Epilepsy & Seizures
Febrile seizures
Jun. 02, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Hemiclonic seizures (non-Jacksonian) are a type of unilateral convulsive seizure, mainly affecting infants and young children. Their most typical presentation is in hemiconvulsion-hemiplegia-epilepsy where hemiclonic status epilepticus is followed by permanent hemiplegia. They also occur in Dravet syndrome, metabolic and electrolyte derangements, age-related benign childhood epilepsies, and, more rarely, in idiopathic generalized epilepsy. They manifest with entirely one-sided, unilateral, clonic convulsions from onset without the focal elements and without the marching of Jacksonian seizures. Hemiclonic status epilepticus is common. Ictal EEG shows discharges that are mainly generalized from the start, but have significant emphasis over one hemisphere. Hemiclonic seizures have not been considered in the updated ILAE classifications and need re-assessment with video-EEG documentation and other modern brain imaging technologies. In this article, the author reviews historical aspects, clinical manifestations, pathophysiology, EEG, differential diagnosis, and management of patients with hemiclonic (non-Jacksonian) seizures.
• Non-Jacksonian hemiclonic seizures mainly affect infants and very young children. | |
• Clinically, non-Jacksonian hemiclonic seizures are unilateral from onset or at least lack the typical focal onset and marching of typical Jacksonian hemiclonic seizures. | |
• Hemiclonic status epilepticus is common. | |
• Interictal EEG mainly shows generalized or unilateral spike-and-wave or polyspike-and-wave discharges that occur either spontaneously or following overbreathing or photic stimulation. | |
• Ictal EEG does not show focal rolandic discharges, but spike or spike-and-wave discharges, identical to those seen in typical generalized epilepsies, appear on the contralateral hemisphere. | |
• The underlying mechanisms of non-Jacksonian hemiclonic seizures are assumed to be similar to those of generalized epilepsies but are limited to one hemisphere as opposed to a focal mechanism. | |
• There is a need for better characterization of hemiclonic (non-Jacksonian) seizures with video-EEG correlations and newer imaging technologies. | |
• Termination of prolonged hemiclonic seizures is a medical emergency because of the risk for permanent hemiplegia and subsequent epilepsy. |
Historical note. The first account of focal clonic seizures progressing to hemiclonic seizures can be found as early as 1050 BC in the twenty-fifth Babylonian cuneiform tablet devoted to miqtu (a disease in which the person loses consciousness and foams at the mouth) (75; 09):
In the time of his possession, while he is sitting down, his (left) eye moves to the side, a lip puckers, saliva flows from his mouth, and his hand, leg and trunk on the left side jerk (or, twitch) like a (newly)-slaughtered sheep, it is miqtu. If at the time of the possession his mind is consciously aware, (the demon) can be driven out; if at the time of the possession his mind is not so aware, (the demon) cannot be driven out. |
Every student of neurology is familiar with “Bravais-Jackson seizures” or “Jacksonian seizures” and postictal hemiparesis (Todd hemiparesis). Bravais was the first to describe focal clonic evolving to hemiclonic seizures and postictal hemiparesis, which he called "hemiplegic epilepsy” (10). Jackson, who described the same phenomena of “convulsions beginning unilaterally,” reasoned that there must be localized movement representation in the cerebral cortex, and provided a new concept of epileptogenesis (38). Furthermore, Jackson accepted that the initially unilateral convulsive phenomena could spread to involve both sides of the body, whereas Bravais believed that this was incompatible with his entity, unless the generalization was a very rare event in the sufferer (25). Todd described "epileptic hemiplegia" of what is today known as Todd paralysis (70).
Non-Jacksonian hemiclonic seizures were first mentioned by Gibbs (34) and Lennox (46) with no particular details. However, it was Henri Gastaut and his associates in Marseilles who provided superb descriptions of the clinical and EEG manifestations of non-Jacksonian unilateral and hemiclonic seizures (hemi grand mal and hemiclonic seizures) (32; 30; 58; 59; 23).
Hemiconvulsive seizures, or one-sided convulsions, include all, tonic or clonic, epileptic phenomena that involve all or part of the skeletal musculature of one side of the body only (thereby excluding "adversive" seizures, which result from the contraction of bilateral synergetic muscles). Clinical observation and simultaneous cinematographic, electroencephalographic and electromyographic recording of thousands of spontaneous and induced seizures have convinced us of the existence of at least two major types of non-Jacksonian hemiconvulsive epilepsy: | |||
• One-sided or "hemi-grand mal” | |||
• Hemiclonic seizures. | |||
The "hemiconvulsive seizures" concern all epileptic convulsions--tonic or clonic--interesting only one side of the body. The "Jacksonian epilepsy" is one of the aspects of this type of epilepsy, the others being: | |||
• "Hemi grand mal": typical grand mal seizures, but strictly localized to one side of the body and, electroencephalographically, to the contralateral hemisphere. | |||
• "Hemiclonic seizures": clonic phenomena often beginning with an "oculoclonic crisis,” spreading to the whole ipsilateral half of the body. On the EEG, on the contralateral hemisphere, two to three c/s slow waves and eight to 10 waves predominating in the posterior region are observed. | |||
The hemiclonic seizures occur specifically in children and in two main groups of epileptics: | |||
• Those with neurologic, radiological or electroencephalographical signs of focalization (among whom are found some with " hemiconvulsion-hemiplegia-epilepsy syndrome"). | |||
• Those without any sign of cerebral focalization, but suffering from petit mal or grand mal (32). | |||
Nomenclature and classification. Hemiclonic seizures are profoundly featured in the 1970 ILAE classification as influenced by Gastaut (29):
111. Unilateral or predominantly unilateral seizures: Seizures in which the clinical and electrographic aspects are analogous to those of the preceding group (generalized bilateral symmetrical seizures), except that the clinical signs are restricted principally, if not exclusively, to one side of the body, and the electroencephalographic discharges are recorded over the contralateral hemisphere. Such seizures apparently depend upon a generalized or at least very diffuse neuronal discharge that predominates in, or is restricted to, a single hemisphere and its subcortical connections. They are characterized by clonic, tonic or tonic-clonic convulsions, with or without an impairment of consciousness, expressed only or predominantly in one side. Such seizures sometimes shift from one side to the other but usually do not become symmetrical. | ||
Ictal EEG may show: | ||
(i) partial discharge very rapidly spreading over only one hemisphere (corresponding with only contralateral seizures) | ||
(ii) discharges generalized from the start but considerably predominant over one hemisphere, susceptible to change from one side to the other at different moments (corresponding to alternating seizures) | ||
(iii) partial discharge, susceptible to change, from time to time, in morphology and topography (from area to area and, sometimes, from one side to the other) | ||
Interictal EEG may show: | ||
(i) focal contralateral discharges | ||
(ii) bilateral and synchronous symmetrical or asymmetrical discharges of spike and wave or polyspike and waves | ||
(iii) focal discharges, susceptible to change, from time to time in morphology and topography. | ||
Hemiclonic and unilateral seizures have been detailed by Oller-Daurella and Dravet and also by Roget in the first edition of the “blue guide” (59; 53).
Subsequent ILAE classifications and proposals do not consider unilateral seizures other than hemiclonic seizures from ipsilateral propagation of focal clonic seizures to neocortical areas (26). A related “hemiconvulsion-hemiplegia-epilepsy syndrome” is recognized as an epileptic syndrome (08; 18).
According to the latest 2014 ILAE epilepsy diagnosis manual, hemiclonic seizures are focal, and they are classified among those with elementary motor features that involve a stereotyped contraction of a muscle or group of muscles (18). Such motor features may be predominantly convulsive (rhythmic jerking (clonic activity)) and may occur alone or in combination with tonic activity (eg, hemiclonic that is rhythmic jerking (clonic activity) involving only one side of the body). Conversely, generalized convulsive seizures are typically bilateral and symmetric although variants with asymmetry including head and eye deviation can be seen (18). According to Dravet and Seino, “these variations reflect our lack of knowledge of the true physiopathology of generalized and unilateral seizures…. In practice, no clear-cut borderline exists between generalized and unilateral clonic seizures, in many cases, making it difficult if not impossible to classify them” (23).
Hemiclonic seizures are not mentioned in the updated ILAE positional paper on the classification of seizure types (28) and epilepsies (63). Also, hemiclonic status epilepticus is not considered in the latest ILAE definition and classification of status epilepticus (71).
Hemiclonic seizures are unilateral clonic seizures manifesting with repetitive, brief, and rhythmic, movements of rapidly alternating contractions and relaxations of muscles on one side of the body. Onset of hemiclonic seizures may be either:
• focal with typical Jacksonian march (Jacksonian or Bravais-Jackson hemiclonic seizures), |
A typical Jacksonian hemiclonic seizure starts with focal unilateral clonic convulsions in the face and thumb and march in an ordinary anatomical fashion to neighboring motor regions to the arm and down the leg to the foot or follow a reverse manner from the foot to the leg, up the arm, and to the face (74; 60; 30; 47; 26; 55).
This article is mainly dealing with two types of non-Jacksonian hemiclonic seizures that are (a) focal with non-Jacksonian march or (b) entirely unilateral without evidence of focal elements. According to Gastaut and associates:
These are the most frequent and the most striking variety of unilateral attacks” (30). They vary in duration but usually last in the order of several minutes or hours. Consciousness is often little, if at all, impaired and is only abolished in the most intense and long-lasting seizures. The attack is characterized by a series of unilateral jerks, which, from the very beginning, may affect all muscles of one side of the body. More frequently, they begin in one limb or in the head (then almost always in the eyes) before becoming truly unilateral. Attacks may begin with oculoclonus and end in conjugate lateral deviation of the eyes or, at times, of clonic movements of the head and eyes, which then usually diffuse to include the palpebrae, the ipsilateral face, and finally, the upper and lower limbs of that side. The spread is usually rapid, but on occasion it is slow, and the attack may be confused with a Jacksonian somatomotor seizure. | |
Sometimes the initial myoclonus moves from region to region in an orderly topographical spread over a period of 20 to 40 seconds. It may, for example, disappear in the face while appearing in the arm, only to disappear from the arm and involve the leg. Such a typical Jacksonian pattern, however, then becomes interrupted by the frank massive unilateral myoclonus, except for attacks in which one or several similar convulsive patterns repeat in succession before the seizure becomes truly unilateral. | |
Most frequently, however, the myoclonus persists in each of the successively involved regions until it affects the entire side of the body, where it remains for minutes, hours, or even days. Continuous variations of amplitude, frequency, and distribution of the jerks then take place and produce combinations that are most disconcerting to the uninformed observer. For instance, myoclonus may be infrequent but intense in the face, more rapid and of lower amplitude in the arm, and so fast and fine in the leg that it appears to be fused into tetanic contraction. This situation may then suddenly change, the myoclonus perhaps disappearing in the arm but continuing in the leg and face where they become infrequent but synchronous and of marked intensity. This instability of spatial distribution becomes even more evident if the jerks suddenly transfer to a part of, or to the entire, contralateral side of the body. Depending on whether or not they also persist on the side first involved, such propagation may produce either a generalized tonic-clonic seizure or an alternating hemiclonic epileptic seizure. The jerks occasionally appear in the nonhomologous contralateral limb before they vanish from the given limb, giving rise to somewhat spectacular crossed myoclonic epileptic seizures affecting, for instance, one leg and the contralateral arm. Immediately following the seizure, the side involved exhibits flaccid hemiparesis of variable duration, with or without a Babinski sign. This is transitory except after severe and prolonged attacks, usually with coma, when they may be succeeded by permanent residual spastic hemiparesis. | |
Also included by some among the unilateral seizures are the erratic seizures of the newborn. They consist of tonic or clonic convulsions localized to a given part of the body or affecting different parts of the body in asynchronous and asymmetrical fashion (30). |
Roget and Dravet with associates give the following description of non-Jacksonian hemiclonic seizures:
When observed, the onset is variable. Sometimes hemiclonic seizures present as repeated clonic jerks or twitching of the eyeballs, followed by a deviation of the head to the same side. These are followed rapidly by hemifacial myoclonus that spreads to the upper and lower limbs. In other cases, the clonic jerks involve the entire side from the onset. In some cases the clonic phase is preceded by a general discomfort, with abdominal pain, pallor and loss of contact. Many children are discovered while already convulsing. More often, the seizures are of long duration (minutes, hours, even days). In such cases the jerks show continuous changes in frequency, rhythm and amplitude in the different body areas involved. This gives a pattern of erratic seizures. They may, for instance disappear in the lower limb while continuing in the upper limb or the face and then spread to the entire side of the body again, thereby creating patterns of considerable complexity. At times they affect both sides of the body, either simultaneous as generalized seizures, or successively, as alternating epileptic seizures, also called “see-saw seizures” or “crises a bascule.” They also can affect one upper limb and the opposite lower limb together as “crossed seizures.” Another eventuality consists of a seizure reduced for some minutes to the loss of consciousness with only slight jerks localized to a small muscular segment or with lateral deviation and slight clonic jerks of the eyelids. | |
The state of consciousness is variable and often difficult to assess. Either initial complete loss of consciousness occurs, or consciousness is little if at all impaired and may fluctuate while the seizure develops. Most often there is a loss of contact, and the clouding of consciousness increases with the duration of the seizure, as do the autonomic manifestations (hypersalivation, pallor, cyanosis, vomiting). The end of the seizure usually is sudden, immediately followed by a flaccid hemiplegia, with or without Babinski sign, in the side involved; this is of variable duration, depending in part on the duration of the seizure. This aspect is difficult to determine in a child in coma with unilateral status epilepticus…. Spontaneous postictal recovery is progressive with an agitation phase whereas, after seizure interruption with a drug injection, the child may sleep quietly. However, when the seizure has been protracted (> 1 hour) there is a risk for a definite hemiplegia, first flaccid then spastic; this sequence constitutes the hemiconvulsion-hemiplegia syndrome, usually followed by epilepsy and manifesting as hemiconvulsion-hemiplegia-epilepsy syndrome (58; 23). |
Non-Jacksonian hemiclonic seizures mainly affect infants and very young children, though they may rarely occur in adults. An autopsy case of hemiconvulsion-hemiplegia-epilepsy syndrome, manifesting as cerebral hemiatrophy in an elderly man, has been reported (37).
Prognosis is variable, depending on the underlying etiology and syndrome. Prolonged hemiclonic seizures and hemiclonic status epilepticus may cause permanent hemiplegia. Fatal outcome has been reported (05; 39).
Vignette 1. A 16-year-old boy presented with two generalized seizures (42). A CT head scan was normal. He had no history of seizures, including absences or early morning myoclonus. There was a family history of epilepsy, although no further details were known. Two weeks after the generalized seizures, he presented again, with hemiclonic jerking of the right upper trunk, arm, and face of several hours’ duration. The jerking movements were rhythmic and lasted from a few seconds to a few minutes, with normal consciousness between episodes of jerking. Synchronous video-EEG recording showed generalized 3 to 4 Hz spike and polyspike wave discharges. The hemiclonic jerking was terminated with intravenous diazepam. Subsequently, the patient was treated with sodium valproate instead of carbamazepine, with full remission of generalized and hemiclonic seizures.
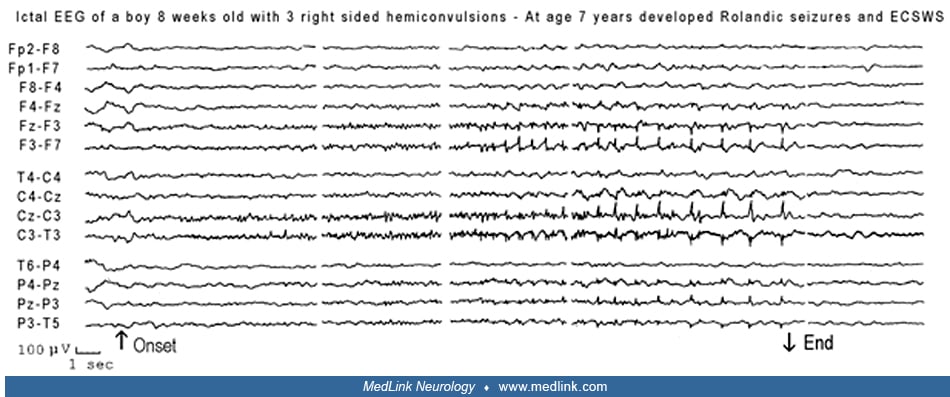
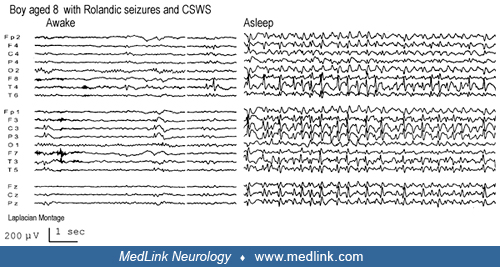
Vignette 2. This is an interesting case of a child with hemiclonic seizures at age 8 weeks (benign infantile seizures) and later at 7 years of age (rolandic seizures) that progressed to epileptic encephalopathy with continuous spike-and-wave during sleep (55).
This boy, born in December 1987, had three right-sided convulsions that mainly involved the face and upper limbs at 8 weeks of age. An EEG 6 days later showed during sleep “a classical focal electrical seizure in the left central regions.”
No clinical manifestations were noticed, but the child was lying in his mother’s arms.
Treatment with valproate was initiated. He did not have any additional seizures, and his next EEG 6 months later both during awake and sleep was normal. Brain MRI was also normal. Valproate was stopped 10 months later.
No further seizures occurred, and he developed well until the age of 7 years old, when he had three seizures. "His hands started shaking, and if he is holding something he will drop it. He is unable to speak during these and trembles. They last for 1 to 2 minutes, and he does not lose consciousness during them. Two occurred on the same day, and the second one was precipitated by a knock on the forehead. Following the attack, his right arm is stiff and this resolves after 10 minutes or so.”
A repeat EEG and brain MRI in 1994 were normal.
Treatment with valproate was initiated again, but soon after he had approximately six right-sided hemiclonic seizures mainly involving the face and arm. In April 1995 he was admitted to hospital because of increasing seizures. However, on discharge he had a number of further seizures where he dribbled or was subject to more extensive fits involving his right arm, with the seizures becoming generalized. This settled with the addition of lamotrigine. EEG conducted at this time was severely abnormal.
Subsequent to these events, he continued to have frequent fits. Two of these lasted for approximately 25 minutes. It is interesting that in April 1996, he had a seizure that was different to all others. He woke up at night unable to see. His eyes were deviated, and he was shaking and hysterical. The episode lasted for about 5 minutes. His vision gradually returned and he fell asleep. He also continued having attacks of dribbling from the corner of his mouth and making a clicking noise. This lasted for 2 to 3 minutes and he was well afterwards.
It was at this time, in 1996, that his school work began to be affected. Until this time, he had regularly been at the top of his class, and this no longer was the case. He had difficulty concentrating, sometimes fell asleep at school, and was often too tired to complete his homework.
In October 1996, he was referred to a specialized pediatric neurology clinic and showed some improvement in response to steroids.
Vignette 3. Hemifacial clonic status epilepticus ending with hemiclonic seizures in rolandic epilepsy, though rare, is demonstrated by the following case report.
This 30-year-old engineer had his first seizure at 11 years of age. While sleeping, he was awakened by numbness in his mouth; his tongue was tightened, and he was unable to speak for 1 minute after which he went back to sleep. The next year he had two brief similar nocturnal seizures. In one of these episodes postictally and briefly he could not see his mother’s head or a glass in front of him. Treatment with phenobarbital 50 mg nightly was initiated. Six months later he had a diurnal seizure after some partial sleep deprivation. While skating, he felt that the left side of his tongue was numb and that he could not see well. Within seconds, this was followed by repetitive and continuous left-sided clonic hemifacial spasms involving mouth and eye that ended 40 minutes later with left hemiconvulsions. There was postictal Todd paralysis. In addition, he had frequent right-sided headaches. Brain scan was normal, but EEG showed right centrotemporal spikes that appeared only in two EEGs when he was aged 12 and 13 years. His first EEG, 8 days postictally, had shown mainly right-sided slow waves. He had treatment with carbamazepine for 4 years. No further seizures and no more serious headaches occurred in the next 18 years. He did not suffer from migraine.
The underlying mechanisms of non-Jacksonian hemiclonic seizures are assumed to be similar to those of generalized epilepsies but limited to one hemisphere as opposed to a focal mechanism (27). However, like the generalized onset clonic seizures, very little is known about the localization and pathophysiology of hemiclonic seizures involving one side of the body from onset. They may be grossly asymmetrical, unilateral, generalized onset seizures. The latter, according to the updated ILAE report, “are conceptualized as originating at some point within, and rapidly engaging, bilaterally distributed networks. Such bilateral networks can include cortical and subcortical structures but do not necessarily include the entire cortex. Although individual seizure onsets can appear localized, the location and lateralization are not consistent from one seizure to another. Generalized seizures can be asymmetric” (08).
According to the ILAE core group, the mechanisms of clonic seizures appear to be due primarily to rhythmic excitatory discharges, whereas the clonic phase of GTCS represents the phasing in of seizure-suppressing mechanisms (26).
According to Gastaut and associates, “While Jacksonian motor seizures originate from a rolandic discharging focus, hemiconvulsive seizures seem to result from a subcortico-cortical neuronal discharge transmitted to the totality of one hemisphere,” and “It is probable that this habitually hemi-generalized character of epileptic seizures in very young children is related to a particular state of their cerebral maturation and perhaps as well to a functional insufficiency of their commissures” (32).
The explanation for involvement of one hemisphere is not clear, but may, at least in hemiconvulsion-hemiplegia-epilepsy, involve maturation of brain structures, such as the corpus callosum, or genetic (CACNA1A) factors (04).
The epidemiology is not precisely known. Hemiclonic seizures (non-Jacksonian) mainly affect infants and young children. In one study of 154 cases of focal epilepsy with seizure onset in the first 3 years of life, 57 patients had hemiclonic seizures and eight patients had clonic seizures (53).
Improved emergency care for status epilepticus is important.
The main differential diagnosis of hemiclonic seizures is Jacksonian seizures. Clinically, non-Jacksonian hemiclonic seizures are unilateral from onset or at least lack the typical focal onset and marching of typical Jacksonian hemiclonic seizures. On EEG, (1) they show generalized or unilateral spike-and-wave or polyspike-and-wave discharges, either spontaneously or following overbreathing or photic stimulation; (2) the few patients with hemiclonic seizures presenting focal signs (pointing actually to the temporo-occipital more often than to the rolandic region) also show generalized or unilateral spike-and-wave EEG discharges; and (3) ictal EEG does not show focal rolandic discharges, but spike or spike-and-wave discharges, identical to those seen in typical generalized epilepsies, appearing on the contralateral hemisphere (32).
Hemiconvulsion-hemiplegia epilepsy syndrome. This is characterized by a sequential combination of unilateral or predominantly unilateral clonic seizures (usually of long duration), occurring during the first 2 years of life, mainly during febrile illnesses, and immediately followed by hemiplegia (initially flaccid before becoming permanent and spastic) ipsilateral to the clonic seizures (02). This may complicate a preexisting brain disorder, for example, Sturge-Weber syndrome, agenesis of the corpus callosum, or tuberous sclerosis (31; 03; 04; 69). A case of hemiconvulsion-hemiplegia syndrome that later developed contralateral hemi Lennox-Gastaut syndrome has been reported (52).
See MedLink Neurology article: Hemiconvulsion-hemiplegia-epilepsy syndrome.
Hemiclonic seizures and hemiclonic status epilepticus. Hemiclonic seizures and hemiclonic status epilepticus have also been reported in pyridoxine dependency of infants, hypoglycemia (50), hypocalcemia (23), cobalamine C deficiency (51), and some chromosomal disorders, such as Angelman syndrome (48; 57) and 4p-syndrome (64; 06; 41). The DNA A3243G mutation is the most common cause of mitochondrial encephalomyopathy, lactic acidosis, stroke-like episodes (MELAS), and Prader-Willi syndrome (19; 73). Hemiclonic seizures in complex febrile seizures may be more common than cited (23).
Dravet syndrome. Hemiclonic seizures are frequently reported in Dravet syndrome, but only two of 60 patients of Dravet herself had true hemiclonic seizures (22), corresponding to the description of Gastaut and Broughton (30). They mainly occur in younger children and primarily in the first year of life. Dravet syndrome should be considered in any child under 1 year of age who presents with a prolonged hemiclonic or bilateral tonic-clonic seizure with fever, without any identified underlying cause (76). The other unilateral seizures have different characteristics, such as shorter duration, association of contralateral tonic changes, and ictal EEG abnormalities more limited to one hemisphere. They can alternate from one side to the other, and they are often followed by transient postictal hemiparesis.
Hemiclonic seizures are common in a severe phenotype of a recurrent missense SCN1A mutation, p.Thr226Met (61; 07). This differs from Dravet syndrome because of earlier age at onset, profound developmental impairment, and a distinctive hyperkinetic movement disorder.
See MedLink Neurology article: Dravet syndrome.
Rarely, patients with CACNA1A encephalopathy show acute attacks of headache, hemiconvulsions, and hemiplegia with coma sometimes related to unilateral brain edema (12).
Idiopathic generalized epilepsy. There is a case report of a patient with idiopathic generalized epilepsy manifesting with hemiclonic and generalized tonic-clonic seizures (42).
Epileptic encephalopathy with continuous spike-and-wave during sleep (CSWS). Continuous spikes and waves during slow sleep are a prerequisite for the diagnosis of epileptic encephalopathy with CSWS, which follows three stages of evolution (11; 72; 01). The first stage manifests with infrequent and mainly nocturnal motor focal seizures, often hemiclonic status epilepticus, and EEG with focal and bi-synchronous generalized discharges.
The first seizure is usually nocturnal in half of the cases, and in 40% consists of unilateral convulsions often lasting for more than half an hour, thus, constituting hemiclonic status epilepticus. In the other cases, the first seizure may be focal motor-clonic, generalized tonic-clonic, complex focal, or myoclonic absence.
Rolandic epilepsy, Panayiotopoulos syndrome, and childhood occipital epilepsy of Gastaut. Hemiclonic seizures are frequently reported in patients with rolandic epilepsy, Panayiotopoulos syndrome, and childhood occipital epilepsy of Gastaut. However, these hemiclonic seizures are likely to be of focal onset as documented with video-EEG (66; 13).
In rolandic epilepsy, hemiclonic seizures occur in 10% to 15% of patients, and they are likely purely Jacksonian hemiclonic seizures evolving from an initial focal clonic seizure with Jacksonian march (13). The focal hemifacial clonic jerks are usually the first clinical symptom that may be entirely localized in the lower lip, manifesting with sudden, continuous, or bursts of clonic contractions. Involvement of the ipsilateral eyelids is not unusual. More rarely, ictal symptoms of clonic convulsions may appear nearly simultaneously or spread to the ipsilateral upper extremity. Involvement of the leg is rare. Hemifacial motor seizures may be the only ictal manifestation, but often this is associated with inability to speak, hypersalivation, localized paresthesia, and tonic contraction of the mouth to one side that may progress or appear synchronously with oro-pharyngo-laryngeal symptoms and other buccal symptoms. Any combination of these symptoms is possible although focal clonic seizures may occur alone and also be the only seizure type in the life of a child. Their duration is usually brief, for seconds or 1 to 2 minutes. Hemiconvulsive status epilepticus may occur, and this is more common in younger than older children and those with multifocal spikes. Postictally, patients may quickly return to normality, but others may show transient postictal paresis of the affected muscles.
In 10% to 20% of patients with Panayiotopoulos syndrome, autonomic seizures and autonomic status epilepticus end with hemiclonic seizures (54; 14; 56; 66; 67). These commonly start long after the onset of the ictal discharge and usually long after other clinical manifestations (autonomic disturbances, deviation of the eyes and head). A Jacksonian march from hemifacial to hemiclonic seizures is often, but not always, observed by witnesses. Overall, hemiclonic seizures occurred in 22 (26%) of 86 seizures in 47 patients with Panayiotopoulos syndrome, including two patients with hemiconvulsive status epilepticus (54). According to one report, “generalized” onset clonic seizures were the most common ictal event in patients with Panayiotopoulos syndrome and convulsive status epilepticus, and one third required admission to intensive care units (73). However, it is doubtful these are of generalized onset clonic seizures in this syndrome, which is a focal epilepsy (55). Figure 1 of this paper illustrates the EEG of right hemiconvulsive status epilepticus in a 5.5-year-old boy, with high-amplitude, repetitive spike wave complexes over the left hemisphere (73).
In idiopathic childhood occipital epilepsy of Gastaut, deviation of the eyes, usually following elementary visual hallucinations and often associated with ipsilateral turning of the head, is the most common nonvisual symptom, occurring in approximately 70% of these patients (55; 15). This focal motor seizure may be mild, but more often it is severe and progresses to hemiconvulsions. According to Gastaut and Zifkin, 43% of visual seizures are followed by hemiconvulsions (33).
See MedLink Neurology articles: Panayiotopoulos syndrome and Idiopathic childhood occipital epilepsy of Gastaut.
Diagnosis is based on a careful clinical history. Diagnostic procedures (EEG, brain imaging, and relevant blood, genetic, and chromosomal testing) depend on the suspected etiology.
Interictal EEG. Interictal EEG may show (1) partial discharge rapidly spreading over only one hemisphere (corresponding with only contralateral seizures), (2) discharges generalized from the start but considerably predominant over one hemisphere, susceptible to change from one side to the other at different moments (corresponding to alternating seizures), or (3) partial discharge, susceptible to change, from time to time, in morphology and topography (from area to area and, sometimes, from one side to the other) (29).
Ictal EEG. Ictal EEG findings in non-Jacksonian hemiclonic seizures are variable and best described by Gastaut and Broughton:
Most frequently, they consist of a continuous bilateral and synchronous discharge of slow waves at about 2-3 Hz, often of greater amplitude over the scalp contralateral to the clinical seizure, intermingled with rapid "recruiting" waves at 10 Hz, which sometimes predominate posteriorly. Inconstant and complex wave forms are observed; they are due to continuous changes of phase, amplitude, frequency, and distribution of the 2 components. The discharge, therefore, includes rhythmic slow-wave activity, sometimes notched, or discharges of spike and wave variety that may predominate in anterior, central, or posterior regions but that, like the clinical seizure, frequently shift in maximum amplitude from location to location. At times the discharge and myoclonus are interrupted for several seconds, during which the EEG shows electrical silence over the scalp, contralateral to the convulsion and slow wave activity on the ipsilateral side. | |
At the usually abrupt ending of the seizure, the same signs of cortical extinction with subsequent depression persist for several or dozens of minutes and are associated with the transitory hemiparesis. | |
The hypersynchronous EMG potentials recorded over the muscles affected do not correlate well with the EEG spike and sharp wave potentials. The relationship between the two becomes even less precise when the cortical discharges and peripheral myoclonus vary from region to region, or when both cross over to the contralateral sides. | |
In those unilateral clonic seizures that alternate from one side of the body to the other, the ictal EEG discharge similarly shifts sides to become maximum over scalp areas contralateral to the attack. | |
The ictal EEG during the erratic seizures of the newborn contains shifting focal discharges over the entire scalp (30). The ictal EEG abnormalities of unilateral convulsive seizures are described by the ILAE Neurophysiology Task Force as follows (44): |
The ictal discharge is characterized by rhythmic (2-3/second) bilateral slow waves of higher amplitude over the hemisphere contralateral to the clinical manifestations and intermixed with 10 Hz recruiting rhythms. In other focal unilateral seizures, the EEG pattern can be variable with onset over the frontal or frontal-central regions of one hemisphere, or with bilateral asymmetric onset, but always predominant over the frontal areas. The EEG onset consists of “pseudo-rhythmic” spikes and waves, contralateral to the clinical manifestations, and may be periodically interrupted by a 1 to 2 second flattening of the EEG. In some cases, the unilateral seizure can be preceded by isolated massive jerks related to generalized spike-wave discharges starting several minutes before the unilateral seizure onset. These myoclonic seizures can be on either side in the same patient; this alternating pattern can be a clue for the diagnosis of Dravet syndrome.
Brain MRI. In a report of 10 patients with hemiconvulsion-hemiplegia syndrome, early MRI (days 1 to 7 from status epilepticus) showed hemispheric cytotoxic edema in all patients, extending to the contralateral side for one patient. T2 hyperintensity in the basal ganglia was disclosed in 70% of patients and in the hippocampus in 60% (05). After 1 month (in intermediate and chronic phases), all surviving patients but one showed hemispheric cortical atrophy corresponding to the regions involved during the early stage. Comparing clinical features of patients presenting with “typical” MRI features (ie, strictly unilateral involvement), to those with “atypical” findings (ie, bilateral involvement), the second group presented psychomotor delay before status epilepticus. This series underlines the major value of early MRI for the prompt diagnosis of hemiconvulsion-hemiplegia syndrome, and it shows that involvement of subcortical structures has been underestimated. Hippocampal involvement is not constant (05).
Genetic testing for SCN1A mutations is needed particularly in infants less than 1 year old with two or more prolonged hemiclonic febrile seizures suspected for Dravet syndrome (68; 35). See MedLink Neurology article: Dravet syndrome.
However, the incidence of SCN1A genetic mutation in patients with hemiconvulsion-hemiplegia-epilepsy syndrome is low (43).
Management is dictated by the underlying etiology and epileptic syndrome. Prevention and urgent termination of prolonged hemiclonic seizures and hemiclonic status epilepticus as of generalized convulsive status epilepticus is mandated because of the risk of permanent brain damage and hemiplegia (65; 17; 02; 62). Many medications are used in treating hemiclonic seizures, but polytherapy is often required for severe forms. Valproate, benzodiazepines (clobazam or clonazepam), topiramate, levetiracetam, zonisamide, and stiripentol are the most useful antiepileptic drugs.
In a single case of CACNA1A-related encephalopathy the acute attacks of headache and hemiconvulsions were improved with early administration of corticosteroid pulses and hypertonic solution (12). Ketogenic diet has been beneficial in epileptic encephalopathies with hemiclonic seizures such as Dravet syndrome and epilepsy in infancy with migrating focal seizures (16; 24).
Cannabidiol. There is a growing interest in the use of cannabidiol for the treatment of drug resistant epilepsies like Dravet and Lennox-Gastaut syndrome (20; 21; 45; 49; 62). In a double-blind, placebo-controlled trial, in a 14-week treatment period of patients with Dravet syndrome, cannabidiol was significantly beneficial particularly in convulsive-seizures, with 5% of patients becoming seizure free though associated with higher rates of adverse events (20; 21). In June 2018, Epidiolex (cannabidiol oral formulation) from GW Pharmaceuticals obtained marketing authorization by the FDA for the treatment of Dravet and Lennox-Gastaut syndrome in the United States. More information can be accessed at:http://www.draccon.com/dravet-pipeline. The package insert for Epidiolex contains additional information and can be reviewed at:https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210365lbl.pdf.
Phenobarbital and potassium bromide completely terminated the seizures in a 10- month-old girl with Wolf-Hirschhorn syndrome and right or left hemiclonic convulsions of epilepsy in infancy with migrating focal seizures (36).
There are a number of agents targeted for severe epilepsies with hemiclonic seizures, such as Dravet syndrome. These are TAK935, lorcaserin, clemizole, huperzine analog, ataluren, selective sodium channel modulators and activators, antisense oligonucleotide therapy, and adenoviral vector therapy (62).
Prognosis varies significantly depending on the underlying etiology and syndrome. It is benign in rolandic epilepsy and Panayiotopoulos syndrome but very severe in Dravet syndrome and hemiconvulsion-hemiplegia epilepsy.
Early and appropriate management in order to terminate possibly hemiclonic status epilepticus is important.
This condition mainly affects children.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Henry Hasson MD
Dr. Hasson of Albert Einstein College of Medicine has no relevant financial relationships to disclose.
See Profile
Solomon L Moshé MD
Dr. Moshé of Albert Einstein College of Medicine has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Epilepsy & Seizures
Jun. 02, 2026
Neuropharmacology & Neurotherapeutics
May. 11, 2026
Epilepsy & Seizures
May. 08, 2026
Epilepsy & Seizures
May. 01, 2026
Epilepsy & Seizures
Apr. 30, 2026
Epilepsy & Seizures
Apr. 17, 2026
Epilepsy & Seizures
Apr. 13, 2026
Epilepsy & Seizures
Apr. 08, 2026