


Neuromuscular Disorders
Viral and retroviral myositis
Jun. 16, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
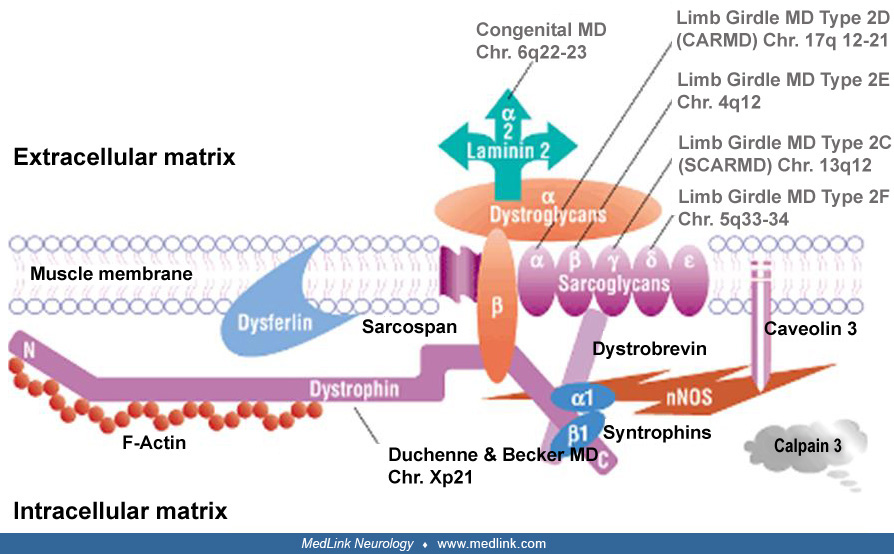
Congenital muscular dystrophies occur with a frequency of 1 in 20,000 births, with a prevalence of 1 in 100,000. They manifest as hypotonia and muscle atrophy at birth, but there is significant clinical and genetic variation. Merosin (laminin alpha-2, also known as LAMA2) is a component of the basal lamina in muscle that is tightly bound to the dystrophin-associated membrane complex and is responsible for about half of the “classical” cases of congenital muscular dystrophy. Brain MRIs of these patients show diffuse white matter signal abnormality, but it does not affect cognition. In this article, the authors describe the clinical and molecular aspects of this disease. Implementation of new care standards, aids in diagnosis, and gene-based therapies that could likely improve the outcome of this disease will also be discussed. The merosin-deficient form of congenital muscular dystrophy will be referred to as LAMA2-related congenital muscular dystrophy (LAMA2-CMD) in this article.
|
• LAMA2 (merosin) is a component of the basal lamina of skeletal muscles and is associated with the most frequent form of congenital muscular dystrophy. | |
|
• Elevated creatine kinase level and white matter signal abnormality on brain MRI are the hallmarks of this disorder. | |
|
• The availability of genetic testing has simplified and expedited the diagnosis of this disorder. | |
|
• Supportive treatment provides long-term clinical benefit. | |
|
• Gene-based research into putative treatments is ongoing. |
Congenital muscular dystrophy was first described in detail by FE Batten in 1903, including three cases with clinical features of muscular dystrophy, which was termed "infantile myopathy" (08). Congenital muscular dystrophy was then referred to as amyotonia congenita to distinguish this group of atrophic muscle diseases from "myotonia congenita" or Thomsen disease. Further histological observations led to the term "congenital muscular dystrophy" in the late 1960s. Since then, various forms have been recognized with or without brain involvement, demonstrating the clinical heterogeneity of congenital muscular dystrophy (06). Congenital muscular dystrophy was traditionally classified as a subgroup of congenital myopathies with an onset in the first 6 months of life by the Research Group on Neuromuscular Diseases of the World Federation of Neurology (65), but with continued advances in our knowledge of the disease, it was found to present within the first 2 years of life (46). Additionally, congenital muscular dystrophies with cerebral involvement (Fukuyama disease, Walker-Warburg syndrome, and muscle-eye-brain disease) were separated into a distinct category of congenital muscular dystrophy associated with glycosylation defects (35). The most prevalent type of congenital muscular dystrophy without clinical involvement of the central nervous system was called "classical" or "occidental" congenital muscular dystrophy (100). Gene discoveries for dystrophin and related membrane or basal lamina proteins revealed the genetic basis of the various forms of congenital muscular dystrophies, including the merosin (LAMA2)-deficient form (98). In light of these genetic findings, muscular dystrophies, including congenital muscular dystrophies, are now best classified according to the genetic and protein defects they harbor (66). Close to half of these cases are related to primary or secondary LAMA2 deficiency (MDC1 A-D) best identified by laminin-alpha 2 staining of muscle biopsies (44). Primary LAMA2-CMD (MDC1A) is an autosomal recessive form of muscular dystrophy due to mutations in the laminin alpha-2 (LAMA2) gene and shows a clinical onset within the first 6 months of life. Congenital inflammatory myopathy, described in the past, has a similar clinical presentation and histological features; therefore, it is a fair assumption that most cases of “congenital inflammatory myopathy” actually represented cases of LAMA2-CMD (78). Late-onset, limb-girdle type of muscular dystrophy has also been reported in patients with partial LAMA2 deficiency with missense mutations in the LAMA2 gene (03; 97). Secondary LAMA2 deficient congenital muscular dystrophy can be associated with glycosylation-related gene defects of fukutin (Fukuyama muscular dystrophy), fukutin-related protein (FKRP) gene (MDC1C), and LARGE gene (MDC1D).
LAMA2 is expressed in many tissue types, including skeletal muscle, cardiac muscle, Schwann cells, and trophoblasts. Patients with LAMA2-CMD experience multisystem involvement. Infants with LAMA2-CMD present with hypotonia and are “floppy” at birth. Early symptoms often include axial weakness and upper, more than lower, limb weakness. Uncommonly, arthrogryposis can be present; rarely, severe respiratory insufficiency may also be present. The infants often have poor suck and cry, multiple joint contractures, and motor development delay (the most common finding). Despite the delay in motor skills, no facial or extraocular muscle weakness or sensory loss is usually noted. Deep tendon reflexes are often decreased, and LAMA2-CMD has associated elevated CK levels (up to five times upper limit of normal, often > 1,000 IU/L). Most of these patients do not achieve independent ambulation (31; 109; 49). In addition, evidence of a demyelinating sensorimotor neuropathy may be present within the first 6 months of life; however, neuropathy is not considered a hallmark feature of this disease (82). Bone density can also be affected (09).
The extent of laminin-alpha2 deficiency dictates disease severity in most cases. In the least severe cases, proximal muscle weakness and poor feeding may be the major concern in the first few months of life (99; 45). Partial loss of laminin-alpha 2 manifests as a late-onset, limb girdle-type muscular dystrophy form of LAMA2-CMD. This can present with similar symptoms as early onset LAMA2-CMD but considerably milder with wider phenotypic variability, and most patients can develop the ability to walk. Complete laminin-alpha2 loss, on the other hand, results in an early-onset congenital form of LAMA2-CMD characterized by severe hypotonia, muscle weakness, skeletal deformity, nonambulatory status, and respiratory insufficiency (70).
Frequently, children have normal intelligence combined with an MRI of the brain exhibiting a diffuse white matter abnormality resembling leukodystrophy. Rarely, patients have intellectual disability with severe brain malformations (58; 59; 93; 80). Partial merosin deficiency due to LAMA2 mutations is also associated with T2 signal abnormalities of the cerebral white matter (10; 77).
Seizures can occur in early-onset LAMA2-CMD and are likely related to neuronal migration abnormalities (105; 31; 69). Work looking at the epileptic phenotype in detail has found it to represent up to one third of individuals with early-onset LAMA2-CMD (109; 68). Natera-de Benito and colleagues provided a detailed investigation of epilepsy in this population, which though a major phenotype with significant impact on the quality of life for both the patient and family, had not been previously characterized in a systematic, well-described cohort of patients (68). They found that the epilepsy: (a) is typically drug-resistant and presents with middle childhood-onset focal seizures that have prominent visual and autonomic features; (b) has a consistent correlation with neuroimaging, suggesting that the extension of the polymicrogyria may serve as a predictor of epilepsy occurrence; (c) was significantly more prevalent in those individuals who had more extensive cortical malformations; and (d) had no association with the patients' motor ability, the size of their white matter abnormalities, or the amount of residual merosin expressed on muscle.
Cardiac abnormalities are predominantly reported in patients with complete LAMA2 deficiency. Initially, only two cases could be found in the literature with cardiac involvement that had partial LAMA2 deficiency (11; 54); however, newer case reports highlight cardiac involvement in partial laminin-alpha 2 chain deficiency also (70). Asymptomatic dilated cardiomyopathy with decreased ejection fraction has been reported in some patients (92; 69). Routine cardiac assessment monitoring in this population is, therefore, important due to the potential for severe presentation, even in those individuals with residual expression of LAMA2.
As the disease progresses, affected individuals can develop facial muscle weakness and macroglossia, which results in typical myopathic facies with a protruded tongue. Weakness of intercostal and accessory muscles results in progressive restriction of the chest wall, decreased lung volume, reduced alveolar gas exchange, and eventually restrictive respiratory insufficiency. Affected individuals also experience skeletal changes such as proximal joint contractures and scoliosis. Scoliosis may result in lumbar and thoracic lordosis, which can interfere with breathing. The most common cause of death in early-onset LAMA2 muscular dystrophy is caused by respiratory tract infection, and 30% of them die within the first decade of life (70).
Most motor dysfunction is noted within the first few weeks, with an initial sharp decline followed by some improvement and the ability to eventually attain some axial functionality. That being said, patients typically do not achieve ambulatory function (31; 86). Morbidity and mortality are usually due to secondary complications of underlying weakness. Most commonly, morbidity and mortality are directly related to respiratory or pulmonary insufficiency secondary to accessory and intercostal muscle weakness, especially in more severe LAMA2-CMD cases. Thus, a notable portion of patients, as high as a third of patients, may need noninvasive ventilatory support (31; 86). Complicating this is swallowing and feeding difficulty, resulting in aspiration, and subsequent complicating pneumonia. Premature death may occur from pneumonia or other respiratory complications. In a series by Pegoraro and colleagues, four of 22 patients died between 5 and 10 years of age (79). Thus, most patients in the London-based Dubowitz Neuromuscular Center study were noted to have gastrostomy tube needs (86). Awareness of the risk of cardiac and CNS complications (including seizures) and early detection can significantly reduce morbidity. A patient with merosin-deficient congenital muscular dystrophy was reported to have malignant hyperthermia in the absence of classic triggering agents (91). The life expectancy in congenital muscular dystrophy is not known but is changing with improved care standards.
A female infant was a product of a nonconsanguineous union born after a normal pregnancy. There was no family history of neuromuscular disease. Poor feeding and failure to thrive were noted at 2 months of age. Examination revealed generalized hypotonia and muscle wasting with hyporeflexia. Baby was otherwise alert and attentive, and no cranial nerve abnormalities were noted. A nerve conduction study at 5 months of age revealed moderately slow motor and sensory nerve conduction responses without conduction block. Needle EMG examination showed grade II fibrillations and polyphasic motor unit potentials. Serum creatine kinase levels were 3275 IU. A muscle biopsy at the time showed muscle fiber necrosis, an increase of connective tissue, and marked lymphocytic inflammation.
Based on the muscle histology, a diagnosis of infantile inflammatory myopathy was made, and the child was started on daily prednisone treatment. She was able to sit at 12 months of age but remained nonambulatory. Her cognitive skills and speech were appropriate for her age. Repeat muscle biopsy at 2 years of age that was immunostained for laminin, dystrophin, and sarcoglycans showed an absence of merosin (LAMA2).
A brain MRI showed abnormal white matter with some posterior dominance and no cortical defect. During a 3-year follow-up, there was no clinical deterioration in her condition, and she did not develop any central nervous system symptoms. Steroid treatment was discontinued. The mother of the child had a subsequent pregnancy where merosin staining from a chorion villus sample revealed that the fetus was not affected. This finding was confirmed by genetic linkage analysis.
More than 10 genes are associated with different congenital muscular dystrophies, elucidating pathologic mechanisms related to processing of proteins (44). Merosin-deficient congenital muscular dystrophy is an autosomal recessive condition due to mutations in the LAMA2 gene on chromosome 6q22-23 (43; 42; 73). The LAMA2 gene is more than 260 kb long and contains 65 exons, with a transcript that is almost 10 kb in size (118). The LAMA2 encodes the laminin-alpha 2 chain, one of the subunits of laminin-211.
The laminins are members of the large basal lamina glycoprotein complex, and they consist of one heavy (alpha) and two light beta and gamma chains. In muscle, the heavy chain is the laminin alpha2-chain (lamininM), or merosin.
Merosin consists of 3110 amino acids and has three globular (VI; IVb; and IVa) and three rod (V; IIIa; and IIIb) domains. Domains I and II participate in the laminin triple-coiled structure. The G domain is the C-terminal part of merosin, which binds either the cell membrane dystroglycan or the integrin (alpha7 beta1) complex (79; 61). In electron microscopic studies of normal muscle, merosin in basal lamina is distributed in zones and localized to the lamina lucida of basal lamina. Merosin molecules form short, fine, cross-bridge fibrils connecting the basal lamina to the muscle plasma membrane (90). The association of laminin with dystroglycan suggests that it may have a role in maintaining the integrity of muscle cell membrane during contraction and stretching (57). Furthermore, laminins are known to affect cell adhesion, cell migration, and muscle regeneration (117; 101; 50). Massive muscle fiber degeneration occurs in the early stage of LAMA2-CMD and may contribute to the severe dystrophic changes in muscle seen in early infancy (40). The loss of muscle‐matrix interaction plays a central role in the pathogenesis of LAMA2-CMD: mouse model intramuscular administration of laminin 111 reduces apoptosis, increases fiber size, and improves muscle strength by stabilizing the interaction between the muscle membrane and extracellular matrix in Lama 2–deficient mice (85; 20). Apoptotic cell death has been implicated in muscle fiber necrosis, and antiapoptotic agents show some benefit in mouse models of disease (24; 32). The amount of residual laminin alpha 2 correlates with the phenotype severity in transgenic mice.
Laminins are present in most tissues, and merosin is found in skeletal muscle and also in skin, heart, peripheral nerve, brain, and placenta. In the peripheral nervous system, merosin is localized to the endoneural basal membrane, where it is likely to bind the dystroglycan complex and alpha6 beta4 integrin on the surface of Schwann cells (112). Interestingly, expression of an agrin minigene (agrin binds to basement membrane and to alpha-dystroglycan) restored muscle function in a transgenic mouse model (dy mouse models) for congenital muscular dystrophy (63). An AAV9 vector containing miniagrin (AAV9-Mini-Agrin) has been found to be beneficial in extending lifespan and improving muscle pathology in the dy mouse (83). It is well established that laminins play a role in peripheral nerve myelin formation, and a myelin abnormality is likely to be responsible for the slow nerve conduction velocity in patients with congenital muscular dystrophy. In laminin alpha-2 null mutant mouse (dy3K/dy3K), nerve myelination occurred, but Schwann cells lacked basal lamina in the spinal roots, suggesting that basal lamina is not an absolute requirement for myelination in vivo. In this mouse model, the motor conduction velocity of the sciatic nerve was significantly reduced, probably due to small axon diameter, thin myelin sheath, and patchy disruption of the basal lamina at the nodes of Ranvier (67). A single case report showed a severe loss of myelin in peripheral nerves, which was associated with a single base pair mutation that affected the laminin-laminin binding (76). However, no myelin sheath defect was found in a few nerve biopsies from other patients (96).
The mutations seen in LAMA2 gene are mainly single base pair changes or small deletions, causing loss of function in 95% of cases (36). These mutations are scattered throughout the gene, but about one third are localized in the C-terminal domain (79; 17; 73). In a cohort of 26 patients from Portugal, one third had exon 56 deletion (74). Total absence or partial loss of merosin has been found in cases of congenital muscular dystrophy with similar clinical severity. Single-point missense mutations can produce partial merosin deficiency and a milder late-onset muscular dystrophy (03; 73). A case report indicated that even a loss-of-function mutation in the LAMA2 gene caused only a mild form of disease, suggesting the existence of potential disease modifiers (81). In other cases, partial merosin deficiency is unrelated to LAMA2 gene mutations and may be a secondary effect of abnormalities in other basal lamina components such as fukutin (39). In general, nonsense mutations in LAMA2 cause more severe phenotype than missense or splice site mutations (31).
LAMA2 gene mutations associated with dystrophic muscle pathology, like congenital muscular dystrophy, have been found in mice (dy and dy2J) (110). The dy mouse lacks merosin completely, develops progressive weakness by 4 weeks of age, and dies within 2 to 3 months. The dy2J mutant mouse has a truncated merosin protein corresponding to a less severe clinical course. Studies in dyW LAMA2 congenital muscular dystrophy mouse models have suggested upstream immunomodulator gene upregulation (eg Lgals3/Galectin-3), resulting in excessive proinflammatory response (including lymphocytic infiltration and increased pro-inflammatory macrophages) that was reduced with Galectin-3 inhibition (TD-139) (16).
The relationship between brain abnormalities and merosin deficiency is not well understood. Diffusion-weighted images from the brain suggest that there is an increase in water content in the cerebral white matter (01), a theory also supported by MR spectroscopy (52). Extensive white matter abnormality exists in almost all complete or partial merosin-deficient patients; cortical migrational abnormalities, polymicrogyria, cortical heterotopia, or cerebellar hypoplasia can be found in some patients (93; 80). The white matter abnormalities seen on MRI resemble the demyelinization characteristic of some leukodystrophies, but histological studies failed to document myelin abnormalities (96). Studies have demonstrated that LAMA2 stimulates oligodendrocytes to extend elaborate membrane sheets using an integrin-linked kinase (13). Merosin has been localized to the basement membrane of capillaries in the mature brain, which suggests that disruption of the blood-brain barrier and serum extravasation are responsible for the abnormal white matter signal on MRI (106). The dy mouse also shows loss of merosin from the brain capillaries; however, MRI of the brain does not reveal any white matter abnormalities (21). Dysfunction in agrin, a basal lamina glycoprotein crucial for the formation and maintenance of neuromuscular junctions, potentially contributes to the impaired neuromuscular transmission described in LAMA2-CMD (49). A review further details the correlation between brain dysfunction in LAMA2-CMD from human case reports and mouse models (05).
LAMA2-CMD seems to occur in all races, presenting within the first 24 months of life. The prevalence for all congenital muscular dystrophies is estimated between 2.5 and 4.7 per 100,000 live births. A European study revealed that LAMA2-CMD constitutes approximately 10% to 30% of total congenital muscular dystrophy cases in that population (49), whereas it is the most common congenital muscular dystrophy affecting approximately four in 500,000 children (70) in another report. It was noted in one study that, of the 53 clinically and genetically defined congenital muscular cases, LAMA2-CMD was the third largest group (about 10%) after collagen VI-related (19%) and dystroglycan-related (12%) congenital muscular dystrophy (14). Similar results were seen in an Italian study with a congenital muscular dystrophy point prevalence of 0.563 per 100,000. Mutations were identified in 65%, and of those, 24.11% had merosin deficiency, with the other two major causes being collagen VI deficiency (20.24%) and dystroglycan deficiency (40.18%) (34). In Japan, Fukuyama muscular dystrophy appears more prevalent than LAMA2-CMD (99).
Prenatal diagnosis is available from chorionic villus biopsy (25). Immunohistochemical staining of merosin in chorion villi is informative in cases with complete LAMA2 deficiency but is not reliable in cases with partial deficiency. Prenatal diagnosis is also possible based on mutation detection or linkage analysis (36). Protein and DNA analysis can be used either independently or in combination to provide an accurate prenatal diagnosis (102).
Congenital muscular dystrophy can be distinguished from the congenital myopathies (eg, structural defects of muscle or metabolic myopathies) based on large elevations of creatine kinase levels and muscle histology. Congenital muscular dystrophy should be a consideration in the phenotype of a “floppy infant.” Lack of encephalopathy, normal cranial nerve findings, decreased or absent deep tendon reflexes, and decreased muscle bulk are important clues for differentiating congenital muscular dystrophy from central hypotonia. Spinal muscular atrophy (type I) and congenital myasthenia can manifest within the first month of life, whereas LAMA2 congenital muscular dystrophy is less severe and is usually recognized later. The presence of high creatine kinase levels is diagnostic of a congenital muscular dystrophy. Nerve conduction studies and EMG can help differentiate a congenital muscular dystrophy from spinal muscular atrophy and myasthenia. Rigid spine syndrome is a rare congenital muscular dystrophy characterized by early rigidity of the spine and respiratory insufficiency and has been associated with mutations of a selenoprotein (SEPN1) gene on chromosome 1p35-36 (62).
Congenital muscular dystrophies are a heterogeneous group of early-onset muscular dystrophies with or without clinical signs of central nervous system involvement. The presence of structural abnormalities in the central nervous system suggests a deficiency in one of the glycosyltransferases that may cause Fukuyama muscular dystrophy, Walker-Warburg disease, or "muscle-eye-brain disease” (MEB) (35). The distinguishing features between these subgroups are severe brain involvement and dysmorphic features. Interestingly, a secondary decrease of LAMA2 occurs in Fukuyama muscular dystrophy but not in Walker-Warburg syndrome (107; 114). About one half of occidental congenital muscular dystrophies are caused by LAMA2 deficiency, whereas the rest are related to defects in integrins, alpha-actinin, or have unknown etiology (71; 22). Inherited congenital muscular dystrophy with no LAMA2 abnormality was linked to chromosome 4p in some families (87). It must be noted that partial LAMA2 deficiency can be responsible for early-onset congenital muscular dystrophy, as well as late-onset muscular dystrophy (most likely limb-girdle type) (03; 95). In these cases, a white matter abnormality is also present on MRI. Late-onset muscular dystrophy with partial LAMA2 deficiency can occur in adults, but some of these cases are secondary and not linked to the LAMA2 gene (10; 41). Ambulatory ability was associated with a missense, single-point mutation or premature termination in an exon with potential for alternative splicing, all resulting in a partial deficiency (73).
If congenital muscular dystrophy is suspected in an infant, serum creatine kinase levels will confirm primary muscle involvement, demonstrating CPK up to five times the upper limit of normal, commonly more than 1000 IU/L (89; 49). Patients with congenital myopathies due to structural defects in the muscle fibers usually do not have high creatine kinase. Nerve conduction studies and needle EMG are useful to differentiate congenital muscular dystrophy from motor neuron disease or myasthenia. Nerve conduction study shows mild to moderate delay in motor nerve conduction (104). Brain MRI is highly useful to exclude severe brain malformation, especially if the patient has dysmorphic features. A diffuse white matter abnormality is almost always diagnostic of a LAMA2-CMD when combined with a high CK level. Occasionally, more severe brain abnormalities have also been found (93; 80). Lower limb MRI has also been used more frequently, demonstrating fatty replacement patterns, but may mimic other muscular dystrophies (69; 38; 94). Muscle biopsy frequency has declined with the advent of genetic sequencing. Specific immunostaining for dystrophin and associated protein complex, including LAMA2, can assist in the diagnosis. Antibodies to both the C-terminus and the central domains of LAMA2 are required to recognize partial merosin deficiencies. If a muscle biopsy is unavailable, LAMA2 staining of a skin biopsy can be informative (88).
Genetic testing for LAMA2 gene mutations has become available. O'Grady and colleagues evaluated 85 congenital muscular dystrophy cases over the last 20 years using conventional approaches of biopsy and immunohistochemistry compared to next-generation sequencing and found that with conventional testing, 51 (60%) remained undiagnosed (72). Twenty-eight of the 51 consented to next-generation sequencing, resulting in 11 confirmed diagnoses. Using a combination of approaches, 51% of patients received a genetic diagnosis. As the cost of genetic testing continues to decrease and its availability increases, earlier and more reliable diagnosis is possible (73).
No effective therapy is available for LAMA2-CMD. There is a “consensus statement” published on care standards that is applicable for this form of congenital muscular dystrophy (108; 46). Supportive treatment includes gastric tube placement for feeding, scoliosis surgery, and treatment of respiratory illnesses. Physical therapy may help prevent muscle contractures. Health-related quality of life and the factors that impact it are key components of therapy (15). In the future, urine miRNA analysis may help in the staging of disease (64). Multiple future therapies are being investigated.
No clinical trials with steroids have been reported for LAMA2-CMD. Therapeutic approaches tried in the animal models of the disease have shown mixed results. In a rodent model of LAMA2-CMD, the inhibition of apoptosis by overexpression of Bcl2 improved the clinical outcome (33; 19). Ablation of myostatin (a muscle growth inhibitory factor) did not provide benefit in a mouse model (53). Various compounds are under investigation that can induce a “read-through” of stop codons in the laminin-alpha2 gene (02; 60). Exon-skipping technology with novel oligomers can partially rescue laminin2 expression (37). Other preclinical studies using proteosome inhibitors, agrin molecule and a laminin-alpha 1 residue are under investigation in animal models (28; 12; 56). Various treatment trials with losartan (an angiotensin-2 type 1 receptor blocker), alpha-7 integrin overexpression, and laminin-111 (an embryonic form of laminin) showed clinical efficacy in LAMA2 deficient mice and hold promise in future treatments for congenital muscular dystrophy (18; 23; 04; 103). Metformin was shown to increase weight gain, energy efficiency, muscle function, and histology in female mouse models (to some extent in males) of LAMA2-CMD (27).
Preclinical treatment approaches. Approaches successfully used in other neuromuscular disorders are also being investigated for LAMA2-CMD. Different approaches for developing a therapy for LAMA2-CMD have been devised and primarily fall into three categories. The first aims to stabilize or restore the structure and function of the basement membrane as well as its interactions with adjacent cells. The second aims to modulate cellular events caused by LAMA2 loss. The third group targets the genetic defect in LAMA2-CMD, either through mRNA processing and translational read-through, using oligonucleotides or small molecules, respectively, or correcting the causative mutation using CRISPR/Cas9 system.
In the first group, intramuscular administration of laminin 111 resulted in membrane stabilization and functional muscle improvement in Lama 2–deficient mouse models (85). Laminin-111 protein was upregulated in muscles and nerves of Lama2dy2J/dy2J mice (47). Specifically, it rescued myelination and nerve conduction velocities in peripheral nerves.
In the second group, mini-agrin was shown to ameliorate LAMA2-deficient phenotype in animal models (84). An AAV9-Mini-Agrin vector was tested in a mouse model of the disease and improved muscle pathology and lifespan (83). AAV insertion of shortened associated αLN linker protein DNA demonstrated increased strength and sciatic myelination (55). Omigapil, an apoptosis inhibitor (related to GAPDH and SIAH1 enzymes), has also shown a safety and reduction in muscle tissue loss in LAMA2-CMD (115; 26); clinical trials are ongoing in early phases.
In the third group, AAV-based gene replacement therapy has been difficult due to the large gene size. However, Packer and colleagues created an AAV-deliverable “microlaminin” gene based on precedence from micro-dystrophin (75). It demonstrated successful microlaminin expression as well as improved muscle strength but did not address all disease phenotypes. Antisense oligonucleotide-based exon skipping has been another technique used in muscular dystrophies; removal of a stop codon promotes merosin re-expression and resulted in mild improvement in a mouse model of LAMA2-CMD (04). Kemaladewi and colleagues delivered CRISPR-Cas9 genomic editing components using AAV and demonstrated the ability to correct a splice site mutation in exon 2 and restore truncated merosin to full length in a mouse model (48). Another example using CRISPR/Cas9d activation system confirmed earlier work (30) and targeted the Lama1 gene promoter delivered by adeno-associated virus (AAV9) (47).
Detailed information on both the underlying pathomechanisms and strategies for therapy development that address each of these areas and animal models for testing has been published in a series of review articles (111; 70; 116; 07; 29; 51; 82; 113).
Future investigations also include upstream targets and proinflammatory pathway modulation. Mouse models (dyW) treated with TD-139, a Galectin-3 inhibitor (see section on biological basis), suggest reduced inflammatory and fibrosis activity and increased muscle contraction related gene expression (16).
Malignant hyperthermia can occur in merosin-deficient congenital muscular dystrophy; therefore, caution should be taken during general anesthesia.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Neil Datta MD
Dr. Datta of Harvard Medical School has no relevant financial relationships to disclose.
See Profile
Partha Ghosh MD
Dr. Ghosh of Boston Children's Hospital and Harvard Medical School received honorariums from Sarepta as an ad hoc advisory board member and speaker and an honorarium from CVS Caremark as a consultant.
See Profile
Aravindhan Veerapandiyan MD
Dr. Veerapandiyan of University of Arkansas for Medical Sciences has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuromuscular Disorders
Jun. 16, 2026
Neuromuscular Disorders
May. 27, 2026
Neuromuscular Disorders
May. 21, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 24, 2026