, strong, or very strong and weighted accordingly with weights of 25, 50, and 100, respectively. An equivalent method would be to weight these as 1, 2, and 4 points, respectively. Strong indicators included early systemic signs (eg, cataract, diarrhea, and neonatal cholestatic jaundice) and neurologic features (eg, intellectual impairment, psychiatric disturbances, ataxia, spastic paraparesis, and dentate nuclei abnormalities at MRI). Very strong indicators included tendon xanthomas and a sibling affected with cerebrotendinous xanthomatosis. A total score of 100 or more (or equivalently, 4 points) warrants serum cholestanol assessment, and either an elevated cholestanol level or a total score of 200 or more (or equivalently, 8 points), with at least one very strong or four strong indicators, warrants CYP27A1 gene analysis. Using this tool (albeit retrospectively), the mean age at diagnosis could be reduced from 36 years to 11 years.
In cases of clinical suspicion, molecular analysis is recommended despite normal plasma cholestanol levels because multiple cases have been reported where plasma cholestanol is not elevated but in which there are prominent cholesterol-rich xanthomas (03; 37; 167).
Neuroimaging. Neuroimaging (CT and especially MRI) illustrates cerebral, cerebellar, and spinal cord atrophy; T2-weighted/FLAIR hyperintensities involving subcortical, periventricular, and cerebellar white matter; and the brainstem and dentate nuclei (104; 71; 88; 137).
Typically, there are lesions in the cerebellar dentate nuclei (hyperintense on T2-weighted images and hypointense on T1-weighted images) associated with diffuse white matter abnormalities (01; 73; 117; 104; 161; 92; 90; 71; 88; 137).
This axial T2-weighted MRI of a 42-year-old man with cerebrotendinous xanthomatosis shows bilateral hyperintense lesions involving the dentate nuclei and deep cerebellar white matter. Clinical presentation: He had a history of ...
MRI of a patient with cerebrotendinous xanthomatosis shows T1-weighted (A, arrow) and T2-weighted (B, arrow) hyperintensities in the dentate nuclei. (Source: Nie S, Chen G, Cao X, Zhang Y. Cerebrotendinous xanthomatosis: a comp...
T1-weighted/FLAIR hypointensities consistent with cerebellar vacuolation and T1-weighted/FLAIR/susceptibility-weighted hypointense alterations compatible with calcifications are seen in the dentate nuclei in a small subgroup of patients (92; 71; 88; 137). Cerebellar vacuolation is a useful biomarker of disease progression and unsatisfactory response to therapy, whereas the absence of dentate nuclei signal alteration is a good prognostic indicator (92).
MRI abnormalities may also involve the spinal cord and, in particular, may show signal changes affecting mainly the lateral corticospinal tracts (71).
This axial T2-weighted spinal MRI from a 27-year-old man with cerebrotendinous xanthomatosis shows signal changes mainly affecting the lateral corticospinal tracts. Clinical presentation: This man presented with a 3-year histor...
Treatment initiation at a younger age markedly decreases evident progression on serial MRI studies (136; 137). It can reverse and even prevent the development of neurologic symptoms.
Diffusion tensor imaging and tractography can (1) detect changes in the brain when the conventional MRI is unremarkable and (2) provide potential neuroimaging biomarkers for monitoring treatment response in cerebrotendinous xanthomatosis, even when conventional MRI remains unchanged (28).
PET imaging of the brain may show hypometabolism in the cerebrum (especially in the frontal and temporal lobes) (104).
This PET image reveals hypometabolism in the cerebrum (especially in the frontal and temporal lobes) in both the sagittal section (A) and the axial section (B). (Source: Nie S, Chen G, Cao X, Zhang Y. Cerebrotendinous xanthomat...
Multinodular thickening of the nerve roots and trunks of the lumbosacral plexus may be detected without gadolinium enhancement on MRI and MR neurography (66).
Multifocal, hypoechogenic nerve thickening of peripheral nerves and nerve roots may be demonstrated using high-resolution ultrasound (119).
Xanthomas. MRI of tendon xanthomas generally shows fusiform enlargement of the tendon, particularly the Achilles tendon (104; 79). The xanthomas are relatively isointense compared to muscle, and there may be evidence of lipid infiltration of the tendon (79).
Coronal T1-weighted MRI of the lower leg shows bilateral Achilles tendon xanthomas (long arrows) and multiple other tendon xanthomas around both ankles (short arrows). (Source: Koroglu M, Karakaplan M, Gündüz E, et al. Cerebrot...
Axial fat-saturated T2-weighted MRI of the left ankle shows diffuse enlargement of the Achilles tendon (arrows) with the characteristic speckled appearance (dotted arrows). (Source: Koroglu M, Karakaplan M, Gündüz E, et al. Cer...
Histology of tendon xanthomas shows accumulation of xanthoma cells and dispersed lipid crystal clefts (104; 79).
Photomicrograph of a surgically resected xanthoma demonstrating cholesterol clefts (asterisk) and multinucleated giant cells (arrow) (Hematoxylin and eosin x 200). (Source: Koroglu M, Karakaplan M, Gündüz E, et al. Cerebrotendi...
Histology (hematoxylin and eosin, 100x) of a tendon xanthoma in a patient with cerebrotendinous xanthomatosis reveals accumulation of xanthoma cells (fine arrows) and dispersed lipid crystal clefts (coarse arrows). (Source: Nie...
Histology (hematoxylin and eosin, 200x) of a tendon xanthoma in a patient with cerebrotendinous xanthomatosis reveals accumulation of xanthoma cells (fine arrows) and dispersed lipid crystal clefts (coarse arrows). (Source: Nie...
Although not needed in most clinical situations, PET imaging, if conducted, may show unusually high radioactivity in the xanthomas (104).
Diagnostic testing for cerebrotendinous xanthomatosis. Genetic analyses or determination of serum cholestanol levels should be used to diagnose cerebrotendinous xanthomatosis (135).
Dried bloodspot testing should facilitate detection in newborns (135).
Biochemical testing. Cerebrotendinous xanthomatosis is caused by a deficiency of the mitochondrial enzyme sterol 27-hydroxylase, with resulting cholestanol and cholesterol accumulation in virtually every tissue. Plasma cholestanol should be measured in any patient with infantile chronic diarrhea or jaundice, juvenile cataract, learning disability or autism spectrum disorder, pyramidal signs, cerebellar syndrome, or peripheral neuropathy (04).
The main laboratory abnormalities that distinguish cerebrotendinous xanthomatosis from other conditions with xanthomas include the following:
|
• High plasma and tissue cholestanol concentration. The cholestanol level is typically three to 15 times higher than mean levels in unaffected individuals and can range from 1.3 to 15 mg/dL. There are rare cases where plasma cholestanol is not elevated but in which there are prominent cholesterol-rich xanthomas (03; 37; 167). | |
|
• Normal to low plasma cholesterol concentration (usually 115 to 220 mg/dL). | |
|
• A low cholesterol-to-cholestanol ratio. In the appropriate clinical setting, this can be diagnostic, although additional confirmatory genetic testing is still recommended. |
Some additional studies are abnormal and may be useful in certain circumstances, but they are currently less commonly used for general clinical diagnosis and management.
|
• Measurement of intermediates in bile acid biosynthesis. Levels of intermediates in bile acid biosynthesis (eg, 7α-hydroxycholesterol, 7α-hydroxy-4-cholesten-3-one, and 7α,12α-dihydroxy-4-cholesten-3-one) are also elevated, although they have not typically been measured in clinical settings. Measurement of 7α-hydroxy-4-cholesten-3-one and 7α,12α-dihydroxy-4-cholesten-3-one may be useful in certain circumstances; for example, measurement of 7α,12α-dihydroxy-4-cholesten-3-one enables sensitive dried bloodspot testing for cerebrotendinous xanthomatosis. | |
|
• Urine testing. Urine testing of bile alcohols and their conjugates using fast ion bombardment–mass spectroscopy can be performed after a positive serum cholestanol result. | |
|
• Bile analysis. Cholestanol is present at high concentrations in the bile of affected individuals (4% to 11% vs. less than 1% in normal individuals). A low concentration of chenodeoxycholic acid is observed, along with high concentrations of bile alcohols (conjugated with glucuronic acid). | |
|
• CSF analysis. Increased concentration of cholestanol and apolipoprotein B in cerebrospinal fluid resulting from changes in the blood-brain barrier. This is not commonly measured for diagnosis in clinical settings. |
Other laboratory abnormalities that are not diagnostic include the following:
|
• Increased plasma lactate concentration. | |
|
• Increased brain lactate concentration (by MR spectroscopy). |
Genetic testing. Sequence analysis of the CYP27A1 gene detects mutations in 90% to 100% of affected individuals.
Phenotypic heterogeneity may occur among affected siblings (64; 80; 89). In one family, one sibling had the spinal xanthomatosis phenotype and the other had a very mild form of cerebrotendinous xanthomatosis manifesting as minor tendon xanthomatosis and gastrointestinal complaints (64). However, biochemical analysis revealed normal levels of serum cholestanol and relatively mild elevations of the bile acid precursors 7α-hydroxy-4-cholesten-3-one and 7α,12α-dihydroxy-4-cholesten-3-one. Nevertheless, both siblings had compound heterozygous variants in CYP27A1. The atypical biochemical findings illustrate that cholestanol is not a perfectly sensitive test for cerebrotendinous xanthomatosis. Therefore, patients with symptomatology consistent with cerebrotendinous xanthomatosis should have bile acid precursor biochemical testing and molecular analysis (64).
Electrophysiology studies. Decreased nerve conduction velocities as well as abnormal somatosensory, motor, brainstem, and visual evoked potential may improve or resolve with chenodeoxycholic acid therapy.
Dual-energy x-ray absorptiometry (DEXA) studies. Osteoporosis is common, so consideration should be given to screening for this with DEXA of the femoral neck and lumbar spine.
Other diagnostic findings. Large paranasal sinuses may be noted on brain imaging or skull x-rays.
Histology of xanthomas. Histological findings from tendon xanthomas show foamy macrophages and lipid crystal clefts (30).
Arrowheads indicate foamy macrophages. (Source: Chun MY, Heo NJ, Seo SW, et al. Case report: Cerebrotendinous xanthomatosis with a novel mutation in the CYP27A1 gene mimicking behavioral variant frontotemporal dementia. Front N...
Arrowheads indicate foamy macrophages and arrows indicate lipid crystal clefts. (Source: Chun MY, Heo NJ, Seo SW, et al. Case report: Cerebrotendinous xanthomatosis with a novel mutation in the CYP27A1 gene mimicking behavioral...
Potential for newborn screening. Because delayed diagnosis and treatment worsens the prognosis, cerebrotendinous xanthomatosis is an excellent candidate disorder for newborn screening using recently developed methods for newborn dried bloodspot analysis (36; 35; 21; 136; 69; 34; 147). Unfortunately, this has not yet been implemented.
|
• Beginning treatment as early as possible is essential for preventing neurologic damage and deterioration from cerebrotendinous xanthomatosis. | |
|
• Once significant neurologic pathology has been established, the effect of treatment is limited, and progressive neurologic deterioration may continue even despite treatment. | |
|
• Long-term chenodeoxycholic acid treatment normalizes bile acid synthesis and improves neurophysiologic findings and other clinical manifestations. | |
|
• Cataract extraction is typically required in at least one eye by 50 years of age. | |
|
• Monitoring plasma cholestanol levels is recommended during treatment. |
Age at diagnosis and early treatment initiation (at birth, where possible) have the biggest impact on treatment outcomes (135; 110).
Beginning treatment as early as possible is essential for preventing neurologic damage and deterioration from cerebrotendinous xanthomatosis (157; 04; 39; 80; 122; 110). Once significant neurologic pathology has been established, the effect of treatment is limited, and progressive neurologic deterioration may continue even despite treatment (157; 47; 110). Indeed, patients who started treatment after the age of 25 years had worse outcomes, were significantly more limited in ambulation and more cognitively impaired, and were more likely to continue to deteriorate despite treatment (157).
Chenodeoxycholic acid. The recommended therapy for patients with cerebrotendinous xanthomatosis is chenodeoxycholic acid, which inhibits the 7-alpha-hydroxylation of cholesterol (a normal negative feedback mechanism), thereby reducing the formation of cholestanol and the excretion of bile alcohols in feces and urine. With chenodeoxycholic acid treatment, there is typically stabilization of the xanthomas and often stabilization or improvement in brain imaging (24).
Sagittal ankle MRI, using a "true FISP" sequence shows focal fusiform thickening of both Achilles tendons, which corresponds to xanthoma accumulations in cerebrotendinous xanthomatosis (arrowhead). "True FISP" is the Siemens tr...
FLAIR sequence shows bilaterally symmetrical hyperintensity in the cerebellar white matter and the dentate nucleus (arrowhead). The left image was taken before therapy, and the right image after 9 months of chenodeoxycholic aci...
T2-weighted MRI shows bilaterally symmetrical hyperintensity in the cerebellar white matter and the dentate nucleus (arrowhead). The left image was taken before therapy, and the right image after 9 months of chenodeoxycholic ac...
A nationwide, multicenter study in Turkey that evaluated the long-term effects of treatment in 86 genetically confirmed patients with cerebrotendinous xanthomatosis receiving chenodeoxycholic acid for greater than or equal to 6 months, showed that early diagnosis and initiation of chenodeoxycholic acid therapy significantly improve neurologic outcomes in cerebrotendinous xanthomatosis (166). There was a critical age for the start of treatment to achieve optimal neurologic outcomes: patients diagnosed before 28 years all had neurologic stabilization or improvement, whereas patients diagnosed later showed therapeutic benefits but had a significantly higher rate of disease progression. Chenodeoxycholic acid effectively stabilized or improved pyramidal and cerebellar symptoms, although myoclonus and parkinsonism were less responsive, and psychiatric symptoms showed an even lower treatment response, with psychosis being the most refractory finding. Chenodeoxycholic acid therapy produced a strong and sustained reduction in cholestanol levels, although biochemical response to treatment did not always correlate with clinical improvement. A longer diagnostic delay and the presence of anxiety and pyramidal/cerebellar symptoms were associated with poorer outcomes.
The importance of early treatment initiation is emphasized by two siblings reported by Koyama and colleagues (80). Two siblings with cerebrotendinous xanthomatosis had differing clinical features at diagnosis and different responses to chenodeoxycholic acid treatment; they shared a paternally inherited c.1420C > T mutation (p.Arg474Trp) and a maternally inherited novel c.1176_1177delGA mutation, predicting p.(Glu392Asp*20). The proband, a 32-year-old man, had chronic diarrhea, bilateral cataracts, and xanthomas, and developed progressive neurologic abnormalities, including ataxia, spastic paraplegia, and cognitive decline during a 5-year period, despite normalization of serum cholestanol after initiation of chenodeoxycholic acid treatment. Serial brain MRI studies over this period revealed pronounced progressive atrophy in the cerebellum, in addition to expanding hyperintense lesions on T2-weighted images in the dentate nuclei, posterior limb of the internal capsule, cerebral peduncles, and inferior olives.
Baseline and 5-year follow-up axial, T2-weighted, brain MRI of the proband, a 32-year-old Japanese man with cerebrotendinous xanthomatosis treated with chenodeoxycholic acid. Serial brain MRI studies over this period revealed p...
Brain 99mTc-ethyl cysteinate dimer (99mTc-ECD) single photon emission computed tomography (SPECT) revealed marked cerebellar hypoperfusion, as well as mild frontoparietal hypoperfusion.
Brain 99mTc-ethyl cysteinate dimer (99mTc-ECD) single photon emission computed tomography (SPECT) of the proband, a 32-year-old Japanese man with cerebrotendinous xanthomatosis treated with chenodeoxycholic acid. SPECT revealed...
In contrast, his 30-year-old sister presented with chronic diarrhea, cataracts, xanthomas, and intellectual disability but had no other neurologic symptoms at the time of diagnosis. Chenodeoxycholic acid treatment lead to improvement of cognitive function, and there were no characteristic CTX-related MRI features during a 5-year period.
Baseline and 5-year follow-up axial, T2-weighted, brain MRI of the proband's younger sister, a 30-year-old Japanese woman with cerebrotendinous xanthomatosis treated with chenodeoxycholic acid. Unlike the case of her older brot...
Brain 99mTc-ECD SPECT revealed mild frontoparietal hypoperfusion, without abnormal cerebellar hypoperfusion.
Brain 99mTc-ethyl cysteinate dimer (99mTc-ECD) single photon emission computed tomography (SPECT) of the proband's younger sister, a 30-year-old Japanese woman with cerebrotendinous xanthomatosis treated with chenodeoxycholic a...
Both siblings had an excellent and sustained biochemical response to chenodeoxycholic acid.
The changes in levels of serum cholestanol during a 5-year period in the proband (solid line) and his younger sister (dashed line). Both siblings had an excellent biochemical response to chenodeoxycholic acid. (Source: Koyama S...
However, the proband, who had more advanced neurologic manifestations at the time of treatment initiation developed progressive neurologic abnormalities, including cognitive decline, whereas his sister avoided these adverse outcomes and showed steady improvement in verbal and performance IQ.
Serial Wechsler Adult Intelligence Scale-III scores in the proband and his younger sister. Legend: Thick solid line: verbal IQ (VIQ) in the proband; thick dashed line: VIQ in his younger sister; thin solid line: performance IQ ...
Chenodeoxycholic acid is a lifetime replacement therapy that, if initiated early, can considerably improve outcomes because it may be capable of arresting, or in some cases, reversing the pathophysiological process in cerebrotendinous xanthomatosis (135). Clinical improvement of individuals with cerebrotendinous xanthomatosis following treatment with chenodeoxycholic acid was first reported by Berginer and colleagues (18). Chenodeoxycholic acid is effective in the long-term treatment of cerebrotendinous xanthomatosis, with an acceptable safety profile (148). Long-term chenodeoxycholic acid treatment (750 mg/day in adults) normalizes bile acid synthesis (leading to disappearance of abnormal metabolites from serum, bile, and urine), normalizes plasma and CSF concentration of cholestanol by suppressing cholestanol biosynthesis, and improves neurophysiologic findings and other clinical manifestations, including osteoporosis (97; 50; 98; 148). In a study of treatment with chenodeoxycholic acid over 11 years, nerve conduction velocities normalized after 4 months of treatment and subsequently remained stable; motor-evoked potentials and sensory-evoked potentials slowly and continuously improved; and clinical manifestations stabilized, even though neurologic deficits did not improve (98; 60). The contrast between two untreated siblings whose symptoms progressed and a third treated sibling whose symptoms stabilized suggests that treatment is beneficial.
Although chenodeoxycholic acid treatment seems to stabilize or improve clinical symptoms in most patients, sudden interruption of chenodeoxycholic acid may lead to irreversible neurologic complications. Chenodeoxycholic acid withdrawal produces significant increases in cerebrotendinous xanthomatosis biomarkers and necessitates rescue therapy in most cases (77).
Although chenodeoxycholic acid has been approved for gallstone dissolution, the U.S. Food and Drug Administration (FDA) has not yet approved it for cerebrotendinous xanthomatosis, despite long-term efficacy and safety data (47; 110). Instead, chenodeoxycholic acid has an orphan drug designation by the FDA. Drug company manipulation resulted in dramatic escalation (500%) in the price for the drug, compromising the health of affected individuals, sometimes with very negative neurologic consequences (114; 115; 23).
Monitoring plasma cholestanol levels is recommended during chenodeoxycholic acid treatment (135).
Cholic acid. Cholic acid (the other primary bile acid) is currently used as an alternative therapy in patients who cannot tolerate chenodeoxycholic acid therapy due to its negative effects. Presumably, cholic acid should have negative feedback effects to those of chenodeoxycholic acid on the classic pathway of bile acid synthesis. However, there is presently no consensus on the value of cholic acid therapy alone (135; 110). Nevertheless, the U.S. Food and Drug Administration has approved cholic acid as a treatment for cerebrotendinous xanthomatosis (110). Cholic acid reduces cholestanol levels in CSF and blood while also reducing bile acid synthesis and excretion of bile alcohols in the urine (110). Outcomes with cholic acid therapy are reportedly indistinguishable from those mediated by chenodeoxycholic acid therapy and are associated with significantly fewer adverse effects (110). Cholic acid treatment also improves liver function, which is likely to contribute to secondary pathology, including neurologic dysfunction.
HMG-CoA reductase inhibitors. Inhibitors of HMG-CoA reductase alone or in combination with chenodeoxycholic acid are also effective in decreasing cholestanol concentration and improving clinical signs (112; 153). However, because of clinical evidence that HMG-CoA reductase inhibitors may induce muscle damage and even rhabdomyolysis, caution is required in the use of these drugs (48).
Other treatment considerations. Riluzole has been proposed as a potential adjunctive therapy, but the only anecdotal case utilized this in conjunction with chenodeoxycholic acid, so its marginal benefit is not clear (155).
Other possible treatments include LDL apheresis, but the results are controversial (94; 17; 42).
Liver transplantation, although not yet performed in individuals with cerebrotendinous xanthomatosis, remains a possibility.
Management of disease complications. Cataract extraction is typically required in at least one eye by 50 years of age (74).
Surgical resection of xanthomas is possible to some extent, but complete removal is often not possible because the tendon itself may be enlarged as a yellow firm hypertrophic neoplasm (79). Endoscopic resection of tendon xanthoma in the elbow or ankle is less invasive than open resection (103).
Symptomatic management. Symptomatic treatments for epilepsy, spasticity, and parkinsonism have been utilized (74). Parkinsonism in individuals with cerebrotendinous xanthomatosis is poorly responsive to levodopa, whereas an antihistamine drug, diphenylpyraline hydrochloride, had an excellent effect in three individuals (107). Occasional patients may show improvement in tremor with ventral intermediate nucleus-deep brain stimulation (118)
Handwriting and drawings of the dominant left upper extremity in a 42-year-old left-handed man with multiple cerebrotendinous xanthomatosis-related sequelae: motor examination pre-DBS placement and 6 months post-DBS VIM placeme...
The clinical status of pregnant women with cerebrotendinous xanthomatosis does not typically deteriorate during pregnancy, and elevated serum cholestanol concentration slightly decreases (12; 87; 15). Perhaps this occurs because the liver of the fetus partially compensates for the defect in bile acid synthesis of the pregnant woman. Treatment with chenodeoxycholic acid should not be discontinued during pregnancy (46).
In a case series of nine affected women with 19 pregnancies, mothers continued chenodeoxycholic treatment in 11 pregnancies, whereas mothers did not receive chenodeoxycholic treatment in eight pregnancies (162). In the 11 pregnancies in which chenodeoxycholic treatment was continued, no complications were reported, and newborns were born at or near full-term with normal birth weight and Apgar scores. In the eight pregnancies in which mothers did not receive chenodeoxycholic treatment, two newborns experienced elevated bilirubin soon after birth. One woman who stopped treatment during pregnancy deteriorated neurologically while off treatment.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Douglas J Lanska MD MS MSPH
Dr. Lanska of the University of Wisconsin School of Medicine and Public Health has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurobehavioral & Cognitive Disorders
Jun. 17, 2026
Neurogenetic Disorders
Jun. 01, 2026
Neurobehavioral & Cognitive Disorders
May. 20, 2026
Neurogenetic Disorders
May. 08, 2026
Infectious Disorders
May. 01, 2026
Neurogenetic Disorders
Apr. 30, 2026
General Child Neurology
Apr. 29, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026