

Epilepsy & Seizures
Febrile seizures
Jun. 02, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Epilepsy is a common serious disorder of the brain characterized by recurrent epileptic seizures. Differential diagnosis depends on distinguishing between nonepileptic events that resemble epileptic seizures, epileptic seizures that are provoked and do not indicate a diagnosis of epilepsy, and epilepsy, which implies the presence of an epileptogenic disturbance in the brain even when seizures are not occurring. Treatment is based on the diagnosis of specific epileptic seizure types and, when present, specific epilepsy syndromes. Pharmacotherapy is the treatment of choice. In 2024, the International League against Epilepsy (ILAE) formally requested that the old term “antiepileptic drug” (AED) be replaced with the more specific term “antiseizure medication” (ASM) (70). Sixty percent to 70% of patients have seizures that can be controlled with antiseizure medications. Alternative therapies include, but are not limited to, surgical treatment, which is highly effective for specific types of pharmacoresistant epilepsy; neuromodulation, including vagus nerve stimulation, responsive neurostimulation, and deep brain stimulation; the ketogenic diet; and behavioral approaches. The health burden of epilepsy results not only from epileptic seizures but also from the social, psychological, and neurologic consequences of these seizures, which can include disabling morbidity and increased mortality. In this updated article, the author stresses the importance of referral to a full-service, multidisciplinary epilepsy center when trials of two antiseizure medications fail.
|
• Epilepsy is the most common serious primary disorder of the brain: 10% of people will have at least one seizure in a lifetime. One third of these will develop epilepsy, and approximately 1% of the world’s population has active epilepsy. | |
|
• Epilepsy is not a benign condition: it accounts for 1% of the global burden of disease due to disability and premature death, equivalent to lung cancer in men and breast cancer in women. | |
|
• The global burden of disease attributed to epilepsy is comparable to depression, dementia, and substance abuse. | |
|
• The primary treatment for epilepsy is antiseizure medications, which can control disabling seizures in approximately two thirds of people with epilepsy. | |
|
• People with epileptic seizures that are not controlled by medication are at greater risk for irreversible disability and increased mortality and should be referred to a full-service epilepsy center. |
The earliest medical writings of antiquity mention epileptic seizures, and some accurately ascribed them to disorders of the brain. However, the dramatic intermittent nature of this affliction caused it to be shrouded in mysticism for many centuries. In medieval Europe, it was known as the falling sickness or the sacred disease and was often ascribed to possession by evil spirits (87). Later, it was generally considered to be a psychiatric condition, until a biological basis was seriously considered in the mid-19th century. The introduction of effective treatment at about the same time, including antiseizure medications such as bromides and barbiturates, as well as surgical intervention, did much to alleviate the suffering of many persons with epilepsy, but stigma and misconceptions surrounding the diagnosis of epilepsy have persisted in all cultures and still contribute greatly to the disabilities associated with this disorder. There are several comprehensive textbooks on epilepsy (21; 67; 81; 11; 23; 82; 97; 26). In 2012, the Institute of Medicine Committee on the Public Health Dimensions of the Epilepsies published a comprehensive report with recommendations concerning epilepsy care, prevention, and surveillance (29).
|
• Classifications have evolved over the years. | |
|
• Epileptic seizures are still classified as focal or generalized. | |
|
• The epilepsies are also divided into focal, generalized, and combined but have a multifactorial classification that also considers seizure type, etiology, comorbidity, and syndrome when present. | |
|
• Epilepsy is defined as an enduring predisposition to generate epileptic seizures. |
Epileptic seizures. An epileptic seizure has been defined by the International League Against Epilepsy (ILAE) as “a transient occurrence of signs and/or symptoms due to abnormal excessive or synchronous neuronal activity in the brain” (35). The clinical manifestations are extremely variable and depend on the cortical areas involved. Epileptic seizures are usually self-limited, lasting a minute or two, and may be followed by a period of postictal cerebral depression manifested clinically as diffuse or localized neurologic deficits. Epileptic seizures may be reactive, reflecting a natural response of a normal brain to transient insults such as head trauma, fever, or alcohol withdrawal, or they may result from more permanent pathological processes within the brain, consisting of a chronic condition, epilepsy.
The classification of epileptic seizures has continued to evolve. The 1981 International Classification of Epileptic Seizures (13) divided the clinical manifestations into partial seizures, which begin in a part of one hemisphere, and generalized seizures, which begin in both hemispheres simultaneously (Table 1). Partial seizures that do not produce impairment of consciousness were referred to as simple partial seizures. When consciousness is impaired, they were referred to as complex partial seizures. The signs and symptoms of simple partial seizures are determined by the function of the cortical area involved and were divided by this International Classification into motor, sensory, autonomic, and psychic phenomena. Simple partial seizures without motor manifestations that precede more obvious ictal events are commonly referred to as auras. Abnormal discharges that begin in a discrete area of the brain and give rise to partial epileptic seizures can propagate over time. Thus, simple partial seizures can evolve into complex partial seizures, and both can evolve into generalized convulsive seizures, referred to as secondarily generalized seizures.
|
I. Partial (focal, local) seizures | ||
|
A. Simple partial seizures | ||
|
1. With motor signs | ||
|
B. Complex partial seizures | ||
|
1. Simple partial onset followed by impairment of consciousness | ||
|
C. Partial seizures evolving to secondarily generalized seizures | ||
|
1. Simple partial seizures evolving to generalized seizures | ||
|
II. Generalized seizures (convulsive or nonconvulsive) | ||
|
A. Absence seizures | ||
|
1. Typical absences | ||
|
B. Myoclonic seizures | ||
|
III. Unclassified epileptic seizures | ||
|
| ||
Generalized seizures can be major motor, including generalized tonic-clonic or grand mal seizures, as well as purely tonic and purely clonic generalized seizures. Other generalized seizure types include:
|
• Absences, consisting of a lapse of consciousness, at times associated with minor movements such as eye blinking or facial twitching; absences can be brief (usually less than 10 seconds), with sudden onset and offset, and associated with a characteristic 3-per-second spike-and-wave EEG pattern (typical, or petit mal absences), or they can be of longer duration with some postictal confusion and less regular EEG features (atypical absences); | |
|
• Myoclonic seizures, consisting of brief, sudden bilaterally synchronous muscle jerks without impaired consciousness; | |
|
• Brief tonic seizures, consisting of slightly more prolonged bilaterally synchronous tonic contractions, with or without impairment of consciousness and often causing the patient to fall; | |
|
• Atonic seizures, consisting of bilaterally synchronous loss of tone, with or without impairment of consciousness, also resulting in falls. |
When atonic, brief tonic, and occasionally myoclonic seizures result in falls (drop attacks, astatic seizures), they can cause serious injury and extreme disability. Auras, by definition, do not occur before these generalized seizures; however, prodromal symptoms due to systemic disturbances that herald the onset of generalized seizures may be reported.
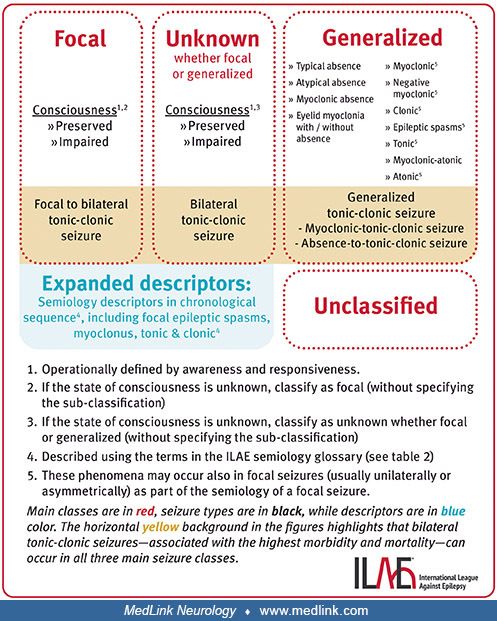
The ILAE has revised the International Classification of Epileptic Seizures multiple times over the years (19; 20; 05; 07; 78; 34; 33; 79; 04). The 1981 classification was based predominantly on EEG and seizure semiology because there was insufficient information on pathophysiology and anatomical substrates to create a more scientifically based mechanistic classification. The ILAE attempted to identify epileptic seizure types that can be considered to represent unique diagnostic entities based on specific criteria, including pathophysiologic mechanisms, neuronal substrates, response to antiseizure medications, ictal EEG patterns, propagation patterns, postictal features, and epilepsy syndromes in which they occur (20). In a subsequent ILAE classification, generalized seizures are viewed as “originating at some point within, and rapidly engaging, bilaterally distributed networks” (05). Such bilateral networks can include cortical and subcortical structures but not necessarily include the entire cortex. Although individual seizure onsets can appear localized, the location and lateralization are not consistent from one seizure to another. Generalized seizures can be asymmetric.” Focal epileptic seizures are viewed as “originating within networks limited to one hemisphere. They may be discretely localized or more widely distributed. For each seizure type, ictal onset is consistent from one seizure to another, with preferential propagation patterns that can involve the contralateral hemisphere. In some cases, however, there is more than one network and more than one seizure type, but each individual seizure type has a consistent site of onset. Focal seizures do not fall into any recognized set of natural classes based on any current understanding of the mechanisms involved.” Most recently, the formal classification of epileptic seizures divides them broadly into those with focal onset, those with generalized onset, and those with unknown onset.
Each of these is divided into those with motor onset and those with nonmotor onset. In addition, focal onset seizures require added indication of whether conscious is preserved, or whether consciousness is impaired. Classification is dependent on initial signs and symptoms, with the knowledge that seizures can evolve; however, determination of impaired awareness is made if the impairment occurs at any time during the seizure. The major change in the 2025 classification, from the 2017 classification, is the emphasis on noting preserved or impaired consciousness for focal and unknown seizure types. Consciousness is operationally defined by awareness and responsiveness.
Changes in recent classifications from the 1981 classification include:
|
• Neonatal seizures are not listed, and the ILAE now has a specific classification for neonatal seizures. | |
|
• Absence seizure classification has been simplified; however, myoclonic absence seizures and eyelid myoclonia have been added. | |
|
• Epileptic spasms have been added. This includes infantile spasms, but it is recognized that epileptic spasms may also occur in older children and adults. | |
|
• Myoclonic atonic (previously called “myoclonic astatic”) seizures have been added. | |
|
• Seizure types described previously as only generalized are now acknowledged to also occur as focal events, including epileptic spasms, tonic, clonic, atonic, and myoclonic seizures. | |
|
• Secondarily generalized seizures are now referred to as focal to bilateral tonic-clonic seizures. |
In 2021, the ILAE also published a modification for neonatal seizures (74). All neonatal seizures are focal and are motor, nonmotor, or without behavioral correlates.
Epilepsy. The ILAE has defined epilepsy conceptually as “a disorder of the brain characterized by an enduring predisposition to generate epileptic seizures and by the neurobiological, cognitive, psychological, and social consequences of this condition. A definition of epilepsy requires the occurrence of at least one epileptic seizure” (35). This definition represented a change from previous definitions in that it defines epilepsy not as recurrent epileptic seizures but as a condition where seizures reflect the existence of an enduring epileptogenic disturbance with the potential to generate further seizures. Reactive (acute symptomatic, provoked) seizures due to transient insults of an otherwise normal brain that resolve spontaneously, or when the cause is successfully eliminated, do not warrant a diagnosis of epilepsy. Objections to this conceptual definition of epilepsy were raised over concern that, taken literally, it could lead to inappropriate diagnoses based on a single seizure (03). It is necessary to clarify, therefore, that this is intended to be an essential definition of what epilepsy is, absolutely--a condition in which seizures occur in an abnormally epileptogenic brain, as opposed to seizures that are a natural, albeit abnormal, response of a normal brain to transient insult. Although this definition conforms to what physicians actually do in practice, ie, attempt to determine whether there is an enduring predisposition to recurrent seizures, as an absolute or ideal concept, it is not necessarily easy to apply in all circumstances. There is, therefore, a need for specific operational definitions of epilepsy for different purposes.
As a result, the ILAE has suggested an operational clinical definition of epilepsy that can be practically applied (Table 2).
|
A. Epilepsy is a disease of the brain defined by any of the following conditions: | |
|
1. At least two unprovoked (or reflex) seizures occurring greater than 24 hours apart. | |
|
2. One unprovoked (or reflex) seizure and a probability of further seizures similar to the general recurrence risk (at least 60%) after two unprovoked seizures, occurring over the next 10 years. | |
|
3. Diagnosis of an epilepsy syndrome. | |
|
B. Epilepsy is considered to be resolved for individuals who had an age-dependent epilepsy syndrome but are now past the applicable age or for those who have remained seizure-free for the last 10 years, with no seizure medicines for the last 5 years. | |
|
| |
This operational definition is important not only for providing practical guidelines for diagnosing epilepsy in the presence of only a single seizure, but it recognizes reflex epilepsy even though unprovoked seizures do not occur, and it establishes a time frame for determining when epilepsy can be considered resolved, although it does not suggest that these patients are cured. This operational definition also emphasizes the current view that epilepsy is a disease (22). It is additionally important to maintain the portion of the conceptual definition, which identifies the neurobiological, cognitive, psychological, and social consequences of epileptic seizures, which are important parts of the diagnostic and therapeutic approaches to people with epilepsy. The ictal events encountered in disorders associated with reactive seizures, such as benign febrile seizures, are usually generalized; however, they may be focal when preexisting localized cerebral disturbances make one area of the brain more epileptogenic than others (for instance, old cerebral contusions common in alcoholics may cause them to have focal alcohol-withdrawal seizures).
The 1989 International Classification of Epilepsies and Epileptic Syndromes (14) divided epilepsy disorders into those due to inherited epileptogenic cerebral dysfunction (genetic or idiopathic epilepsy) and those due to specific structural abnormalities, which may be genetic (such as tuberous sclerosis) or acquired (secondary or symptomatic epilepsy) (Table 3). When a disorder is presumed to be symptomatic epilepsy, but the specific underlying pathological substrate has not been identified, it was referred to as cryptogenic. Epilepsy disorders were further subclassified as partial or generalized, depending on whether the underlying disturbance is believed to be localized to a part of the brain or diffusely distributed over both hemispheres. This classification also recognized undetermined epilepsy disorders when there is insufficient information for classification and disorders associated with reactive seizures, referred to as situation-related epilepsies. Disorders characterized by ictal events that are induced by specific stimuli (eg, photogenic epilepsy, reading epilepsy, startle epilepsy) were referred to as reflex epilepsies.
|
I. Localization-related (focal, local, partial) | |||
|
A. Idiopathic (genetic) | |||
|
1. Benign childhood epilepsy with centrotemporal spikes | |||
|
B. Symptomatic (secondary) | |||
|
1. Temporal lobe epilepsies | |||
|
C. Cryptogenic, defined by: | |||
|
1. Seizure type | |||
|
II. Generalized | |||
|
A. Idiopathic (genetic) | |||
|
1. Benign neonatal familial convulsions | |||
|
B. Cryptogenic or symptomatic | |||
|
1. West syndrome (infantile spasms, Blitz-Nick-Salaam Krämpfe) | |||
|
C. Symptomatic (secondary) | |||
|
1. Nonspecific etiology | |||
|
a. Early myoclonic encephalopathy | |||
|
2. Specific syndromes | |||
|
a. Epileptic seizures may complicate many disease states. | |||
|
III. Undetermined epilepsies | |||
|
A. With both generalized and focal seizures | |||
|
1. Neonatal seizures | |||
|
B. Without unequivocal generalized or focal features | |||
|
IV. Special syndromes | |||
|
A. Situation-related seizures (Gelegenheitsanfälle) | |||
|
1. Febrile convulsions | |||
|
| |||
A 2006 ILAE report (20) also developed criteria for identifying specific epilepsy syndromes as discrete diagnostic entities, which include the epileptic seizure type associated with it, age of onset, progressive nature of the disorder, interictal EEG abnormalities, associated interictal signs and symptoms, pathophysiologic mechanisms, anatomical substrates, etiologic categories, and genetic basis. The 2010 ILAE report eliminated the terms “idiopathic,” “symptomatic,” and “cryptogenic,” but it recommends the use of the following terms when appropriate (05):
|
• Genetic: These disorders are “the direct result of a known or presumed genetic defect(s) in which seizures are the core symptom of the disorder, but the possibility that environmental factors contribute to expression of the disease is not excluded.” | |
|
• Structural-metabolic: In these conditions, “there is a distinct other structural or metabolic condition or disease that has been demonstrated to be associated with a substantially increased risk of developing epilepsy in appropriately designed studies.” These disturbances can be acquired or genetic (eg, tuberous sclerosis). | |
|
• Unknown cause |
With respect to classification of the epilepsies, the 2010 ILAE report recognized that the former dichotomies of idiopathic versus symptomatic and focal versus generalized are false, and it recommended that they be abandoned. Rather, syndromes can be characterized according to many different features, including “age of onset, cognitive and developmental antecedents and consequences, motor and sensory examinations, EEG features, provoking or triggering factors, and patterns of seizure occurrence with respect to sleep.” A list of accepted electroclinical syndromes and other epilepsies, organized by age, is shown in Table 4.
|
Electroclinical syndromes arranged by age at onset* | |
|
Neonatal period | |
|
• Benign familial neonatal epilepsy (BFNE) | |
|
Infancy | |
|
• Epilepsy of infancy with migrating focal seizures | |
|
Childhood | |
|
• Febrile seizures plus (FS+) (can start in infancy) | |
|
Adolescence – Adult | |
|
• Juvenile absence epilepsy (JAE) | |
|
Less specific age relationship | |
|
• Familial focal epilepsy with variable foci (childhood to adult) | |
|
Distinctive constellations | |
|
• Mesial temporal lobe epilepsy with hippocampal sclerosis (MTLE with HS) | |
|
Epilepsies that do not fit into any of these diagnostic categories can be distinguished firstly on the basis of the presence or absence of a known structural or metabolic condition (presumed cause), and then on the basis of the primary mode of seizure onset (generalized vs. focal). | |
|
Epilepsies attributed to and organized by structural-metabolic causes | |
|
• Malformations of cortical development (hemimegalencephaly, heterotopias, etc.) | |
|
Epilepsies of unknown cause | |
|
Conditions with epileptic seizures that are traditionally not diagnosed as a form of epilepsy per se. | |
|
• Benign neonatal seizures (BNS) | |
|
* The arrangement of electroclinical syndromes does not reflect etiology. | |
|
| |
In 2001, the ILAE recommended a diagnostic scheme for describing epileptic disturbances in individual patients that deviated from the previous taxonomic approval (19). This is not, however, a classification. The scheme consisted of five axes: an optional semiological description of ictal signs and symptoms, seizures as diagnostic entities, epilepsy syndromes, etiology, and an optional designation of impairment. Most recently, the ILAE has used this approach to introduce a new classification of the epilepsies (79). This classification consists of three levels. The first level is the seizure type, as defined in the “Classification of Seizures. The second level is the epilepsy type, which is divided into focal epilepsy, generalized epilepsy, combined generalized and focal epilepsy, and unknown epilepsy type. The third level is the epilepsy syndrome, when this can be made. A unique feature of this classification is that etiology is considered at each level, etiology being divided into structural, genetic, infectious, metabolic, immune, and unknown.
Although now recognizing the group of syndromes referred to as idiopathic generalized epilepsies, this classification emphasizes that they are more correctly referred to as genetic generalized epilepsies. It is recommended that the term “benign” be replaced with “self-limited” and “pharmacoresponsive” where appropriate. Epileptic encephalopathies are redefined as “developmental and epileptic encephalopathies,” applicable to changes occurring as a result of epilepsy at any age. Finally, it is acknowledged that comorbidities need also to be identified in individual patients.
The ILAE has now also published more detailed descriptions of syndromes (95), including definitions of those with onset in neonates and infants (100), childhood (84), and at variable ages (76), as well as a definition of idiopathic generalized syndromes (45). An ILAE diagnostic manual can be found at the following website: EpilepsyDiagnosis.org.
The genetic epilepsies are usually benign and many resolve in late adolescence or early adulthood, although we now know that this is not always the case; Dravet syndrome, for instance, is far from benign. The prognosis for epilepsies with structural-metabolic causes depends on the underlying pathological substrate. Patients with seizures due to diffuse brain damage usually have other neurologic and cognitive impairment that contributes to their disability. Such patients may require institutionalization because of intractable epileptic seizures, consequences of the underlying neurologic disorder, or both. Patients with epilepsies due to single well-circumscribed lesions may have their seizures easily controlled with antiseizure medications or be essentially cured by surgical resection. It is now accepted that some epilepsies can be progressive disorders and that seizures themselves, under certain circumstances, can injure the brain or induce enduring dysfunction such as psychosis or other behavioral disturbances (85). Overall, 30% of patients with a diagnosis of epilepsy have seizures that are poorly controlled by antiseizure medications, and many of the remainder have occasional seizures or neurologic or mental disturbances that could be due to the underlying substrate, repeated epileptiform discharges, or side effects of antiseizure medications. Sudden unexplained death in epilepsy (SUDEP) is a rare complication of epilepsy, and its cause and risk factors are largely unknown (37). Mortality rate from SUDEP and other causes is increased when seizures are poorly controlled (83), and death usually occurs in the prone position (58). The risk of SUDEP is dramatically increased for patients with uncontrolled bilateral tonic-clonic seizures who sleep alone (86). It is recommended that risk of SUDEP be discussed with patients (16). Stigma and limitations placed on persons with epilepsy by society are also a significant cause of psychosocial disability and may otherwise negatively impact quality of life. The International Consortium for Health Outcomes Measurement (ICHOM) is developing an international set of outcomes and measurement methods for routine practice for children and adults with epilepsy (61; 62).
|
• Epileptic seizures reflect altered excitatory and inhibitory activities in the brain that generate hypersynchronization. | |
|
• Many poorly understood mechanisms lead to the initiation of epileptic seizures and even less is known about why they stop. | |
|
• Multiple acquired and genetic factors contribute to the development of epilepsy. |
Epileptic seizures are presumed to result from an imbalance of excitatory and inhibitory influences within a neuronal aggregate, which predisposes to hypersynchronous discharges. The primary disturbances are believed to exist in the cerebral cortex. In the case of generalized epilepsies, however, abnormal as well as normal subcortical afferent input to susceptible epileptogenic cortex may also play an important role; the clinical manifestations of some generalized seizures involve diencephalic or brain stem-mediated phenomena. Recurrent epileptic seizures can result from disturbances in neuronal interconnections and neurotransmitter function, particularly involving the excitatory and inhibitory amino acid neurotransmitters glutamate and GABA. In acquired epilepsy, evidence exists that neuronal cell loss induces aberrant synaptic reorganization, which enhances excitatory, and to some extent inhibitory, influences and predisposes to spontaneous hypersynchronization. Similar mechanisms have not yet been demonstrated for the genetic epilepsies. Less is known about why seizures stop than about how they begin. The termination of ictal events is an active process involving, in part, purine and peptide neurotransmitters, particularly adenosine and the endogenous opiates. These mechanisms, in turn, contribute to the appearance of postictal symptoms. Many seizures occur with specific periodicity (02), and elucidating the reasons for this may lead to novel approaches to treatment.
The causes of epilepsy are multifactorial, involving both genetic and acquired factors. There are three potential genetic contributions to the appearance of an epileptic disorder: (1) variations among individuals in their susceptibility to epileptic symptoms given a transient epileptogenic insult or to chronic epilepsy given a specific structural lesion are in large part genetically determined; (2) some specific cerebral disturbances that give rise to structural-metabolic epilepsies are genetically determined, such as tuberous sclerosis and phenylketonuria; and (3) genetic epilepsies reflect genetic impairment in cerebral excitability and synchronization. Specific epilepsy genes are being rapidly identified, and most are sporadic mutations that are not inherited (88; 47).
Acquired lesions that give rise to structural-metabolic epilepsies can be bilateral and so diffuse that seizures appear to be generalized from the start, or the lesions may be localized to smaller areas of the brain, giving rise to focal seizures or generalized seizures with focal features. Common pathological substrates of structural-metabolic epilepsies include malformations of cortical development, hippocampal sclerosis, neoplasms and other tumors, and cerebral damage due to trauma, vascular accidents, and infection.
Much has been learned about the causes of focal epilepsy from examination of brain tissue removed as treatment for refractory seizures. The International League against Epilepsy has issued a recommendation for a comprehensive neuropathologic work-up of epilepsy surgery brain tissue (10).
|
• Ten percent of the population will have at least one epileptic seizure in a normal lifetime. | |
|
• One third of people who have an epileptic seizure will have epilepsy. | |
|
• Over 1% of the population has active epilepsy. | |
|
• Epilepsy accounts for 1% of the global burden of disease. |
The U.S. Centers for Disease Control and Prevention determined that 1.2% of the United States population, 3 million adults and 470,000 children, reported active epilepsy in 2015 (99). The incidence is approximately 50 per 100,000 per year. The figures are considerably higher, however, in areas with inadequate access to medical care or increased exposure to environmental hazards, such as in underdeveloped countries and inner-city areas of the United States. The age-related incidence varies depending on the epilepsy condition, but it is greatest within the first few years of life and again in the elderly. The accumulated risk of having at least one epileptic seizure over an 80-year lifespan is 1 in 10 (41). According to the World Health Organization, epilepsy accounts for 1% of the global burden of disease, based on disability-adjusted life years - that is, years of productivity lost as a result of disability or premature death. This is equivalent to the disease burden of lung cancer in men and breast cancer in women. Among primary disorders of the brain, epilepsy ranks with depression and other affective disorders, Alzheimer disease and other dementias, and substance abuse (63). Note that these data represent a gross underestimate of the true global burden of disease represented by epilepsy as conditions where a structural-metabolic cause was known were classified by that cause and not as epilepsy.
|
• Many causes of acquired epilepsy are preventable through public health efforts. | |
|
• There are no treatments that prevent epileptogenesis after a potential epileptogenic insult. |
Many acquired causes of epilepsy are preventable (89). Inadequate prenatal care, unsanitary conditions that result in intracerebral infections (particularly with cysticercosis, tuberculosis, and malaria), unacceptable risk of cerebral trauma from automobile accidents, violent crime, and armed conflict, and the widespread use of intravenous recreational drugs contribute significantly to the incidence of epilepsy worldwide. Controversy exists over whether isolated reactive epileptic seizures, such as febrile seizures, produce subsequent epilepsy. There is reliable evidence, however, that certain epileptic conditions are progressive. To cause epilepsy, closed-head trauma must be severe and involve loss of consciousness. The risk of posttraumatic epilepsy in these patients is increased with prolonged amnesia, skull fracture, and intracranial hemorrhage. Although prophylactic treatment with antiseizure medications following head trauma or intracranial surgery reduces the risk of early seizures, there is no evidence that it prevents the development of chronic epilepsy. There are no interventions that can prevent the development of epilepsy after a potentially epileptogenic insult, but the search for antiepileptogenic therapies is an active field of basic research.
|
• Nonepileptic seizures can be due to systemic, neurologic, or psychiatric conditions. | |
|
• Epileptic seizures due to transient insults of a normal brain that do not predispose to recurrent seizures are not epilepsy. |
|
• An important part of the differential diagnosis is the search for an underlying treatable cause (see Etiology). | |
|
• Often an underlying cause is not found, or is not treatable, or treatment does not stop seizures – in this case, treatment and prognosis depend on diagnosis of a syndrome or on the seizure type. |
Diagnosis in epilepsy requires first determining that an ictal event in question is epileptic. Many systemic, neurologic, and psychiatric conditions are associated with intermittent symptoms that can be mistaken for epilepsy, including syncope, hyperventilation, toxic and metabolic disturbances, cardiovascular disorders, sleep disorders, paroxysmal dyskinesias, hyperekplexia, hemifacial spasms, paroxysmal vertigo, trigeminal neuralgia, migraine, transient global amnesia, psychogenic seizures, (now proposed to be called functional/dissociative seizures) (44), episodic dyscontrol, and psychiatric dissociative states. Once it is determined that the ictal event was epileptic, it is necessary to determine the presence of an enduring epileptogenic abnormality before a diagnosis of epilepsy can be made. Reactive (acute symptomatic, provoked) seizures are a natural response of the normal brain to transient insult and do not recur once the insult is removed. These events do not constitute an enduring epileptogenic abnormality and should not be diagnosed as epilepsy. If an enduring predisposition to generate epileptic seizures is identified or suspected, the next diagnostic step is to search for an underlying treatable cause. If such a cause (eg, a brain tumor) is successfully treated and seizures no longer exist, the patient should not be given a diagnosis of epilepsy. When seizures persist, either because an underlying treatable cause has not been found or treatment has not resulted in the remission of seizures, a diagnosis of epilepsy is warranted. Diagnostic evaluation is then aimed at determining what type(s) of seizures are occurring and whether the clinical features constitute a recognized epilepsy syndrome.
The most important part of the differential diagnosis of epilepsy is the history, including a detailed description of the ictal events, preferably by an observer, as well as by the patient. For patients with one or more seizures, the age of onset, occurrence of specific seizure types, and, occasionally, a family history of epilepsy will help make a diagnosis of a specific age-related genetic epilepsy. Risk factors such as intrauterine or perinatal insults, serious head trauma, intracranial infections, and febrile seizures in infancy are important when a structural-metabolic epilepsy is suspected. It is also important to document the possibility of previous unrecognized ictal events by determining whether the patient has ever had episodes of impaired consciousness or strange behavior for which there is no memory, daydreaming as a child, or possible unrecognized nocturnal seizures that might be suspected by awakening with a bitten tongue, a wet bed, or muscle soreness. Patients presenting with a single generalized tonic-clonic seizure are usually not aware of the fact that previous ictal events may be much less pronounced. For instance, patients with juvenile myoclonic epilepsy are rarely aware that their early morning myoclonic jerks are abnormal and will not offer this information unless asked specifically. Usually, it is possible to arrive at a fairly accurate diagnostic hypothesis before embarking on detailed diagnostic testing. Differential diagnosis between nonepileptic and epileptic seizures, however, particularly psychogenic nonepileptic seizures, can be particularly difficult, and the latter are often misdiagnosed in the community. Consequently, when seizures do not respond to appropriate trials of two antiseizure medications, patients should be referred to an epilepsy center, not only to determine whether the habitual seizures are indeed epileptic, but also whether the patient might be a candidate for surgical treatment or other alternative therapies, including experimental drug trials. Early complete control of epileptic seizures provides the best opportunity for avoidance of adverse psychological and social consequences that lead to irreversible disability.
The initial patient and family interview is also essential in order to determine the predicament caused by the occurrence of epileptic seizures, to address concerns and misconceptions, and to establish a plan to achieve the best quality of life outcome. This would include discussion of the risks associated with the occurrence of subsequent seizures. Patients with seizures associated with impaired consciousness need to be warned of the risk of serious injury or death for themselves or others if appropriate precautions are not taken. They should be advised not to enter into situations where a seizure without warning could pose an undue risk, such as swimming alone, climbing into high places, and operating dangerous machinery, including driving. In some states, this may also require that the patient be reported to the appropriate authorities.
|
• EEG | |
|
• MRI | |
|
• Additional testing depends on the type of epilepsy and intended treatment. |
The EEG remains the most important diagnostic test for epilepsy (52; 71). The pattern and location of interictal epileptiform abnormalities not only help to make a diagnosis of epilepsy but also help to characterize the type of epileptic disorder or epileptic syndrome. The interictal EEG alone, however, is not sufficient because 10% to 20% of patients with epilepsy do not demonstrate interictal epileptiform EEG abnormalities, whereas 2% to 3% of patients without epilepsy may show these abnormal transients. The likelihood of finding an interictal epileptiform EEG abnormality depends on the type of epilepsy, whereas the likelihood that an interictal epileptiform transient indicates the existence of an epileptic condition depends on its topographic morphological and temporal features. Interpretation of the interictal EEG, therefore, requires considerable specialized expertise. A definitive diagnosis of epilepsy can be made if a seizure occurs during an EEG recording and electrographic ictal discharges can be correlated with habitual clinical signs and symptoms. Some types of ictal events can be precipitated in the EEG laboratory when necessary or occasionally occur spontaneously during routine EEG testing, but continuous long-term EEG recording with video monitoring is the most effective way to evaluate habitual seizures. Long-term monitoring for epilepsy is commonly used for differential diagnosis and for identifying candidates for surgical treatment. When precise localization of a resectable epileptogenic region is required for surgical resection, long-term monitoring can involve the use of intracranial depth or subdural electrodes. Continuous EEG in critically ill patients is becoming an important approach for diagnosing unrecognized seizures (42; 43).
Routine neurologic and laboratory examinations are useful for identifying an underlying treatable cause of epilepsy. Unless an unequivocal diagnosis of a genetic epilepsy is made, the use of CT or MRI is obligatory to look for a structural cerebral lesion. MRI is the structural imaging procedure of choice except for patients in whom a disorder associated with small calcifications is expected (09). Neuropsychological testing is useful for characterizing functional deficits for psychosocial prognosis and rehabilitation (94). Localizing information may also be obtained from these tests if surgical resection is contemplated. Functional imaging such as PET, SPECT, MRI, MRS, and MEG can also provide confirmation of a localized functional deficit. Genetic testing is important for some patients, particularly children (77), but increasingly in adults as well (49).
The current debate about classification of seizures and epilepsy has caused some confusion for clinicians. It is important to understand that classification of groups of patients is important for academic purposes such as gene discovery, epidemiological studies, and basic research, but is not necessarily relevant to management in individual patients where the objective is to diagnose the epileptic seizure type and, when possible, an epilepsy syndrome, which will then inform further workup for specific causes, treatment, and prognosis. Clinicians caring for individual patients are advised to diagnose, not classify. A diagnostic manual is being prepared by the ILAE for this purpose.
|
• Confirm the existence of epileptic seizures. | |
|
• Confirm an enduring condition. | |
|
• Identify treatable underlying causes. | |
|
• Diagnose a syndrome if possible. | |
|
• Control seizures with antiseizure medication. | |
|
• Address comorbid conditions and maximize quality of life. | |
|
• Refer to a comprehensive epilepsy center if two medication trials fail. |
The American Academy of Neurology has updated the 2014 evidence-based performance measures to act as guidelines for physicians to measure quality of care for epilepsy (Table 5).
|
1. Counseling for women of childbearing potential with epilepsy | |
|
2. Comprehensive epilepsy care center referral or discussion for patients with intractable epilepsy | |
|
3. Quality of life assessment for patients with epilepsy | |
|
4. Quality of life outcome for patients with epilepsy | |
|
5. Depression and anxiety screening for patients with epilepsy | |
|
6. Seizure frequency for patients with epilepsy | |
|
Additional current guidelines from the ILAE and other organizations can be found at www.ilae.org/guidelines. | |
Treatment with antiseizure medications is warranted when a diagnosis of epilepsy is made. Single seizures, therefore, usually are not treated unless there is sufficient additional evidence that an epilepsy condition exists. The American Academy of Neurology has issued evidence-based guidelines for management of a single unprovoked seizure in adults (53). If the diagnosis is equivocal, treatment is instituted, and no further seizures occur, it is impossible to know whether the drug is needed. Usually, after 1 or 2 years of seizure freedom, whether the initial diagnosis is definitively made or not, medication is tapered and withdrawn to determine whether an epilepsy condition (still) exists. At this point, the risk of a subsequent seizure is likely to be as high as it was when the drug was first prescribed. Consequently, prescribing a drug in the absence of a definitive diagnosis does not resolve the problem but merely postpones it. When this situation is discussed with the patient or family, and the risk of a subsequent seizure is clearly presented, there will usually be agreement to wait and see whether another seizure occurs and, thus, whether treatment is actually necessary. Treatment begins with a single drug, which is increased until seizures stop or adverse side effects occur. If the first drug is unsuccessful, a second is added, and the first may then be tapered and discontinued, with monotherapy the preferred goal. The choice of antiseizure medication depends on the seizure type and the diagnosis of a specific syndrome if possible (36). The ILAE has published guidelines for treating seizures in the neonate (73), and special considerations may be appropriate for treartnent of seizures in the elderly (72). Cost, side effect profile, and patient preference for a dosage schedule are usually more important considerations. Pharmacotherapy is designed to maximize function and reduce disability.
In some patients, seizures may occur in clusters or evolve into status epilepticus, a condition where seizures recur continuously without return to consciousness. For generalized tonic-clonic seizures and some other types, this can be a life-threatening condition and demands immediate treatment. Patients who tend to cluster or who are at high risk for status epilepticus can be given a fast-acting benzodiazepine as rescue medication that can be administered rectally, and more recently intranasally, to prevent progression (46).
Because recurrent disabling seizures, particularly during critical periods of childhood, adolescence, and early adulthood, can result in irreversible social and psychological compromises, which could prevent rehabilitation if seizures are eventually stopped, the goal of pharmacotherapy for epilepsy should be no seizures and no side effects, as soon as possible. This requires that the physician know which drugs are most effective against the patient’s seizures, and syndrome if known, so that appropriate pharmacotherapy trials can be instituted. Dosages can be escalated quickly with some drugs, but for others it is important to go slowly at first in order to avoid dose-related side effects that may prevent the patient from continuing with what otherwise might have been an effective therapy. Although serum levels are available for most drugs and are useful as a guide, published therapeutic ranges are based on population averages and may not pertain to individual patients. Consequently, effect, rather than serum level, should be the endpoint of a drug trial. If seizures are controlled below the published therapeutic range, the patients can be continued on a lower dose. If seizures continue but no adverse side effects are experienced, the dose can be increased even though the serum level is above the published therapeutic range. When intolerable dose-related side effects occur, leaving the dose as is, or lowering it slightly, can result in tolerance over several days.
If the side effects persist, however, and seizures are not controlled, then the dose should be lowered, and a second-choice drug should begin. Most epileptologists recommend increasing the dose of the second drug to a therapeutic level before attempting to taper and discontinue the first drug. If seizures are controlled with the second drug, discontinuation of the first drug is not absolutely necessary, although it is advisable because monotherapy reduces problems that arise as a result of drug interactions, and it is more cost-effective than polytherapy. If seizures are not controlled by the second drug, discontinuing the first drug can sometimes permit the second drug to be increased further with beneficial effects. Although this process can be repeated as necessary to find a single drug or drug combination that can control seizures without side effects, if seizures are not responsive to adequate trials of two appropriate antiseizure medications, it is highly likely that seizures are medically refractory (55). A few studies, however, have suggested that a significant number of patients might experience prolonged periods of seizure freedom even after many trials of antiseizure medications have failed (12; 59). Nevertheless, because early control of seizures reduces the risk of irreversible psychological and social disability, it is recommended that physicians refer their patients to a full-service, multidisciplinary epilepsy center after failure of two drug trials, particularly if seizures are interfering with school, work, or interpersonal relationships (21; 29). These epilepsy centers can not only provide specialized diagnostic and therapeutic approaches, but they are equipped to deal with adverse psychological and social consequences of epilepsy that can be a major cause of disability (66; 56). The International League against Epilepsy has now formally defined medically refractory epilepsy, which essentially consists of failure of two appropriate drug trials (54).
In 2008, the U.S. Food and Drug Administration carried out an analysis of antiseizure medication trials and concluded that these agents elevated the risk of suicidality (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3001021/), resulting in a formal warning. The American Epilepsy Society and the Epilepsy Foundation both issued statements in response, acknowledging the increased risk of depression and suicidality associated with epilepsy (for a variety of reasons not necessarily related to drug treatment) but urging patients to continue to take their antiseizure medications as prescribed. A subsequent study concluded that antiseizure medication use does not increase the risk of suicide for patients with epilepsy, but it does increase the risk in patients with depression (01). A more recent study, however, refutes this finding (17).
Serum drug levels are useful once a therapeutic dose of a drug has been obtained as a baseline to assess reasons for a seizure breakthrough or appearance of side effects in the future. They are also useful when a second drug is being added, to determine which drug might be responsible for the appearance of side effects, or during seizure recurrence or when noncompliance is suspected. Although serum levels are useful when changes occurring in the patient’s status could be attributed to the antiseizure medication, there is no need to repeat them routinely. Adverse side effects are determined clinically and not on the basis of serum levels. Some side effects, however, may be insidious and difficult to diagnose, and it is important not to substitute sedation or cognitive dysfunction for seizures—particularly in children, who often do not complain of drug side effects. Parents and physicians can mistakenly consider reversible drug toxicity to be a necessary consequence of the epilepsy disorder.
In 2007, the American Epilepsy Society issued a position statement on the substitution of generic antiseizure medications, voicing concern that breakthrough seizures and side effects might occur given the lack of sufficient evidence that the bioavailability of generic drugs is the same as brand-name medications. Since then, two studies have demonstrated bioequivalence between generic and brand-name medications (90; 75). As a result, the American Epilepsy Society has issued a position statement confirming that the Federal Drug Administration standards for bioequivalence are appropriate for persons with epilepsy but supporting ongoing research by the Federal Drug Administration to study factors such as extended-release products, tablet or capsule color and shape, and nocebo effect, related to the substitution of generic antiseizure medications in adults and children (93). Since then, a German study has reported increased risk of seizures when the manufacturer is changed (57).
With frequent seizures, adequate trials of two drugs could be performed in weeks, whereas with infrequent seizures it may take months or years. Even a few seizures a year, however, is unacceptable; if disabling seizures persist despite adequate trials of at least two appropriate antiseizure medications, patients should be referred to a full-service, multidisciplinary epilepsy center where a more complete diagnostic evaluation can be performed and either experimental drugs, surgical treatment, or other alternative therapies can be options. The American Academy of Neurology recommends localized surgical resections for medically refractory mesial temporal lobe epilepsy, and surgery can also be beneficial for many forms of neocortical epilepsy due to well-circumscribed lesions (28). Early surgery is advised to avoid the development of irreversible disabling adverse psychological and social consequences of epileptic seizures, as well as increased morbidity and mortality (25; 18). Corpus callosum sections can abolish disabling drop attacks; hemispherectomies or large multilobar resections can yield gratifying results in patients, usually infants and small children with severe generalized seizures due to diffuse lesions limited to one hemisphere; and surgery is recommended for gelastic epilepsy with hypothalamic hamartoma. In these surgically remediable epilepsies, early intervention provides the best opportunity to avoid psychosocial disability (25). A semi-invasive approach, laser interstitial thermal therapy, is now also being used for small discrete lesions in which the epileptogenic tissue is ablated using a thin probe inserted through a twist drill hole (51; 96). Although results are not as good as open resection, a second procedure can be performed if the patient is not seizure-free.
Other alternative therapies, such as vagus nerve stimulation (65) and the ketogenic diet (92), are also available at specialized epilepsy centers and can be effective when medication and standard surgical approaches are ineffective or inappropriate. Deep brain stimulation was shown to be effective and has now been approved in the United States (31). Responsive cortical stimulation has been approved in the United States (64; 08). External trigeminal nerve stimulation has also demonstrated effectiveness in patients with refractory focal seizures (15). There are now three randomized controlled trials supporting the effectiveness of cannabidiol in Dravet syndrome and Lennox-Gastaut syndrome (69; 98). Therapeutic complications resulting from comorbidity, the necessity for treatment with other pharmaceutical agents, surgery, or problems peculiar to neonates, infants, and young children at one extreme, and the elderly at the other extreme of age, also often require referral to a full-service, multidisciplinary epilepsy center.
The Institute of Medicine (IOM) has published a major review of the public health dimensions of the epilepsies (48), which includes specific recommendations designed to close the data gap and prevent epilepsy, as well as improve health care and community services, raise awareness, improve education, and strengthen stakeholder involvement (Table 6).
|
• Validation and implementation of standard definitions and criteria for epilepsy case ascertainment, health care and community services use and costs, and quality measurement | |
|
• Continuation and expansion of collaborative surveillance efforts | |
|
• Development and evaluation of prevention efforts for epilepsy and its consequences | |
|
• Improvement in the early identification of epilepsy and its comorbid health conditions | |
|
• Development and implementation of a national quality measurement and improvement strategy for epilepsy care | |
|
• Establishment of epilepsy center accreditation and an Epilepsy Care Network | |
|
• Improvement in health professionals’ education about the epilepsies | |
|
• Improvement in the delivery and coordination of community services | |
|
• Improvement in and expansion of educational opportunities for patients and families | |
|
• Provision of information to media to improve awareness and eliminate stigma | |
|
• Coordination of public awareness efforts | |
|
• Continuation and expansion of Vision 20-20 working groups and collaborative partnerships | |
|
• Engagement of people with epilepsy and their families in education, dissemination, and advocacy for improved epilepsy care and services |
Early identification and treatment of epilepsy remains a problem. One third of newly diagnosed patients with epilepsy in the United States remain untreated for up to 3 years (50). A major concern in the management of people with epilepsy is the fact that the percentage of patients whose seizures are not controlled by medication has not changed appreciably with the introduction of many antiseizure medications. Less than 1% of people with epilepsy in the United States whose seizures are not controlled by medication are referred to full-service epilepsy centers, and those who are referred for surgical treatment receive surgery on average of 22 years after the onset of epilepsy (06), often too late to prevent irreversible psychological and social disability. Studies indicate that there has been an increase in hospital admissions for intractable epilepsy over the past two to three decades, but not in the number of therapeutic surgical procedures performed (30; 80). This is attributed to failure to refer patients with intractable epilepsy to full-service epilepsy centers that offer surgical therapy. These centers also offer other alternative treatments and access to experimental drug and device trials, as well as psychological and social services of great benefit to people with epilepsy. All patients with epilepsy who have failed two trials of antiseizure medications and continue to have seizures that interfere with work, school, or interpersonal relationships deserve a consultation at a multidisciplinary epilepsy center (24).
Epilepsy syndromes can be simplistically divided into mild and severe forms. The mild forms, which for the most part consist of age-related genetic conditions, are easily treated by primary care physicians or general neurologists who can recognize the syndromes and institute the appropriate medication. In many cases, seizures will remit spontaneously, and in some cases, they are so benign no treatment is necessary. The severe forms, however, can also be simplistically divided into those that are remediable and those for which no successful treatment exists. The latter are often associated with extensive brain damage and other neurologic and cognitive disturbances. These patients require supportive care that can improve their lifestyle, but they will likely remain disabled. The remediable epilepsies, on the other hand, can be successfully treated in most cases with appropriate interventions, most commonly surgery, but specialized pharmacotherapy or alternative treatments can also be beneficial for some. The distinction between remediable and nonremediable severe epilepsy requires the expertise of epilepsy specialists at full-service, multidisciplinary epilepsy centers.
Pregnancy is associated with many concerns for women with epilepsy, the most important of which is the teratogenicity of antiseizure medications. A child has an increased risk of birth defects if either parent has epilepsy, regardless of medication, presumably due to genetic disturbances associated with the tendency to develop epilepsy. No "safe" antiseizure medication exists, and in general, these agents double the risk of birth defects. Valproate and topiramate are more likely to cause neural tube defects than other commonly used antiseizure medications, but this problem can be diagnosed in utero. Only trimethadione has a sufficiently high risk to be contraindicated in women who may become pregnant. The teratogenic risk is decreased by folic acid, and all women of childbearing age on antiseizure medications should take 1 to 2 mg of this vitamin daily. The teratogenic risk increases with higher doses of antiseizure medications and the use of two or more medications. In addition, valproate use during pregnancy is associated with an increased risk of cognitive impairment at 3 years of age (60). Valproate and topiramate are not recommended for use in women of childbearing age. When a woman with epilepsy on an antiseizure medication desires to conceive, it is appropriate to attempt to reduce medication, institute monotherapy, or if she has been seizure-free for several years, taper and discontinue medication. Once conception has occurred, however, it is generally not advisable to change medication because the most serious teratogenic effects occur during the first trimester, and seizures themselves pose a risk to the fetus. However, the effect of valproate on cognition most likely continues throughout pregnancy.
Seizures can worsen or improve during pregnancy. Clearance of antiseizure medications increases and serum levels can fall, but the dosage of medication need not be increased unless there is an associated increase in seizures. Because of decreased protein binding during pregnancy, the free levels that enter the brain may remain therapeutic. Following delivery, serum levels of antiseizure medications can rise, and medication should be decreased if toxic effects occur. There is no contraindication to breastfeeding for women taking antiseizure medications unless the infant develops an adverse drug reaction or becomes sedated and is unable to suck. These and other issues concerning the management of women with epilepsy are addressed in an American Academy of Neurology Practice Parameter (38; 39; 40) and an Executive Summary of the International League against Epilepsy (91).
Antiseizure medication regimens should be maintained while patients are anesthetized. Many antiseizure medications are available as intravenous preparations for use during anesthesia.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Jerome Engel Jr MD PhD
Dr. Engel of the David Geffen School of Medicine at the University of California, Los Angeles, has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Epilepsy & Seizures
Jun. 02, 2026
Neuropharmacology & Neurotherapeutics
May. 11, 2026
Epilepsy & Seizures
May. 08, 2026
Epilepsy & Seizures
May. 01, 2026
Epilepsy & Seizures
Apr. 30, 2026
Epilepsy & Seizures
Apr. 17, 2026
Epilepsy & Seizures
Apr. 13, 2026
Epilepsy & Seizures
Apr. 08, 2026