






















Neurobehavioral & Cognitive Disorders
Mental status examination
Jun. 17, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Globoid cell leukodystrophy, or Krabbe disease, is an autosomal recessive, and it is rapidly progressive fatal lysosome disease when it occurs in infancy. Globoid cell leukodystrophy is caused by variants in the GALC gene that reduce galactosylceramidase (GALC) activity, leading to psychosine accumulation, cerebral white matter degeneration, and peripheral neuropathy. The disease usually begins between the ages of 3 and 6 months with ambiguous symptoms, such as irritability or hypersensitivity to external stimuli, but soon progresses to severe mental and motor decline. Patients are initially hypertonic with hyperactive reflexes, but they later become flaccid and hypotonic. Blindness and deafness are common. Patients with late-onset forms, including adult-onset, may present with blindness, spastic paraparesis, and dementia. Saposin A deficiency is a rare cause of Krabbe disease. Brain MRI has characteristic features that depend on the age of onset of the disease (infantile, juvenile, or adult). Optic nerve and cauda equina enlargement and enhancement are common, as is midbrain atrophy. The presence of peripheral nerve enlargement detected by ultrasound strongly supports the diagnostic possibility of Krabbe disease (61). Newborn screening and presymptomatic hematopoietic stem cell transplantation have not yielded clear benefits.
|
• Globoid cell leukodystrophy may occur at any age, but the infantile type is the most common. | |
|
• Typical MRI changes that vary according to each phenotype suggest the diagnosis. | |
|
• The adult-onset type is the most prevalent in certain populations, such as Japan. | |
|
• Hematopoietic stem cell transplantation in presymptomatic infants only mitigates the disease and is not the optimal therapy it was once hoped to be. | |
|
• Measuring psychosine (galactosylsphingosine), a major offending metabolite in Krabbe disease, in dried blood spots helps in diagnosis and differentiation of infantile from later onset variants and in monitoring of disease progression and response to treatment. |
Globoid cell leukodystrophy, or Krabbe disease, was described in 1916. Danish neurologist Knud Haraldsen Krabbe (1885-1961) reported the clinical and neuropathologic description of five cases that appeared to represent a new disease entity (57).
Previous neuropathologic studies by American neuropathologist William Norton Bullard (1853-1931) and American neuropsychiatrist and neuropathologist Elmer Ernest Southard (1876-1920), working at Boston City Hospital in 1906, and German pathologist Rudolf Beneke (1861-1946) in 1908, however, had already described the "diffuse gliosis" of brain that was later characterized as "diffuse brain-sclerosis" in Krabbe patients (11; 08).
In 1924, English neurologist James Stansfield Collier (1870-1935) and Scottish neuropathologist Joseph Godwin Greenfield (1885-1958) used the term "globoid cells" to describe the phagocytic cells that appeared to be unique to this disorder (21). German neuropathologist Julius Hallervorden (1882-1965), a former member of the Nazi Party (ie, the National Socialist German Workers' Party) who had knowingly performed much of his research on the brains of executed prisoners and participated in the action T4 euthanasia program, suggested that these globoid cells may contain kerasin or cerebroside (39).
Biochemical and histochemical studies confirmed the presence of cerebroside in globoid cells (09; 05), and galactocerebroside was the only glycolipid that could produce globoid cells when injected into the central nervous system of experimental animals (03). Analytical biochemical studies of total brain lipids did not show an increase in galactosylcerebroside in this disease, but rather a lowering of total cerebroside and sulfatide and a reduced sulfatide-to-cerebroside ratio (102). Only a fraction of brain lipids enriched in the specialized globoid cells showed increased galactosylceramide (05). In 1970, Malone reported a deficiency of leukocyte galactosylceramide beta-galactosidase in a Krabbe disease patient (72); this was confirmed by Suzuki and Suzuki, who demonstrated the enzyme deficiency in the brain, liver, and spleen of three patients with Krabbe disease (100). Psychosine, a related glycolipid, was suggested to be the toxic metabolite responsible for the pathogenesis of this disorder (76; 99). The gene for the galactosylceramidase (GALC) enzyme has been mapped to chromosome 14 (120), and the cDNA has been cloned by Chen and colleagues (17).
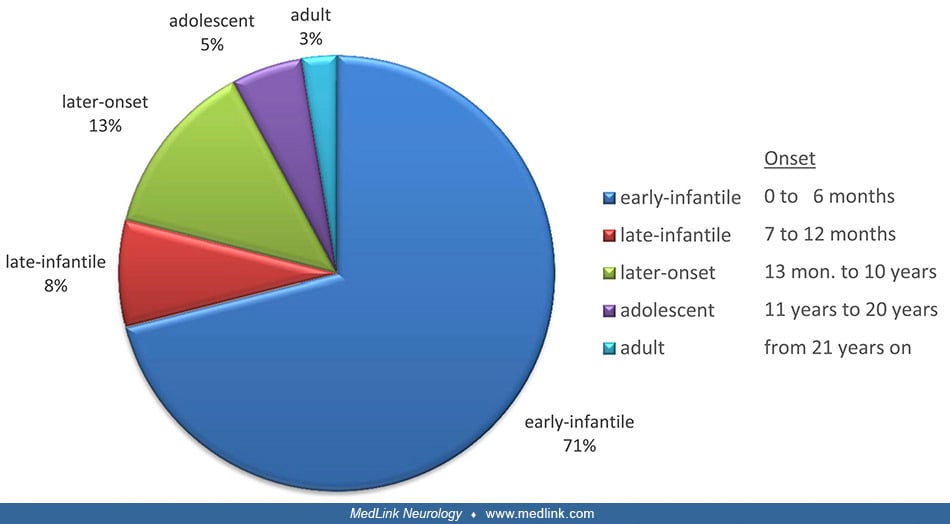
Initial clinical reports of Krabbe disease described the classic early infantile presentation with onset by 12 months, irritability, feeding difficulty, neurologic regression, and death in early childhood (57). The early infantile presentation, which accounts for over 70% of globoid cell leukodystrophy, begins before 6 months of age in most cases, and in 25% of cases, prior to 3 months of age (69; 110; 59). This form is rapidly progressive, and irreversible brain damage occurs unless timely hematopoietic stem cell transplantation is performed (although hematopoietic stem cell transplantation does not prevent the progression of peripheral nerve disease) (36).
Delayed motor milestones and subsequent regression of previously acquired milestones are typical.
Categories are represented by symbols. The symbols for each category are linked together for better identification. (Source: Krieg SI, Krägeloh-Mann I, Groeschel S, et al. Natural history of Krabbe disease: a nationwide study i...
Patients commonly develop spasticity early in the disease course, followed by feeding difficulties requiring placement of a gastrostomy tube, loss of fixation, and epileptic seizures late in the course.
Many acquire the ability to walk, albeit with reduced quality of performance, but then there is progressive loss of ambulation progressing to no ambulation but with some retained ability to control movements of the head; the decline from ambulation with reduced quality to no ambulation but with some retained head control occurs over a wide span of time from several months to up to 10 years later.
|
• Stage 1: Sensitivity to noise and light, periodic fever without infection, stagnation and then regression of development, and increased tone with normal deep tendon reflexes, irritability, stiffness, and feeding difficulties. | |
|
• Stage 2: Permanent opisthotonos, tonic flexion of arms and extension of legs, loss of nearly all previously acquired skills, myoclonic jerks, seizures, optic atrophy, blindness, and hypertonicity. | |
|
• Stage 3: Immobile, decerebrate, hypotonic, and unable to feed. |
However, the disease progresses in a continuum, as was demonstrated in a prospective study of a large cohort of children presenting at 0 to 5 months of age (07). Children may rarely present in the neonatal period, but this is an uncommon occurrence (69); a prenatal onset was first suggested when a 5-month-old fetus was noted to have the characteristic neuropathologic findings (26). Cerebrospinal fluid protein is usually elevated (69).
Much less common is a “late-infantile” (sic) variant for which the age of onset is between 19 months and 4 years, in early childhood but well beyond infancy (66). Affected children have normal intelligence or are only moderately retarded during the first years but then gradually develop ataxia, weakness, spasticity, and later dysarthria. Cases with onset after 12 months have less peripheral nerve involvement and slower disease progression (06). Visual loss with the early onset of optic atrophy, mental regression, occasional seizures, deafness, and normal peripheral nerves are the characteristic findings. A slowly progressive spinocerebellar degeneration associated with peripheral neuropathy, but without visual loss or dementia, has been reported (106). As in other forms of globoid cell leukodystrophy, CSF protein is usually, but not always, elevated (66). Peripheral neuropathy may be of the demyelinating type or predominantly axonal (71). These can very rarely have relatively high residual galactocerebrosidase activity (58).
Late-onset variants of globoid cell leukodystrophy have been somewhat arbitrarily defined as juvenile-onset (beginning between 4 and 19 years of age) and adult-onset (20 years of age and older) (110; 28; 63; 30; 42; 80; 88; 75). These patients usually have optic nerve pallor, pes cavus, slowly progressive spastic tetraplegia, and sensory-motor demyelinating polyneuropathy with preserved mental function in approximately one half of the affected individuals (70; 98); cranial CT shows hypodensity in the parieto-occipital white matter, and brain MRI shows symmetric, parieto-occipital predominant, periventricular abnormality with high signal in the pyramidal tracts, eventually with involvement of the splenium of the corpus callosum and optic radiations (92; 23). A detailed description of 20 patients with adult-onset Krabbe disease showed that the corticospinal tract was always affected, but other areas in the brain were also often involved, whereas the genu of the corpus callosum was always normal (22). The pyramidal tracts can be involved in isolation in middle-aged patients (107). The adult-onset form is the most common phenotype in Japan (45). CSF protein is frequently not increased. The lifespan of individuals with the late-onset variant may vary, but survival for over 24 years has been reported (04). Interestingly, 82% of Chinese patients have late-onset Krabbe disease (119).
Untreated children with the early infantile form of globoid cell leukodystrophy rarely live past 2 years of age. The late-infantile variant may have a much more prolonged course, with death usually occurring 1 year to 6 years after onset. In one report, however, the patient was not diagnosed until 34 years of age despite symptoms beginning in the late infantile period (106). In juvenile-onset and adult-onset patients, the disease will frequently have a much more protracted course (63).
A 4-month-old child was brought in for irritability that manifested as a high-pitched cry and poor feeding. The child had prior unexplained fevers associated with episodes of extreme irritability. On examination, the child was hypotonic and kept the hands continuously fisted. Muscle stretch reflexes were reduced, but Babinski sign was present. No optic atrophy was evident. An EEG was only minimally abnormal, with occasional paroxysmal beta waves noted over the vertex. CT scan demonstrated increased attenuation in the thalamus and basal ganglia but decreased attenuation in the white matter. Laboratory studies included serum amino acids, urine organic acids, viral titers for congenital infections, serum lactate, and pyruvate, all of which were normal. CSF examination demonstrated normal cell count, lactate, and pyruvate, but protein was increased to 158 mg/dL. A leukocyte galactosylceramidase level was 0.03 nmol galactose/mg protein per hour with a control of 2.3, confirming the diagnosis of Krabbe disease. The child died of a respiratory infection at 15 months of age.
Globoid cell leukodystrophy is caused by a deficiency of the enzyme galactosylceramidase.
Neuropathology. Krabbe disease is categorized as a leukodystrophy because most neuropathologic changes are noted in the white matter of the brain and in the peripheral nerves. The typical neuropathologic findings in these patients include severe loss of oligodendroglia, myelin, and axons, as well as dense astrocytic proliferation and the presence of "globoid cells."
(Photomicrograph by Jensflorian on July 14, 2011. Creative Commons Attribution-Share Alike 3.0 Unported License, creativecommons.org/licenses/by-sa/3.0/deed.en.)
In the murine model of Krabbe disease (a twitcher mouse), the oligodendrocyte loss is caused by an apoptotic depletion (103). Loss of myelin can also be seen in peripheral nerves; however, typical globoid cells are frequently not observed (73). Although the identification is somewhat controversial, these characteristic globoid cells appear to be specialized macrophages originating from mesoderm (03).
Ultrastructurally, globoid cells may be mononuclear or multinuclear; the latter contain two types of membrane-bound tubules. These tubules are either slender twisted tubules similar to those observed in Gaucher disease or a larger straight or arched variety (116).
Genetics. Krabbe disease is inherited as an autosomal recessive trait.
The gene for galactosylceramidase (GALC) was mapped to chromosome 14 (14q.31) by linkage analysis (120) and confirmed by in situ hybridization using a portion of the human cDNA for this gene (14). Following the difficult purification of the galactosylceramidase enzyme from human urine (18), the cDNA encoding GALC was cloned (17). Sodium dodecylsulfate-polyacrylamide gel electrophoresis of fractions containing the purified enzyme showed bands corresponding to 80 kD, between 50 and 52 kD, and 30 kD, all of which have a similar N-terminal amino acid sequence. This sequence similarity suggested that the 50 kD and 30 kD species are derived from the 80 kD precursor species (17; 18). The cDNA is 3.8 kb in length and contains a 2007 bp open reading frame that codes for 669 amino acids representing an unglycosylated protein with a molecular weight of 72,781 (18). This weight is consistent with the glycosylated 80 kD precursor form identified (17). Confirmation of the deduced amino acid sequence for the cDNA occurred following the purification of the enzyme from human leukocytes and the subsequent cloning using the polymerase chain reaction method (87). The entire gene structure and organization has been determined to consist of nearly 60 kb with 17 exons and 16 introns (67). The promoter region, located at the 149 to 112 nucleotide region from the initiation codon, contains three galactocerebroside-box-like sequences and one YY1 binding site (86). A common polymorphism in the human GALC gene occurs at nucleotide position 1637, where either a T or C may be present. The enzyme derived from the 1637T allele has 1.5- to 2-fold more activity than the enzyme derived from 1637C, which explains the wide range of human GALC gene activity in the general population (24).
Molecular analyses demonstrated at least 73 mutations, including base transitions, polymorphisms, and deletions that are associated with Krabbe disease Human Gene Mutation Database GALC web page. In infantile Krabbe disease patients of Northern European ancestry, approximately 40% to 50% have a mutant allele that has a 30 kb deletion beginning in intron 10 and extending past the 3’ end of the gene (53). In addition to the deletion on this allele, an invariable C →T transition at position 502 exists, which is a polymorphism seen in only about 4% of the population. Improved detection of mutations using comparative genomic hybridization (CGH) to analyze the GALC gene has been described (104). Most of the mutations causing infantile-onset disease are located on the region coding for the 30 kD subunit of the enzyme, suggesting that this subunit is critical for the normal enzyme function (110). Adult-onset cases may also have the 502/del mutation on one allele, but many other mutations have been identified, which appear to occur predominantly in the region coding for the 50 kD subunit (29; 90; 24).
Biochemistry of Krabbe disease. Experimentally, the only glycolipid that could produce globoid cells when injected into the cerebral cortex was galactosylceramide (03). Initial biochemical studies of brains from patients with Krabbe disease demonstrated the accumulation of the glycolipid, galactosylceramide, only within a fraction of brain that was enriched in globoid cells (05). However, total glycolipid and sulfatide levels in white matter from these brains were reduced (05). The association between galactosylceramide and Krabbe disease was confirmed when the enzyme that degrades galactosylceramide, galactosylceramidase, was found to be deficient in the leukocytes, brain, liver, and spleen of Krabbe disease patients (72; 100). Elevated expression of matrix metalloproteinase (MMP-3) and tenascin-C may mediate the production of globoid cells in Krabbe disease (49; 20).
Galactosylceramidase hydrolyses not only galactosylceramide, but also other glycolipids containing a terminal beta-galactose group, such as lactosylceramide, monogalactosyl diglyceride, and galactosylsphingosine (“psychosine”) (76; 111; 109). To properly function, the galactosylceramidase enzyme requires a sphingolipid activator protein, saposin A or saposin C, which helps solubilize the protein-lipid complex (40). Saposin A and saposin C also activate the degradation of galactosylsphingosine (40). A very rare deficiency of saposin A leads to a phenotype identical to that of early-infantile Krabbe disease caused by GALC enzyme deficiency (13). A second lysosomal enzyme, GM1 ganglioside beta-galactosidase, hydrolyzes GM1 ganglioside, and its deficiency is associated with the storage disease GM1 gangliosidosis. Different, but overlapping substrate specificity exists for these two beta-galactosidase enzymes; under certain conditions GM1 ganglioside beta-galactosidase can hydrolyze galactosylceramide, but not psychosine (55).
Although galactosylceramide does not accumulate in the white matter of affected individuals except in the specialized globoid cells (05), psychosine is increased in the brains of patients with Krabbe disease (99), and it is the toxic accumulation of psychosine that causes progressive demyelination and neuronal death. Psychosine has been demonstrated to cause cell death and injury to oligodendrocytes in culture that can be ameliorated by the presence of compounds that activate protein kinase C, activate galactosylceramidase, or bind to psychosine (19). Researchers found that psychosine localizes to detergent-resistant membrane microdomains (lipid rafts), perturbs natural and artificial membrane integrity, and inhibits protein kinase C translocation to the plasma membrane (41). These data strongly suggest that a significant component of psychosine toxicity is achieved through membrane perturbation rather than through protein-psychosine interactions (41). In the twitcher mouse, psychosine seems to accumulate in lipid rafts causing a maldistribution of cholesterol and specific proteins, which leads to a dysfunction of protein kinase C (112). Psychosine also inhibited fast axonal transport through the activation of axonal PP1 and GSK3beta in the axon (15). Abnormal levels of activated GSK3beta and abnormally phosphorylated kinesin light chains were found in nerve samples from a mouse model of Krabbe disease (15). It has been proposed that because of the overlapping substrate specificity of the beta-galactosidase enzymes, only psychosine and not galactosylceramide accumulates in the brains of Krabbe disease patients (99). Toxic psychosine or galactosylsphingosine accumulation may be responsible for the apoptotic depletion of oligodendroglial cells and the abnormal myelination in this disease. Further confirmation of the role of psychosine in Krabbe disease has been obtained by deleting the activity of acid ceramidase in the Twitcher mouse (62). The combination of a single gene defect prevented psychosine elevation in the Twitcher mouse and a marked improvement in its phenotype (62). Psychosine is generated catabolically through the deacylation of galactosylceramide by acid ceramidase. Although bone marrow transplantation normalizes the level of psychosine in the brain, transplanted mice still end up succumbing to the disease (68).
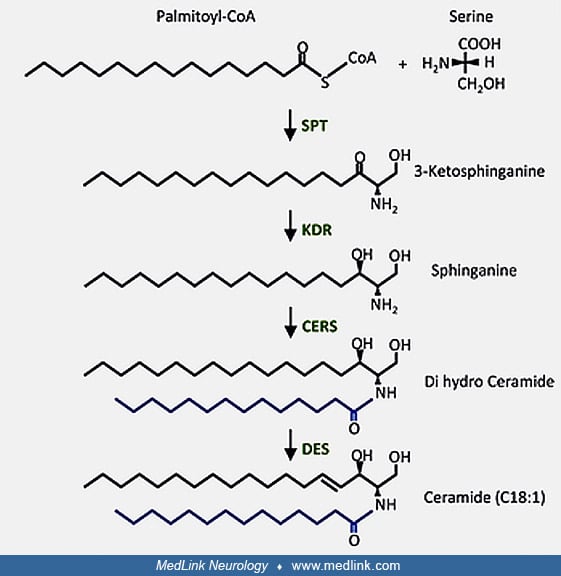
The spectrum of disorders of the lysosomal degradation of glycosphingolipids. Ceramide is synthesized in vivo in multiple steps, by de novo synthesis, hydrolysis of sphingomyelin, or the salvage pathway from sphingosine.
Ceramide, in turn, can be metabolized to various other molecules needed for cellular metabolism, including gangliosides, sphingomyelin, and sulfatide.
The various sphingolipids are closely related from a structural perspective.
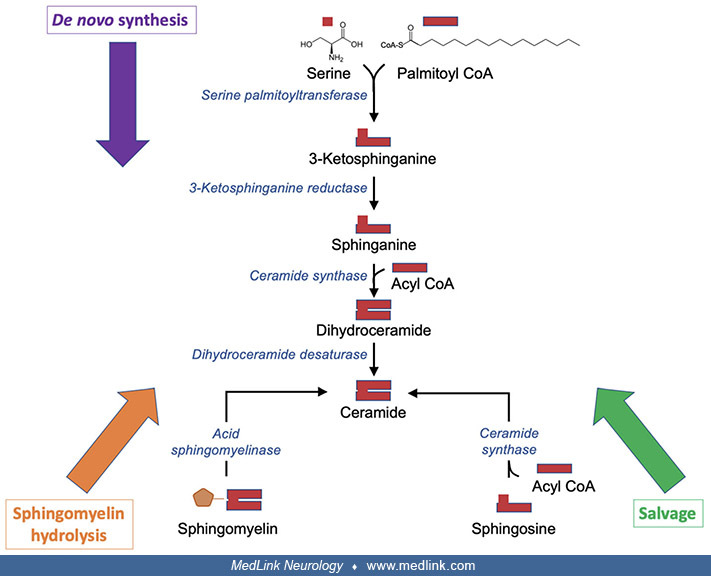
The lysosomal degradation of glycosphingolipids proceeds through multiple enzymatic steps, with specific recognized diseases corresponding to abnormal enzymatic activity at multiple sites in the metabolic pathways (GM1 gangliosidosis, Tay-Sachs disease, Sandhoff disease, sialidosis, Fabry disease, Gaucher disease, metachromatic leukodystrophy, Krabbe disease, and Farber disease.
Abbreviations: ASA, Arylsulfatase A; ASAH1, acid ceramidase; GALC, β-galactosylceramidase; GBA, β-glucocerebrosidase; GLA, α-galactosidase A; GLB1, β-galactosidase; HEXA, β-hexosaminidase A; HEXB, β-hexosaminidase B; NEU3/4, ne...
A schematic of the various sphingolipid metabolic pathways shows the enzymes whose deficiency leads to several diseases. Note that each enzyme is assisted by one or more saposins (Saps): (1) GM2A assists both β-gal and β-hexosa...
As noted above, the enzymatic block in Krabbe disease is in the conversion of galactosylceramide (GalCer) to ceramide through the action of β-galactosylceramidase, which is the gene product of GALC.
Globoid cell leukodystrophy is inherited as an autosomal recessive trait; therefore, it affects males and females equally and appears to be panethnic.
The prevalence of Krabbe disease in the general population is not known, but a report from Sweden estimated the prevalence to be 1.9 per 100,000 population (38), whereas in Japan the estimated prevalence was between 0.5 to 1 per 100,000 population (101). Newborn screening for Krabbe disease in the state of New York yielded a prevalence of infantile Krabbe disease of approximately 1 in 394,000, but it may be higher for later-onset forms (78); the detection of mild or even non-disease-related variants is a complicating issue (79; 78). The early infantile form of Krabbe disease is frequent in the Muslim Arab population in Israel, with a very high prevalence of approximately 6.7 to 10 per 1000 live births (117).
In families previously identified through an affected child or by carrier detection, prenatal diagnosis is the only current method for preventing globoid cell leukodystrophy.
It is often difficult to distinguish among children with the various forms of inherited degenerative diseases. Recognizing the pattern of abnormalities on brain MRI greatly facilitates the differentiation of Krabbe disease from other disorders (91). MRI imaging abnormalities in Krabbe disease vary according to the specific phenotype (01). Metachromatic leukodystrophy (MLD) shares many features with Krabbe disease, but metachromatic leukodystrophy often begins in the second year of life, and the CT and MRI scans are more likely to demonstrate white matter involvement, especially in the frontal regions (113). Canavan disease, a spongy degeneration of white matter, is associated with macrocephaly, hypotonia early in the course of the disease, and normal CSF protein. Canavan disease has been associated with an accumulation of N-acetylaspartic acid in brain and urine and a deficiency of the enzyme aspartoacylase in fibroblasts (74). In Alexander disease, another cause of macrocephaly in infancy, CT images may show more low-intensity abnormalities in the anterior white matter as well as contrast enhancement in the caudate, fornices, and subependymal white matter (43). Another infantile leukodystrophy, Pelizaeus-Merzbacher disease, differs from Krabbe disease by the prominent nystagmus seen early in infancy and the slow development of spastic quadriplegia (118). GM2 gangliosidosis (Tay-Sachs disease) also begins in infancy, but it is characterized by a cherry-red spot in the retina and the presence of early seizures. Other lysosomal storage diseases that begin in infancy (eg, infantile Gaucher disease, Niemann-Pick disease, Sandhoff disease, and the mucopolysaccharidoses) can be distinguished from Krabbe disease by the presence of systemic signs (ie, organomegaly and dysostosis multiplex) (69).
In 2024, after a push from affected families, the U.S. Department of Health and Human Services added infantile Krabbe disease to the Recommended Uniform Screening Panel, meaning that it will now be on the newborn screening list. Newborn screening for Krabbe disease is based on low GALC levels in dried-blood spots.
The debate over whether to test babies for Krabbe disease reflects the limited treatment options for infantile Krabbe disease. Without newborn screening, diagnosis of infantile Krabbe disease is generally made after significant clinical symptoms develop, past when hematopoietic stem cell transplantation (HSCT) can be effective. Although HCST in infancy before clinical disease onset augments survival, mitigates cognitive decline, and produces a somewhat less severe motor phenotype, it has about a 10% risk of mortality within 100 days, and affected individuals still have significant functional impairment (27; 84).
Enzyme and metabolite assays. With a symptomatic proband, the only specific test that will establish a suspected diagnosis of Krabbe disease is the measurement of galactosylceramidase activity in serum, leukocytes, or cultured fibroblasts. Although most patients have galactosylceramidase activity below 5% of mean normal levels, rare patients with high residual activity have been described (58).
Psychosine quantitation is critical for the correct diagnosis of Krabbe disease in newborn screening (37). Measuring psychosine in dried blood spots helps to differentiate infantile- from later-onset Krabbe disease variants, as well as from GALC variant and pseudodeficiency carriers (37).
Molecular genetic testing. Abnormal results from enzyme assays require follow-up molecular genetic testing of GALC (77).
Biomarkers. A variety of other biomarkers can provide clinically useful information for managing patients with Krabbe disease. However, note that any of the usual biomarkers, such as abnormal neuroimaging, brainstem evoked responses, nerve conduction velocities, and elevated CSF protein, may be normal in up to 25% of patients (25).
Neuroimaging. Early in the course of the disease, when spasticity, fever, and irritability become apparent, CT demonstrates increased density in the brainstem, thalami, caudate nuclei, corona radiata, cerebellar cortex, and periventricular and capsular white matter (89).
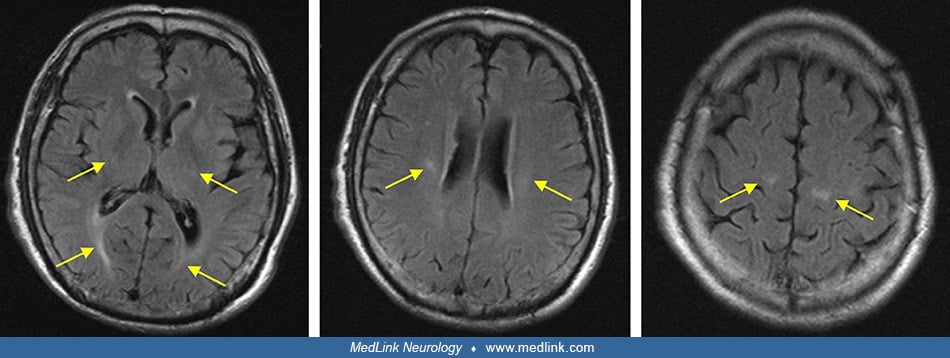
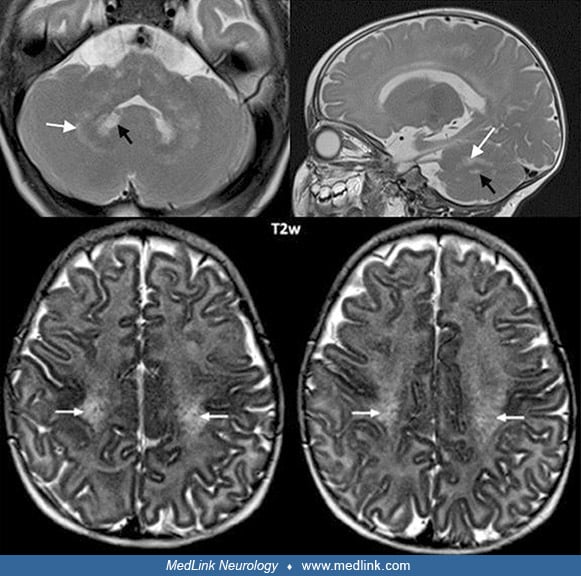
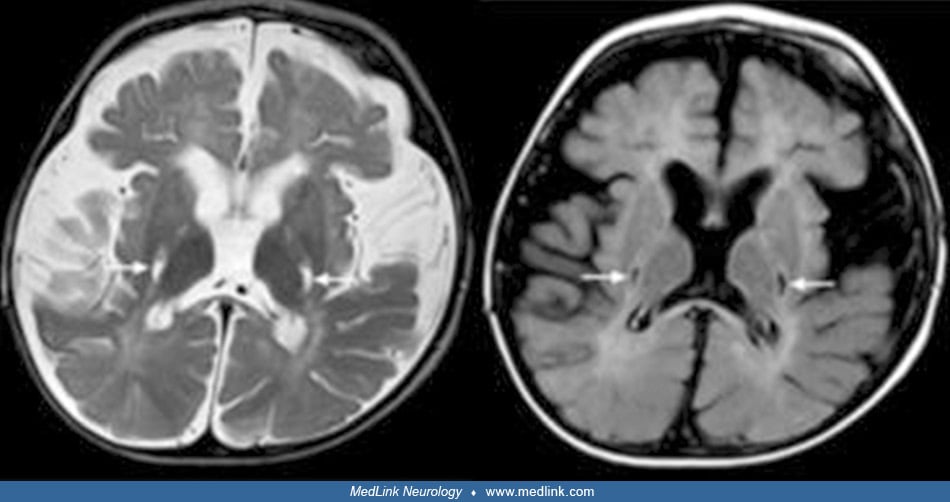
Magnetic resonance imaging. The child suspected of having globoid cell leukodystrophy should have an MRI scan to evaluate myelin development. MRI performed during these initial symptoms shows decreased T1 values and normal or slightly decreased T2 values in the abnormal areas demonstrated by CT scan, and large symmetric plaque-like areas on T1- and T2-weighted imaging in the white matter of the centrum semiovale (89; 01). Optic nerve hypertrophy, thought to be due to extensive gliosis, has been noted in MRI studies of a child with early infantile-onset Krabbe disease (94). As the disease progresses, brain atrophy, decreased attenuation in white matter, and symmetric punctate high-density areas in the corona radiata are noted on CT scan, whereas high attenuation on T2-weighted images and low attenuation on T1-weighted images are present in white matter on MRI scans (89; 01; 105).
Remarkable brain MRI features include enlargement and, sometimes, enhancement of the optic nerves and chiasm, and variably other cranial nerves and the cauda equina (32; 46). Selective involvement of the corticospinal tracts is often also present in the late-onset form (93; 16).
Rarely, brain MRI can be normal, even in the early-onset form (50). Subsequently, other studies performed in infantile-onset Krabbe disease have demonstrated a decreased signal on T2-weighted images from the thalamus and basal ganglia (particularly the globus pallidus) with increased T2-weighted signal in centrum semiovale, corpus callosum, internal capsule, midbrain, and pons (105). All parameters of diffusion-weighted imaging are abnormal, particularly radial diffusivity reflecting the white matter abnormality of Krabbe disease (81). Variable atrophy of the midbrain viewed sagittally may be rather useful and correlates with clinical severity and other neuroimaging aspects (121).
Pattern-recognition approach for MRI. A pattern-recognition approach has been described for MRI in Krabbe disease (59); this approach categorizes MRI results into four patterns: A1, A2, B1, and B2.
Pattern A1. Pattern A1 is present in all early-infantile cases.
|
• Cerebral white matter changes, beginning in the central region. | |
|
• Cerebellar white matter changes and signal changes of the dentate nucleus. | |
|
• Involvement of the corpus callosum and the pyramidal tract. | |
|
• In the early-infantile form, signal change in the dentate nucleus (hyperintense on T2-weighted images) may be an early sign but may not persist. | |
|
• In the advanced stage: global atrophy, diffusely affected white matter, and cystic degeneration of the pyramidal tract. |
Pattern A2. Pattern A2 is seen in children with a late-infantile onset and is very similar to pattern A1, except the dentate nucleus is not affected.
In children affected with this pattern, the cerebellar white matter is affected relatively late during the disease course and demyelination starts in the central region and spreads outward.
Pattern B1. Pattern B1 is seen in all later onset patients (ie, all patients with first symptoms after the first year of life up to age ten years).
Occasional patients with either late-infantile onset or onset beyond 10 years may also show this pattern.
|
• White matter signal changes, primarily in the periventricular parieto-occipital region. | |
|
• The corpus callosum (body and splenium) is affected. | |
|
• Pyramidal tract abnormalities (as in patterns A1 and A2). | |
|
• The cerebellum is not affected. |
Pattern B2. Pattern B2 is seen in an adult patient with a late onset (ie, first symptoms at age 60 years).
|
• Visible abnormalities largely restricted to the pyramidal tract. | |
|
• Some focal white matter abnormalities. |
Atypical findings. Atypically, neuroimaging may show calcifications in the bilateral thalami and dentate nucleus (35).
The "Loes score" for MRI. Assessment of an intermediate outcome of hematopoietic stem cell transplantation can be done with the brain MRI "Loes score," a MRI severity scoring system developed originally for use in patients with adrenoleukodystrophy (65; 82; 59).
(Source: Krieg SI, Krägeloh-Mann I, Groeschel S, et al. Natural history of Krabbe disease: a nationwide study in Germany using clinical and MRI data. Orphanet J Rare Dis 2020;15[1]:243. Creative Commons Attribution 4.0 Internat...
Cerebrospinal fluid. CSF in patients with early and late infantile forms of Krabbe disease shows elevated protein (frequently 75 to 500 mg/dL) with normal cell counts (110; 75). CSF glycolipids demonstrate reduced galactosylceramide and trace amounts of lactosylceramide but no evidence of psychosine (51).
Neurophysiological studies. Electroencephalograms are usually normal during the early stages, but later, the background slows and becomes irregular with frequent multifocal spike waves (48).
In infantile Krabbe disease, electrodiagnostic testing generally shows evidence of severe sensory-motor polyneuropathy of mixed demyelinating and axonal types (33). Nerve conduction velocities are slowed in the early infantile form, consistent with a demyelinating disorder (95), but may be normal in adult-onset patients. Electromyograms frequently show evidence of axonal injury, with a reduced number of high-amplitude motor unit action potentials (MUAPs) and occasional fibrillations. Some patients may show improvement in motor amplitudes on repeat nerve conduction studies after hematopoietic stem cell transplantation done soon after birth (33).
Electron microscopy of eccrine sweat glands. Ultrastructural data suggest that Krabbe disease may be diagnosed based on electron microscopic inclusions in eccrine sweat glands (34); the inclusions appear similar to those seen in Schwann cells, consisting of needle-like and cleft-type inclusions (34).
Benzodiazepines or low-dose morphine have been used for the symptomatic treatment of severe irritability (97). Since this was suggested, though, there has been a shift away from the use of either medication for such indications.
Mouse model. Bone marrow transplantation (BMT) has been used in a murine model of Krabbe disease, the twitcher mouse. The lifespan of the twitcher mouse was prolonged, with remyelination noted in peripheral nerves (115). Donor-derived macrophages could be demonstrated with extensive remyelination in the CNS of mice receiving bone marrow transplantation, and significant increases in galactocerebroside beta-galactosidase levels were noted along with a reduction of galactosylceramide and psychosine in several organs (47; 44).
Human studies. Hematopoietic stem cell transplantation has been performed in several patients with Krabbe disease with some success. In a study of five patients with Krabbe disease (four juvenile- and one infantile-onset) who received hematopoietic stem cell transplantation (HSCT), all the treated patients had significant improvements in their neurologic symptoms (60). The four late-onset cases demonstrated improvement in MRI findings and reversal of protein elevation in CSF.
Hematopoietic stem cell transplantation in infants before disease onset using umbilical cord blood greatly augments survival, mitigates cognitive decline, and produces a somewhat less severe motor phenotype, but has about a 10% risk of mortality within 100 days, and affected individuals still have significant functional impairment, including behavioral and cognitive impairment (27; 84). Although nerve conduction improves in the transplanted patients, they had substantial residual motor deficit (96; 33). In the newborn screening performed in New York State over 8 years, five infants were diagnosed with early infantile Krabbe disease (108): three died--two from transplantation-related complications and one from untreated disease. Two children who received hematopoietic transplantation have moderate to severe developmental delays.
Newborn screening and transplantation in the days after birth remain controversial. A group from Duke University suggests that transplantation before age 30 days provides a significantly better mobility outcome, with 90% able to walk independently or with an assist device and 80% developing normal speech, although 60% required special resources in school (02). However, the New York state experience was much less positive (108). In a study of 18 patients in Pittsburgh, 15 patients were identified prenatally because of family history, and only three infants were identified by state-mandated newborn screening (114). Hematopoietic stem cell transplantation only delayed disease progression and was not curative.
Most patients treated early probably had prenatal-onset and substantial motor impairments with spasticity because of corticospinal tract pathology. In addition, many survivors, particularly if transplanted after 30 days of age, had variable degrees of feeding, vision, hearing, language, and cognitive impairment.
Bone marrow transplantation can be useful for late-onset Krabbe disease (64).
Gene therapy in animal models. Gene therapy using adeno-associated virus has been used in the animal model (83). Early intervention of combined hematopoietic stem cell transplantation and lentiviral-mediated gene transfer to newborn twitcher pups before postnatal day 2 prolonged survival, but it did not prevent ultimate axonal degeneration (31). A combination of more than one form of therapy might be needed (85). In a dog model of the disease, a combination of intravenous and intracerebroventricular injections of AAVrh10 targeted both the peripheral and the central nervous system (12). This approach showed a clear dose response and resulted in delayed onset of clinical signs, extended lifespan, correction of biochemical defects, and attenuation of neuropathology.
The disease appears to begin in utero because globoid cells and galactosylsphingosine accumulation have been reported in the fetus (26; 54). Prenatal diagnosis is available using direct chorionic villus sampling at 8 to 9 weeks, or later in pregnancy using cultured amniotic fluid cells (110).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Douglas J Lanska MD MS MSPH
Dr. Lanska of the University of Wisconsin School of Medicine and Public Health has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurobehavioral & Cognitive Disorders
Jun. 17, 2026
Neurogenetic Disorders
Jun. 01, 2026
Neurobehavioral & Cognitive Disorders
May. 20, 2026
Neurogenetic Disorders
May. 08, 2026
Infectious Disorders
May. 01, 2026
Neurogenetic Disorders
Apr. 30, 2026
General Child Neurology
Apr. 29, 2026
Neuroimmunology
Apr. 26, 2026