

Neurobehavioral & Cognitive Disorders
Mental status examination
Jun. 17, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
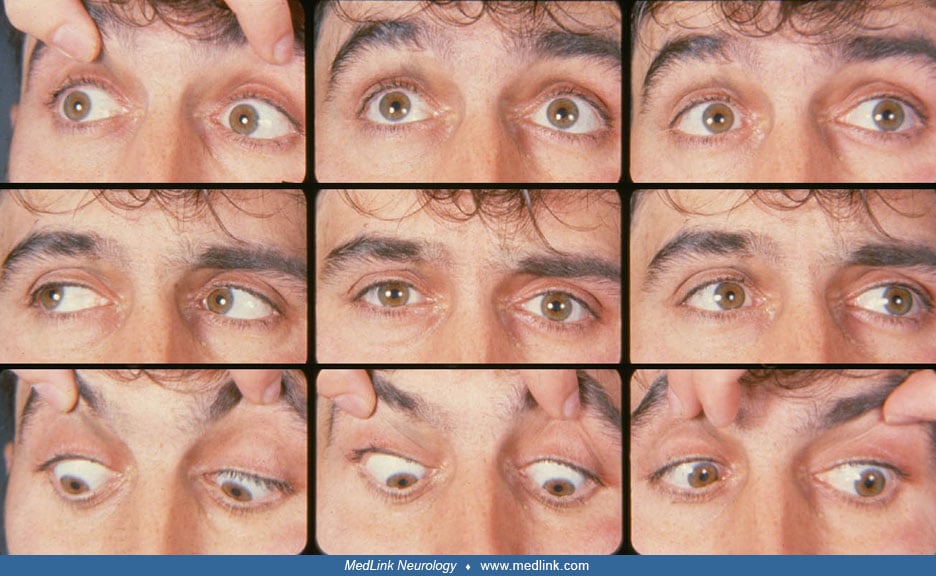
The term “gaze palsy” refers to an impairment in conjugate ocular excursions that is of the same degree in the two eyes and limited to either the horizontal or the vertical plane. The causative lesions will almost always be located in the central nervous system. This article deals with gaze palsies in the horizontal plane; gaze palsies in the vertical plane are covered in a separate article.
In the most severe form of horizontal gaze palsy, neither eye can cross the midline into the opposite field of gaze, and the eyes may actually be deviated toward the intact side, a phenomenon called “gaze deviation.” In less severe horizontal gaze palsies, the eyes may cross part of the way into the opposite field of gaze. In the mild palsies, the eyes have full excursions into the opposite field of gaze, but the saccades in that direction are slow and the eyes will display a jerk nystagmus with its fast phase in the direction of the gaze palsy.
Horizontal gaze palsies are caused by lesions of the central nervous system: the cerebrum, diencephalon, or pons. These gaze palsies are divided into those that arise from lesions rostral to the ocular motor nuclei in the cerebrum or diencephalon (“supranuclear”) and those that lie within the sixth nerve nuclei in the pons (“nuclear”). Supranuclear lesions damage the volitional subsystems (saccades and pursuit) and spare the non-volitional subsystem (the vestibulo-ocular reflex). Nuclear lesions damage the volitional and non-volitional ocular motor subsystems. Therefore, clinical examination of a patient with an apparent gaze palsy or gaze deviation should include not only the testing of saccades and pursuit, but also the testing of the vestibulo-ocular reflex by means of the doll head (doll eye, oculocephalic) maneuver.
Lesions of the extra-axial ocular motor cranial nerves, neuromuscular junction, or extraocular muscles may appear to cause gaze palsies if the eye movement deficits happen to be of the same degree in both eyes and limited to a single plane. Such an occurrence is rare. The notable exception is the acquired extraocular muscle dystrophy known as chronic progressive external ophthalmoplegia (CPEO) attributed to mitochondrial dysfunction. The rare congenital muscular dystrophies may also simulate horizontal gaze palsies. However, close examination of these cases discloses that the impairment in ocular excursions also involves the vertical plane. Myasthenia gravis and botulism may fortuitously simulate horizontal gaze palsies, as will traumatic or inflammatory shortening of extraocular muscles. A prominent example is Graves disease, which may rarely restrict horizontal movement of both eyes to the same degree. These mimickers, and the ways to distinguish them from CNS lesions, will be reviewed later in the article.
|
• Gaze palsies are impaired conjugate excursions of the two eyes. | |
|
• Both eyes must have the same degree of excursional impairment, which can be unidirectional or bidirectional, and occur in either the horizontal or vertical plane. | |
|
• This article covers horizontal gaze palsies; vertical gaze palsies are covered in a separate article. | |
|
• Severe unidirectional palsies may also display conjugate gaze deviation to the side opposite to the direction of the palsy; in those cases, evidence of hemispatial neglect will usually be present. | |
|
• Less severe unidirectional horizontal gaze palsies can be overcome by imploring the patient to look in the opposite direction; these disorders are called gaze preferences. | |
|
• Mild gaze palsies display full excursions but slow saccades or gaze-evoked jerk nystagmus when the eyes are directed toward the palsied side. | |
|
• Gaze palsies are divided into supranuclear (saccades and pursuit impaired and vestibulo-ocular reflex spared) and nuclear (saccades, pursuit, and vestibulo-ocular reflex impaired). | |
|
• Supranuclear horizontal gaze palsies are caused by cerebral lesions or focal seizures; nuclear horizontal gaze palsies are caused by pontine lesions. | |
|
• Symmetrically reduced horizontal ocular excursions are rarely caused by lesions outside the central nervous system. Excursions are often reduced in the vertical plane as well. Clues to a peripheral localization are usually present. |
Conjugate and disconjugate gaze. The term “conjugate gaze” refers to the movements of the two eyes in the same direction. Gaze palsies are disorders of conjugate gaze. They may be complete or incomplete, congenital or acquired, and unidirectional or bidirectional.
The term “disconjugate gaze” refers to movements of the two eyes in opposite directions. It is not pertinent to gaze palsies, in which the impairment is always conjugate. This term is often incorrectly applied to a static condition in which one eye is aimed at the viewed target and the other eye is deviated off-target. The proper term for that condition is “ocular misalignment.” The term “strabismus” is a synonym for ocular misalignment. It is most often used to designate ocular misalignment discovered at birth or in early childhood.
The term “convergence” is used to describe a specific type of disconjugate eye movement in which the eyes move inwardly (toward each other) as the patient alters fixation from a distant to a near target. Convergence is physiological (normal) when used to fixate targets within reading distance. It may be insufficient or excessive in certain disease states. If horizontal gaze is impaired, the brain may automatically activate an intact convergence system in order to bring the adducting eye over to fixate an eccentric target viewed at distance. This phenomenon, which occurs particularly in chronic pontine lesions, is called “convergence substitution.”
Ocular versions and ductions. The term “ocular versions” is applied to conjugate movements of the eyes in the horizontal or vertical plane. Rightward conjugate ocular movements are called “dextroversion” and leftward movements are called “levoversion.” However, these terms are rarely used, being replaced by the more familiar terms “right gaze” and left gaze.”
“Ocular ductions” is the term applied to the excursions of each eye examined one at a time (the unexamined eye is covered). Thus, inward movement of an eye is called “adduction,” outward movement is called “abduction,” upward movement is called “supraduction,” and downward movement is called “infraduction.” If one eye has impaired ductions and the other eye has full ductions, it is appropriate to refer to the impaired eye movement in that one eye as a “ductional deficit,” not a “gaze deficit.”
Gaze impairments and binocular diplopia. Binocular diplopia is double vision that is eliminated by covering either eye. It is virtually always caused by ocular misalignment, as the viewed target is imaged on the fovea of the fixating eye and on the parafoveal retina of the deviating eye. Because gaze palsies impair the horizontal movements of both eyes to an equal degree, the eyes will not be misaligned and diplopia will not occur. However, if convergence is also affected, the eyes may go out of alignment when viewing a target at reading distance, creating diplopia at near.
Selective gaze palsies. Gaze palsies may affect volitional ocular motor subsystems (saccades and pursuit) and spare the nonvolitional subsystem (vestibulo-ocular reflex), a phenomenon called a “supranuclear gaze disorder.” It arises from lesions in the cerebrum or diencephalon that spare the vestibular pathway, which is confined to the brainstem. Gaze palsies may also impair pursuit but spare saccades and the vestibulo-ocular reflex because saccades and the vestibulo-ocular reflex are less vulnerable. The impaired pursuit will be evident as cogwheel rather than smooth conjugate movements because the more robust saccadic subsystem substitutes for the damaged pursuit subsystem to help the eyes catch up with a moving target.
Gaze palsies as gaze-evoked jerk nystagmus. A mild form of gaze palsy may consist of the inability to maintain eccentric gaze, in which the eyes repeatedly drift back toward straight ahead position. That drift will be met by a saccadic effort to take the eyes back into eccentric gaze, setting up an oscillatory cycle called “gaze-evoked jerk nystagmus.”
Gaze palsies as gaze deviations or gaze preferences. Unidirectional horizontal (or vertical) gaze palsies are often accompanied by eccentric positioning of the eyes to the opposite side. The term to describe this phenomenon is “gaze deviation.” If the gaze deviation is mild, intermittent, or easily overcome by commanding the patient to look to the opposite side, the term “gaze preference” is often used.
Supranuclear gaze palsy versus gaze apraxia. The term “apraxia of gaze” has been applied to a variety of gaze disorders by analogy to loss of volitional motor functions with preservation of automatic motor functions in disorders of the parietal lobe.
Gaze apraxia was used in early descriptions of acutely acquired supranuclear gaze palsy because the nonvolitional vestibulo-ocular reflex was preserved in the face of absent volitional movements—saccades and pursuit (20). The gaze palsies in these patients have often extended to the vertical plane. Most patients have had biparietal-frontal ischemic stroke, often in the setting of systemic hypotension. They have also had inattention, earning the diagnosis of Balint-Holmes syndrome.
Apraxia of gaze has also been applied in parkinsonian states, where bradykinesia may cause delayed initiation of horizontal gaze.
Apraxia also appears in a disturbance in toddlers called congenital ocular motor apraxia (COMA). Children with COMA cannot execute horizontal volitional gaze without thrusting their heads in the direction of intended gaze. These distinctive head thrusts are posited to recruit cervical input to initiate gaze. But the thrusts also activate an intact vestibulo-ocular reflex, which will cause the eyes to move opposite to the direction of the head thrusts. As a result, the head thrusts must carry the eyes beyond the intended target. COMA is usually not associated with focal brain lesions. It is believed to be a developmental delay in maturation of volitional ocular motor pathways. Most children outgrow it by the third decade of life.
An acquaintance with the ocular motor pathways helps to understand why lesions in these locations cause horizontal gaze palsies (33; 94; 55; 73; 59).
Two volitional ocular motor subsystems--saccades and pursuit--drive the eyes to the side. The saccadic pathway, which mediates spontaneous refixations and those elicited by patient desire to fixate an eccentric visual target, originates in the frontal and parietal cortex and crosses in the rostral brainstem to synapse on the pontine paramedian reticular formation (PPRF) on the opposite side. The PPRF sends signals to the ipsilateral sixth nerve nucleus. The sixth nerve nucleus contains two sets of neurons--those that directly innervate the lateral rectus muscle and interneurons that connect via the medical longitudinal fasciculus to the contralateral medial rectus subnucleus in the midbrain. The joint firing of these two sets of sixth nerve neurons initiates horizontal gaze ipsilateral to the sixth nerve nucleus and contralateral to the cortical signal source.
The pursuit pathway, elicited by instructing the patient to follow a moving target, originates in the visual cortex, which sends signals to the parietal and frontal cortex. Neurons there send signals that course through the cerebellum and pass through the PPRF without synapsing. The pursuit pathway finally synapses on the sixth nerve nucleus, which mediates horizontal gaze ipsilateral to the sixth nerve nucleus and ipsilateral to the cortical signal source.
The non-volitional pathway--the vestibulo-ocular reflex--originates in the semicircular canals and otoliths, sending signals through the vestibular nerve to the medullary vestibular nuclei. Those nuclei connect directly to the sixth nerve nucleus for horizontal gaze. The vestibulo-ocular reflex moves the eyes in a direction opposite to that of any change in head position in order to keep the eyes stable in space. Stability in space allows the eyes to maintain foveal fixation on the viewed target. Were fixation to change with each shift in head position, visual acuity would be severely degraded every time the head or body moves. A defective vestibulo-ocular reflex will cause blurred vision with minor shifts.
Lesions of the cerebrum and diencephalon cause impairments of volitional eye movements (saccades and pursuit), sparing the reflex eye movements generated by the vestibulo-ocular reflex. Because these lesions lie hierarchically above the sixth nerve (and other ocular motor) nuclei, the gaze palsies are called “supranuclear.” Lesions within the sixth nerve nuclei cause impairment of all eye movement subsystems. Because these lesions lie within the sixth nerve nucleus, the gaze palsies are called “nuclear.”
The direction and plane of the gaze palsies depends on the location of the lesion. Thus, unilateral cerebral hemispheric (mostly parietal) lesions cause contralateral horizontal gaze palsies. Bilateral cerebral hemispheric lesions cause impaired bidirectional gaze palsies in the horizontal and vertical planes.
Unilateral diencephalic lesions may cause contralateral or ipsilateral horizontal gaze palsies and also vertical gaze palsies. If the diencephalic lesions are restricted to the dorsomedial thalamus, vertical gaze palsies will predominate and may occur without horizontal gaze palsies.
Unilateral pontine lesions that involve its tegmentum cause ipsilateral horizontal gaze palsies that involve volitional (saccades, pursuit) and reflex eye movements (vestibulo-ocular reflex). Bilateral pontine lesions cause bidirectional horizontal gaze palsies, but only if the lesions involve the pontine tegmentum. Lesions in the basis pontis tend to spare all eye movements because they are remote from the sixth nerve nuclei and their connections to the midbrain. Pontine lesions typically spare vertical gaze, which is mediated in the midbrain. The sparing of vertical gaze by pontine lesions is an important clinical feature that comes into play in pontine locked-in syndrome, in which vertical gaze may be the only preserved movement (42). Once the lesion extends into the pontomesencephalic junction, vertical gaze palsies will appear.
Cerebral lesions. Acute unilateral cerebral lesions cause contralateral saccadic gaze palsies. The eyes will not, to command, cross to the side opposite to the cerebral lesion. Because the pursuit pathway is ipsilaterally mediated, the patient may be able to move the eyes contralaterally by following a moving target in that direction. The vestibulo-ocular reflex is spared, so the patient’s eyes will move contralaterally on passive movement of the head toward the side of the lesion.
The cerebral lesions that cause contralateral gaze palsies tend to be centered in the parietal rather than the frontal lobe. These lesions are large and usually involve white matter as well as gray matter. Within the first week after an acute ischemic stroke or lobar hemorrhage, the eyes are often deviated toward the side of the lesion, a phenomenon that is far more common with right-sided than left-sided lesions. The frequent concurrence of signs of hemispatial neglect has given rise to the hypothesis that ipsilateral horizontal gaze deviation is a manifestation of motor neglect (46; 60; 80; 21).
Focal seizures, most often originating in frontal or parietal lobes, may give rise to contralateral gaze deviation, together with automatisms, contralateral head turning, and tonic-clonic contralateral face and limb movements (104; 32). During the ictus, none of the eye movement systems will carry the eyes back toward the side of the seizure focus. When the seizure ends, the eyes often drift to the side of the seizure focus and stay there until the post-ictal state has resolved. During the post-ictal state, volitional eye movements may not elicit contralateral gaze, but the vestibulo-ocular reflex will be successful.
Pontine lesions. Acute unilateral pontine tegmental lesions often cause ipsilateral horizontal gaze palsies. Volitional and non-volitional (reflex) gaze is impaired. In some cases, the eyes will be deviated to the side contralateral to the lesion but not to the same extent as occurs with cerebral lesions. A handy clinical epigram that helps to separate cerebral from pontine unidirectional gaze palsies is that in cerebral nonepileptic lesions, the eyes point to the side of the lesion and away from the side of hemispatial neglect or hemiparesis, whereas in unilateral pontine lesions, the eyes point away from the side of the lesion and toward the hemiplegia. Mild or resolving gaze palsies can manifest as slow saccades or a gaze-evoked nystagmus (08; 64).
In pontine gaze palsies, patients may converge their eyes when attempting gaze to the side of the deficit. In that case, both eyes will move inward toward each other, a phenomenon called “convergence substitution” (43). Convergence substitution is automatic but also serves to bring the adducting eye to a visual target displayed in the field of the gaze palsy.
Other clinical features assist in localization of cerebral and pontine horizontal gaze palsies (Table 1) (77; 10; 51; 25). In cerebral lesions, the gaze palsy is unidirectional, whereas in pontine lesions, which frequently affect both sides of the pons, the horizontal gaze palsy is often bidirectional. In cerebral lesions, hemispatial neglect is often present, but the eyes are aligned. By contrast, in pontine lesions there is no hemispatial neglect, but the eyes are often misaligned owing to the concurrence of sixth nerve palsy, skew deviation, or internuclear ophthalmoplegia (22; 88; 102; 48; 25; 107). In cerebral lesions, there may be contralateral hemiparesis and sometimes contralateral hemisensory loss. In pontine lesions, there is also contralateral hemiparesis, accompanied by nystagmus, ataxia, sensory loss on one or both sides of the body, and sometimes altered consciousness.
|
Clinical Feature |
Nonepileptic Cerebral Lesion |
Focal Seizure****** |
Pontine Lesion |
|
Bidirectional |
Rare |
Never |
Common |
|
Vestibulo-ocular reflex |
Spared |
Not spared |
Not spared |
|
Conjugate eye deviation |
Ipsilateral to lesion* |
Contralateral seizure focus in ictal state |
Contralateral to lesion** |
|
Convergence substitution |
Not present |
Not present |
Often present |
|
Hemispatial neglect |
Often present |
Not usually present |
Not present |
|
Hemiparesis |
Contralateral to gaze deviation and contralateral to lesion |
Ipsilateral to seizure focus in post-ictal state |
Ipsilateral to gaze deviation and contralateral to lesion |
|
Nystagmus |
Rarely present*** |
Often present**** |
Sometimes present*** |
|
Skew deviation |
Not present |
Not present |
Sometimes present |
|
Sixth nerve palsy |
Not present |
Not present |
Sometimes present |
|
Internuclear ophthalmoplegia |
Not present |
Not present |
Sometimes present |
|
Ataxia |
Not present |
Not present |
Sometimes present |
|
Head deviation |
Not present |
Contralateral to seizure focus during ictal state |
Not present |
|
Tonic-clonic face and extremity movements |
Not present |
Contralateral to seizure focus during ictal state ***** |
Not present |
|
*Most common with right cerebral hemisphere lesions centered in parietal lobe. Eyes will move past mid position with doll head maneuver. Gaze deviation disappears within weeks of the event. | |||
Cerebral lesions. The acquired lesions most commonly associated with unidirectional gaze palsies are large unilateral parietal-based ischemic strokes or lobar hemorrhages (44; 41; 78; 24). They cause contralateral horizontal gaze palsies and usually ipsilateral gaze deviation within the first week after the event. Vestibulo-ocular reflex can overcome the gaze palsy. Other unilateral hemispheric lesions—abscesses, tumors, and malformations—are less likely to produce gaze palsies, perhaps because the perturbation is less acute. Such chronic lesions may, however, cause gaze palsies if they generate focal seizures.
Focal seizures of frontal or parietal lobe origin cause gaze palsies by a pulsive mechanism. During the ictus, the eyes are conjugately driven to the side opposite the seizure focus. The gaze palsy is ipsilateral to the seizure focus. It cannot be overcome by the vestibulo-ocular reflex and is often accompanied by contralateral gaze and head deviation, as well as contralateral jerk nystagmus and clonic facial and extremity movements (32). In the post-ictal state, the eyes may deviate to the side of the seizure focus; at that point, the vestibulo-ocular reflex may be able to initiate gaze deviation to the opposite side.
Bilateral cerebral lesions may produce bidirectional horizontal gaze palsies, but vertical gaze will be impaired as well. Acute causes are bilateral ischemic or hemorrhagic strokes, hypoxic-ischemic encephalopathy, inflammatory conditions, and multiple metastases. Subacute or chronic causes include Alzheimer disease, Creutzfeldt-Jakob disease, progressive supranuclear palsy, Behcet disease, inflammatory disease, cancer, and Whipple disease.
Among the congenital causes of horizontal gaze palsies, the most common is congenital ocular motor apraxia (COMA). In this condition, children cannot generate horizontal saccades, quick phases of nystagmus, or pursuit (16; 40). The vestibulo-ocular reflex is intact. Because initiation of the volitional horizontal eye movements is impaired in the face of intact reflex gaze, this syndrome was originally called an apraxia. It should more properly be called a “congenital supranuclear horizontal gaze palsy.” Brain imaging is often normal, although some development abnormalities may be found in the cerebrum, brainstem, or cerebellum (71; 29; 13; 90; 40; 62).
A characteristic feature of COMA is blinking in advance of attempted lateral gaze, perhaps in order to interrupt fixation, which may be acting as a brake on saccades. Horizontal head thrusts are also common as part of refixation on an eccentric target. The thrusts carry the head and eyes beyond the target. The intact vestibulo-ocular reflex shifts eyes contraversively and allows them to settle on the viewed target. Such head thrusts are not as common in acquired gaze palsies. In the idiopathic form of this condition, the gaze deficit appears to lessen and even disappear entirely as the child progresses into adulthood.
Ocular misalignment, nystagmus, hypotonia, and developmental motor and speech delay are sometimes present in COMA (82; 40). A vertical gaze palsy may rarely be present, prompting consideration of Joubert syndrome (96), a genetic disorder that usually includes underdevelopment of the cerebellar vermis. These features portend a more intrusive condition that may not resolve.
Pontine lesions. The acquired pontine lesions causing horizontal gaze palsies are Wernicke encephalopathy, ischemic and hemorrhagic stroke, cavernous malformation, infections, primary demyelinating disorders, and neoplasms (88; 92; 64; 70; 76; 102; 79; 23; 51; 48; 25; 68; 56; 58; 107; 65; 87; 28; 83; 50; 30; 35).
By damaging the pons but sparing the midbrain, basilar artery thrombosis may preserve vertical gaze and produce the familiar locked-in syndrome of four-extremity and facial paralysis with relatively intact consciousness. Communication can take place by eliciting the spared vertical gaze movements (42).
A congenital cause of bidirectional horizontal gaze palsies is Mobius syndrome (86). Vertical gaze impairment and facial palsies may be present (100). Other defects include limb and craniofacial deformities, such as missing digits and amputations of the distal limb (66). The Mobius-Poland anomaly includes absence of one pectoral muscle, sometimes with an ipsilateral hand deformity (34; 72). Craniofacial abnormalities include micrognathia, tongue malformations, oligodontia, facial or oral clefts, and palatal or pharyngeal abnormalities (66; 101). Autism, motor weakness, and incoordination are common (101), as well as learning disabilities (54; 61; 95; 67). A genetic basis is based on co-occurrence in monozygotic twins (84; 13), siblings (36; 81; 38), or successive generations (98). One family had associated deletions of the NPHP1 gene for juvenile nephronophthisis on chromosome 2q13 (09).
Congenital gaze palsy is a component of Athabascan brainstem dysgenesis syndrome. Related to Mobius syndrome, it occurs in Navajo and Apache children and consists of bidirectional horizontal gaze palsy, sensorineural deafness, central hypoventilation, developmental delay, swallowing dysfunction, vocal cord paralysis, facial paresis, seizures, and cardiac outflow tract anomalies (47).
Horizontal gaze palsy with scoliosis (105; 01) is a congenital disorder in which patients often use convergence to shift gaze, mimicking convergence spasm (52). Patients also have horizontal pendular nystagmus, developmental delay, microcephaly, seizures, and scoliosis (93; 75; 27; 14; 49; 63). Brain MRI shows brainstem hypoplasia with absence of facial colliculi, a deep midline pontine cleft, and a butterfly configuration of the medulla. Diffusion tensor imaging shows absence of decussating superior cerebellar peduncle and transverse pontocerebellar fibers (85; 91; 05; 97), although the corpus callosum shows normal interhemispheric connectivity (39). There is a defect in a ROBO3 gene, which is related to a gene involved in the guidance of developing axons (53; 14; 17; 02; 03; 37).
Pontine tegmental cap dysplasia manifests bidirectional horizontal gaze palsy, ataxia, deafness, facial palsy, trigeminal numbness, and dysphagia. MRI discloses vermis hypoplasia, reduced volume of the ventral pons and middle cerebellar peduncles, an absent inferior olivary eminence, and a vaulted pontine tegmentum (07). Impaired axon guidance is thought to underlie this rare disorder, as MR tractography shows an abnormal bundle of horizontally oriented axons in the dorsal pons and no decussation in the cerebellar peduncles (15). Boney abnormalities are common with duplication, stenosis, or atresia of internal auditory canals and atresia or stenosis of the vestibulocochlear canals (69).
Midbrain lesions of many kinds may uncommonly cause horizontal gaze palsies by disrupting descending ocular motor inputs to the pons (108; 11; 18). However, signs of midbrain damage are usually also present, including vertical gaze palsies, disturbances of consciousness, ataxia, nystagmus, skew deviation, and third nerve palsy (108; 11; 31).
A 32-year-old man presented with 6 months of new intermittent headache, drop attacks, and occasional horizontal diplopia. His examination showed reduced rightward and leftward gaze in saccades, pursuit, and vestibulo-ocular reflex. He also had gaze-evoked nystagmus in all directions and bilateral facial weakness affecting both the upper and lower face. CT scan showed a large, calcified lesion in the fourth ventricle, which proved on resection to be an ependymoma. On examination 1 month after surgery, rightward gaze had recovered, but leftward gaze remained impaired.
Disorders that affect the ocular motor cranial nerves, neuromuscular junction, and extraocular muscles may fortuitously produce a pattern that looks like a true horizontal gaze palsy. The five major considerations are discussed in this section.
(1) Chronic progressive external ophthalmoplegia. This is a mitochondrial disorder affecting the extraocular muscles that may be evident in childhood or later. It often limits horizontal eye movements symmetrically in the two eyes, but vertical gaze is often impaired as well. Occasional accompanying features include ptosis, retinopathy, neck weakness, and cardiac conduction defects. Orbital MRI typically discloses small-caliber extraocular muscles.
(2) Fisher syndrome and Bickerstaff encephalitis. Fisher syndrome is a peripheral nervous system disorder attributed to antiganglioside antibody myelin destruction. Characterized by ophthalmoplegia, ataxia, and areflexia, the gaze deficits in the two eyes are often so symmetric that a gaze disorder of CNS origin is suspected. That inference becomes even more credible in a variant called Bickerstaff encephalitis, which manifests altered consciousness, nystagmus, ataxia, and other signs of brainstem dysfunction (57; 04). Anti-GQ1b antibodies are often present in serum (19; 103).
(3) Myasthenia gravis. This antibody-mediated post-synaptic disorder may rarely impair eye movements symmetrically in the two eyes and affect horizontal ocular excursions alone. Orbicular oculi, proximal extremity, and bulbar weakness are likely to be present. Acetylcholine receptor, muscle-specific kinase, or lipoprotein receptor-related protein 4 antibodies are often present. In antibody-negative cases, a decremental response may be demonstrated on repetitive stimulation electromyography, and jitter may be demonstrated on single-fiber electromyography.
(4) Botulism. Usually a food-borne toxic post-synaptic disorder, this condition disproportionately limits ocular excursions in the horizontal plane. Clues to the diagnosis are autonomic effects, including copious lacrimation and salivation, as well as dilated pupils and accommodative paralysis. Diagnosis is confirmed by detection of botulinum toxin in food or stool.
(5) Inflammatory orbitopathy. Any inflammatory disorder that affects extraocular muscles may involve both eyes and happen to cause symmetrical eye movement deficits limited to the horizontal plane. The ocular adnexal tissues (eyelids, conjunctiva, lacrimal glands) may show congestive features. Lid retraction or ptosis may be present. Examples are Graves disease and idiopathic orbital inflammation. The deficits in eye movements are due to cicatricial shortening of extraocular muscles that restricts excursions. Diagnosis is based on identifying the clinical features and noting inflammatory imaging signs. Passive ductions, in which the eye is anesthetized and moved by means of grasping the sclera with a forceps, will disclose the restriction.
The most efficient way to approach localization and diagnosis of horizontal gaze palsies is to divide them into unidirectional and bidirectional types.
Unidirectional horizontal gaze palsies. These palsies have three possible localizations:
(1) Contralateral nonepileptic cerebral lesion. Usually based in the parietal lobe, the lesion is frequently of ischemic or hemorrhagic origin. Gaze deviation toward the side of the lesion is common within the first week, together with contralateral hemispatial neglect and perhaps hemiparesis. Vestibulo-ocular reflex overcomes the gaze palsy.
(2) Contralateral frontal lobe epileptic focus. The gaze palsy occurs during the post-ictal state. Even if not observed, the ipsilateral gaze palsy must have been preceded by contralateral gaze deviation, together with head turning and facial and extremity tonic-clonic movements on the side of the eye deviation.
(3) Pontine tegmental lesion. The eyes are often deviated away from the side of the lesion. Vestibulo-ocular reflex does not overcome the gaze palsy; nystagmus, sixth nerve palsy, skew deviation, internuclear ophthalmoplegia, and ataxia are often present.
These unidirectional horizontal gaze palsies are rarely mimicked by non-CNS lesions. The examiner should expect that brain imaging and electroencephalopathy will confirm the diagnosis.
Bidirectional horizontal gaze palsies. These gaze palsies are more difficult to localize and diagnose. The examiner’s first task is to exclude the five imitators of gaze palsy (see Imitators of gaze palsy). Once that is done, it is useful to divide the bidirectional palsies into those of congenital and acquired origin.
Congenital horizontal gaze palsies. Congenital and early childhood-onset horizontal gaze palsies are typically accompanied by other neurologic and systemic abnormalities that allow more precise syndromic characterization. Careful examination discloses that the palsy is usually not limited to the horizontal plane. Considerations include COMA, Mobius syndrome, Athabascan brainstem dysgenesis syndrome, horizontal gaze palsy with scoliosis, and pontine tegmental cap dysplasia. In Mobius syndrome, brain MRI shows a flat fourth ventricular floor because of the absence of the facial colliculi (74; 45), as well as other abnormalities of the posterior fossa in some cases (99). In some cases of familial horizontal gaze palsy with progressive scoliosis, brain MRI shows a hypoplastic pons with absent facial colliculi and a deep ventral sulcus in the pons or medulla, causing a “butterfly” appearance to the medulla (75; 85; 14; 26; 12; 106). Electroretinography is usually normal in the idiopathic form of COMA, but can be abnormal in Kearns-Sayre syndrome, Joubert syndrome, and Refsum disease (89). In Leigh disease, a disorder of mitochondrial dysfunction, horizontal gaze palsies are often present, but excursional deficits are also present in the vertical plane.
Acquired horizontal gaze palsies. Among causes of acquired bidirectional horizontal gaze palsies, Wernicke encephalopathy is an urgent consideration (50). It must be suspected in the context of bariatric surgery, alcoholism, deviant diets, and psychiatric disorders of food intake. Imaging and serology are often negative. Apart from the confusional state, patients typically manifest imbalance and gait ataxia. Horizontal gaze palsies to both sides are often evident, but they may appear in muted form as direction-changing, gaze-evoked nystagmus. Upbeat and downbeat nystagmus and bilateral sixth nerve palsies are the other neuro-ophthalmic manifestations. Osmotic demyelination syndrome (formerly known as pontine myelinolysis) may produce horizontal gaze palsies, but other brainstem and long tract signs overshadow the gaze deficits. Ischemic, hemorrhagic, autoimmune inflammatory, infectious inflammatory, and neoplastic pontine lesions causing acquired bidirectional gaze palsies will often be evident on MRI (06). The exceptions are the small or subtle inflammatory and ischemic lesions, which are often occult to imaging. Additional studies will then be necessary for diagnosis.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Jonathan D Trobe MD
Dr. Trobe of the University of Michigan has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurobehavioral & Cognitive Disorders
Jun. 17, 2026
Neuro-Oncology
May. 27, 2026
Neuro-Oncology
May. 27, 2026
Neuropharmacology & Neurotherapeutics
May. 14, 2026
General Neurology
May. 13, 2026
General Child Neurology
May. 12, 2026
Neuro-Oncology
Apr. 30, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026