Epilepsy & Seizures
Febrile seizures
Jun. 02, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Hyperekplexia is a rare, predominantly hereditary neurologic disorder characterized by pathologic and excessive startle responses. Onset varies from the perinatal period through adulthood. Hyperekplexia may be inherited, sporadic, or acquired and is divided clinically into minor and major forms. Minor forms may present with exaggerated startle reflexes alone, whereas major forms are defined by the clinical triad of generalized stiffness at birth, excessive startle reflexes, and prolonged generalized stiffness related to the startle reflex. In untreated babies, truncal stiffening may cause respiratory impairment and apnea with fatal consequences. Hyperekplexia is the first human disease shown to result from mutations within a neurotransmitter gene. It is primarily caused by inherited mutations in the genes encoding the postsynaptic glycine receptor alpha1 subunit (GLRA1) on chromosome 5q33-35, postsynaptic glycine receptor beta subunit (GLRB) on chromosome 4, and the presynaptic glycine transporter GlyT2 (SLC6A5) on chromosome 11. Uncommon causes of more severe cases are mutations in GPHN on chromosome 14 and ARHGEF9 on X chromosome. Sporadic, nonfamilial cases of hyperekplexia are common and may manifest as ataxia. Prolonged stiffness in infants is terminated with the simple maneuver of forced flexion. Pharmacological treatment of hyperekplexia with clonazepam is often life-altering. In this article, the author details developments in etiology, genetics, clinical manifestations, and treatment.
|
• Hyperekplexia is a rare disorder characterized by exaggerated startle reflexes and hypertonia in response to sudden unexpected stimuli with major and minor forms. | |
|
• It is primarily caused by genetic defects resulting in aberrant glycinergic neurotransmission. | |
|
• In infants, prolonged hypertonia causes respiratory impairment and potentially fatal apnea, though symptoms may be apparent as early as the third trimester of pregnancy. | |
|
• A simple maneuver of forced neck flexion may be lifesaving when prolonged stiffness impedes respiration. | |
|
• Clonazepam is the most effective treatment. |
Excessive startle responses of epileptic or nonepileptic origin have been known for many years (03; 14; 13). See also MedLink Neurology articles: Startle epilepsy and Sleep starts.
The first description of hyperekplexia was published by Kirstein and Silfverskjold in four members of three generations of a Swedish family with violent falls triggered by fright, stress, unexpected stimuli, or surprise (16).
Soon thereafter, Suhren, Kok, and Bruyn described 29 affected individuals in six generations and later coined the term “hyperexplexia” in the monumental work “Hyperexplexia, A Hereditary Startle Syndrome” (18; 38). The term hyperexplexia was later corrected to hyperekplexia (from the Greek word ekplexis, meaning surprise; hyper to emphasize excessive) by Gastaut and Villeneuve in a report describing sporadic cases (12).
It wasn’t until the 1990s that Ryan and colleagues linked the disease to chromosome 5q, later identified by Shiang and colleagues as a mutation in the alpha 1 subunit of the inhibitory glycine receptor (GLRA1) (32; 33; 36). A review and a 60-year follow-up of Kok’s described family reported by Paucar and associates attributed their disease to the R271Q mutation in the GLRA1 gene (29).
“An autosomal dominant congenital disorder resembling stiff-man syndrome” was described in 1972 and was later named hereditary stiff-baby syndrome by Lingam in 1981, though these disorders actually represent the neonatal form of hyperekplexia (20; 42). The term stiff baby syndrome has also been used for ATAD1-related lethal encephalopathy manifesting with hypertonia, absence of spontaneous movements, almost no motor development, and death within the first months of life (45). This phenotype was assigned the name hyperekplexia-4 (HKPX4, #618011) in OMIM despite its genetic, pathophysiologic, and prognostic differences from the other forms discussed in this article (34). It is important to distinguish these and other genetic encephalopathies and epileptic disorders as treatment and prognosis differ.
For the discovery of other genes involved in hyperekplexia (SLC6A5, GLRB, GPHN, ARHGEF9, and others) see the Etiology section.
Hereditary hyperekplexia may manifest as early as fetal life with excessive startles severe enough to wake the mother from sleep during the third trimester aborted by applying pressure through the abdominal wall (19). Shortly after birth, infants present with generalized stiffness, violent startle reflex to ordinary stimuli followed by sustained tonic stiffening of the trunk and limbs, clenching fists, and attacks of a high-frequency trembling. In newborns, the muscle stiffening may cause respiratory impairment or fatal apnea and feeding difficulties. The sudden and sustained movement may be confused for epileptic seizure, though EEG is normal sleep improves or abolishes it. A distinctive clinical feature is a lack of habituation to startling stimuli, which can be tested at the bedside (34). This more severe phenotype is characteristic of the major form of disease.
The startle response is resistant to habituation, but the resultant prolonged hypertonia can be terminated by forced neck flexion known as the Vigevano maneuver.
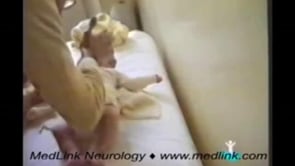
Fifteen-day-old neonate with typical startle response to tactile stimuli in the face and mainly the nose. Please note that marked generalized startle response (typical of hyperekplexia) is triggered each time the face and, main...
Muscle stiffness subsides by the first few years of life, but excessive jerking to external stimulation or excitement persists. Neurodevelopment is typically normal or mildly delayed. Motor delay is most common likely due to hypertonia and improves with improvement in tone. Intellectual disability and autism spectrum disorder are rare. Affected children walk toddling and often seek assistance. Gait disturbance increases when in a hurry, amongst a crowd, or if forced. Stumbling or an unexpected jolt may induce uncontrolled falls (''like a log'') with the risk of serious injuries.
In contrast to the major forms described above, minor forms manifest with excessive but often mild startle responses without hypertonia. Consciousness is maintained, but stiffness persists temporarily. Attacks can become more common during febrile illnesses in children and during emotional stress in adults (39).
Other neurologic manifestations may include sleep disorders and seizures. Sleep myoclonus resembling generalized clonus or repetitive myoclonus is often prominent. The jerks are spontaneous arousals. Sleep terrors, rapid eye movement (REM), and non-REM sleep behavior disorders have been reported (21). Epilepsy is rare but has been reported, especially in recessive mutations (39).
Non-neurologic manifestations of the disease include umbilical and inguinal hernias, presumably due to increased intra-abdominal pressure and congenital hip dislocation of the hip (09).
The prognosis is generally good in treated patients (34). Infants experience recurring apnea until 1 year of age. Therefore, recognition of the neonatal form is important so that treatment can be initiated (43). The exaggerated startle response persists into adulthood (23). Hypertonia and stiffness diminish during the course of the first and second year of life, and tone is usually almost normal by the age of 3 years, though hypertonia may recur in adult life. As with other causes of voluntary motor control loss, certain activities such as swimming and driving could threaten life, so patients should be counseled accordingly.
Cases 1 and 2 are illustrated in video clips of a neonate and an infant with hyperekplexia.
Fifteen-day-old neonate with typical startle response to tactile stimuli in the face and mainly the nose. Please note that marked generalized startle response (typical of hyperekplexia) is triggered each time the face and, main...
Additionally, the second video illustrates the Vigevano maneuver.
Case 3 is illustrated in a video clip of an adult with hyperekplexia presented by Mariani and colleagues https://www.neurology.org/doi/10.1212/WNL.0000000000003766 (23). This was a 47-year-old man with pathologic startles. These are bilateral and synchronous myoclonus triggered only by unexpected specific stimuli and resisting to habituation. No reflex myoclonus was triggered by distal tactile stimuli of the limbs; this is one of the signs that distinguish hyperekplexia from cortical or reticular reflex myoclonus. The other distinguishing signs are the lack of posture/action myoclonus reflexes. Clonazepam markedly reduced abnormal startle and limited falls. Two of his children had similar symptoms. Neurophysiologic polymyograph confirmed pathologic startle. Molecular analysis of GLRA1, encoding the α1 glycine receptor subunit, revealed a c896 G > A (pR299Q) mutation.
Case 4 is from the original study of Mrs. Suhren and associates (38):
|
A boy, aged two years, was admitted to the Pediatric Department on account of bronchitis. The mother volunteered the information that he suffered from a particular disorder that ran in her family. Immediately after the patient's birth his mother knew him to be afflicted because his muscles were hypertonic. For example his fists could not be opened passively without a great deal of force having to be exerted. There also was such a strong spasm of the adductor muscles, that the mother had to put her head between the baby's knees in order to change diapers. There was a conspicuous lack of spontaneous movements. After three months the boy became more active, but immediately reacted with an increase of generalized spasm in flexion to stimuli such as being lifted out of the cradle, or when the cradle was bumped inadvertently. During sleep, the muscles were relaxed in a normal fashion. There were no feeding problems. When six months old, he appeared normal, but his movements were conspicuously stow and he never succeeded in putting his big toe into his mouth. Development was not retarded. In the course of the first year of life he was seen regularly in a Welfare Clinic, where he was noted to show generalized stiffness with adductor hypertonia. After having learned to walk, he stumbled readily, occasionally becoming stiff all over and falling on to his face without extending his arms. As a rule, however, he would fall like any other child. From interviews with other members of the family, evidence was obtained that the above-mentioned peculiar mode of falling was always related to an abnormal startle-reaction. General physical examination revealed no abnormality. On neurological examination an exceedingly well-marked head retraction reflex, a palmo-mental reflex on the left side, and a marked snout reflex were found. The cranial nerves were intact, the ocular fundi were normal and the tendon reflexes were symmetrical. On passive flexion of the left arm at the elbow-joint, an immediate and powerful contraction of the triceps would oppose the movement. He was skillful when playing; when walking however, the slightly abducted and inwardly rotated arms assumed an attitude of slight flexion in the elbow-joints, and the child set his feet widely apart with the knees somewhat flexed. When asked to assume a supine from a sitting position, he would carefully place the extended arms on the floor and let himself down; the legs were always in hyperextension and externally rotated. At the end of a movement of kicking a ball, he would stop abruptly and stand with abducted, internally rotated arms, showing an alternating movement of the forearms. |
Hyperekplexia is a pathologically exaggerated startle reflex. Startle reflex is a normal reticular and cortical reflex elicited in normal newborns and infants. It is a basic alerting reaction consisting of an early automatic motor activation causing blinking, facial grimacing, head flexion, hunching of shoulders, adduction of the arms, and flexion of the trunk and the knees, followed by a late behavioral, autonomic response separated by a brief (200 to 300 ms) period of decreased activity. It appears in infancy at the same time as the Moro reflex, and it becomes more noticeable as the Moro reflex disappears (34).
The inhibitory neurotransmitter glycine is stored in presynaptic nerve vesicles in the brainstem and spinal cord. Normally, glycine is released and binds the α1 subunit of glycine receptors, opening the chloride channel and hyperpolarizing the postsynaptic cell membrane. Glycine is removed from the synaptic cleft by glycine transporters 1 and 2, GlyT1 in astrocytes, and GlyT2 in presynaptic cells, replenishing the presynaptic pool. The scaffolding protein gephyrin binds the β subunit of the glycine receptor at the postsynaptic terminal, and collybistin ensures proper translocation of gephyrin in the postsynaptic cell. Defective glycine receptors, transporters, or their associated scaffolding proteins result in ineffective inhibition and, thus, exaggerated or prolonged excitability (34).
Genetic screening studies have demonstrated that hyperekplexia is genetically heterogeneous. Most cases are of an autosomal recessive or an autosomal dominant trait, with 100% penetrance (39). Rarely, hereditary hyperekplexia is inherited in an X-linked pattern when caused by alterations in the ARHGEF9 gene, and this is usually associated with intellectual disability and epilepsy (17). Two cases of compound heterozygosity have been described with more severe phenotypes than their heterozygous parents (37).
Hereditary hyperekplexia is the first human disease shown to result from mutations within a neurotransmitter gene. It is caused by mutations that disrupt the functioning of inhibitory glycinergic synapses in neuromotor pathways of the spinal cord and brainstem. These mutations cause a variety of dysfunctions of the neuronal chloride channel. Hereditary hyperekplexia is, therefore, regarded as a channelopathy.
Mutations in five genes are known to cause hyperekplexia. GLRA1, encoding glycine receptor subunit alpha1; SLC6A5, encoding the presynaptic sodium- and chloride-dependent glycine transporter 2 (GlyT2); GLRB, encoding glycine receptor subunit beta; GPHN, encoding the glycinergic clustering molecule, gephyrin; and ARHGEF9, encoding collybistin (39).
Neonatal and occasionally prenatal onset of hyperekplexia occurred in all 97 individuals with genetically confirmed hyperekplexia (61 cases had mutations in GLRA1, 24 cases in SLC6A5, and 12 in GLRB) (39). Conversely, the cardinal feature in 35 gene-negative cases was presentation after the first month of life (P < 0.001) (08). Patients with GLRB and SLC6A5 mutations were more likely to have developmental delay (RR1.5 P < 0.01; RR1.9 P < 0.03) than those with GLRA1 mutations; 92% of GLRB cases reported a mild to severe delay in speech acquisition. Ninety percent of neonates with hyperekplexia due to SLC6A5 mutations have multiple serious apneas, and cognitive development is impaired in two thirds.
Seven individuals with hyperekplexia displayed lower pain thresholds than 14 healthy age- and sex-matched controls for all of the quantitative sensory tests, and conditioned pain modulation was significantly reduced in hyperekplexia. The authors of this study concluded that their findings demonstrate increased pain sensitivity and impaired central pain modulation in hyperekplexia patients, supporting the importance of glycinergic neurotransmission for central pain modulation in humans (44).
Sporadic, nonfamilial cases of hyperekplexia are common and may manifest as ataxia (30). Some cases may be congenital, idiopathic, or associated with brain pathology, usually in the brainstem or rarely in the supratentorial compartment (11). Sporadic hyperekplexia plus syndrome are cases with additional features of other system involvement (25; 05).
Hyperekplexia may be a symptom of neurometabolic disorders such as asparagine synthetase deficiency characterized by congenital microcephaly and severe global developmental delay, intractable seizures, or hyperekplexia (35), or molybdenum cofactor deficiency (22).
Transient acquired hyperekplexia has been reported in an adult as the presenting symptom in a top of the basilar stroke, with improvement after thrombectomy (04).
This is a rare disorder with onset often manifested from the intrauterine life or from birth and any time from neonatal period to adulthood. Both sexes are equally affected. Hereditary hyperekplexia has been identified in over 70 pedigrees from Europe, Japan, Canada, the United States, and mostly in northern European descendants (41). Hyperekplexia affects approximately one in 40,000 people in the United States (26).
According to a report of 90 cases of hyperekplexia with an identified genetic cause, homozygous deletions of exons 1 to 7 are predominantly seen in people with Turkish backgrounds (n=16/17, p< 0.001) (40). In contrast, the dominant point mutation R271 is seen in people of Asian, Caucasian, and African-American heritage (n=19), but not in people with Arab or Turkish ethnicities (p< 0.001). Cultural practices appear to influence the inheritance patterns, and a Caucasian founder is postulated for R271 mutation (40).
Because the disorder may be anticipated in the offspring of an affected parent, early treatment with clonazepam may be expected to prevent the attacks of stiffening as well as the rigidity of affected infants. This should avoid the fatalities that are not uncommonly described in this age group, and that are related to respiratory arrest as a consequence of episodic extreme stiffening.
Affected families should seek genetic counseling.
Hyperekplexia in the neonatal period may be misdiagnosed as congenital stiff-person syndrome, startle epilepsy, myoclonic seizures, neonatal tetany, cerebral palsy, and drug (phenothiazine) toxicity. Accurate recognition of hyperekplexia in a newborn is important for appropriate and potentially lifesaving treatment.
In normal people, excessive startle may be related to tension or lack of sleep. It may be present in isolation in individuals described as "hyperstartlers." Whether these people actually suffer from the minor form of hyperekplexia is unclear (01).
Startle epilepsy usually occurs in patients with diffuse encephalopathy or widespread hemispheric damage (28). It is mainly related to frontal epileptogenic abnormalities. There is no history of similar symptoms in close family members, and other features of epilepsy are usually present. See MedLink Neurology article: Startle epilepsy.
Conditions such as jumping, latah, imu, or myriachit present with excessive startle associated with echolalia, echopraxia, and forced obedience. These disorders are also genetically determined and probably represent an unusual tic syndrome. For many years, these disorders have been misinterpreted as behavioral. In different cultures, various psychosocial manifestations are superimposed on a basically similar and probably identical pathophysiological process (01; 02). Excessive startle has also been described in about 20% of patients with tics.
Paroxysmal extreme pain disorder (previously familial rectal pain syndrome) is another condition that sometimes may be misdiagnosed as hyperekplexia (10).
Symptomatic hyperekplexia can be seen in underlying brain or brainstem damage. Neurodegenerative conditions manifesting with hyperekplexia include gangliosidoses, leukodystrophies, and other neurometabolic disorders (34).
Hyperekplexia may also be a symptom of Coffin-Lowry syndrome, an X-linked semi-dominant condition with learning difficulties, epileptic seizures, and dysmorphism caused by mutations in the gene, RSK2 (27). Hyperekplexia may also occur in “progressive encephalomyelitis with rigidity and myoclonus” associated with glycine receptor alpha 1 antibodies, and it may be manifested with rigidity, painful muscle spasms, and brain stem signs (07).
The medical history provides the most important clues to the diagnosis. In early childhood, spastic quadriparesis has often been suspected. Neonates and infants with rigidity, episodic tonic spasm, apnea, aspiration, and near-miss sudden infant death should be evaluated for hyperekplexia. A misdiagnosis of epilepsy is common.
The diagnosis is based primarily on three clinical features (41):
|
(1) Generalized stiffness immediately after birth | |
|
(2) Excessive startle reflex to unexpected (particularly auditory) stimuli without impairment of consciousness | |
|
(3) Short period of generalized stiffness following the startle response, during which voluntary movements are impossible |
The nose tap test is the most useful test. Tapping the tip of the nose of an unaffected baby will elicit no response, but in hyperekplexia there is an obvious startle response, which is repeated each time the nose is tapped (43).
Fifteen-day-old neonate with typical startle response to tactile stimuli in the face and mainly the nose. Please note that marked generalized startle response (typical of hyperekplexia) is triggered each time the face and, main...
Molecular genetic testing of GLRA1, GLRB, and other implicated genes confirms the clinical diagnosis. Early genetic testing for symptomatic neonates and possibly preconception counseling for those at risk for GLRB and SLC6A5 mutations are recommended because of the more challenging associated clinical phenotype (39).
Brain MRI is generally normal.
EEG is generally normal, including during episodes of startle. Background slowing and even generalized attenuation may occur as a result of apnea, corresponding to the bradycardia and cyanosis. Nerve conduction studies are normal, but somatosensory evoked potentials are giant.
Polymyography can confirm a pathologic startle.
There is a dramatic response to clonazepam (0.01 to 0.1 mg/kg/day for infants and children; 0.8 mg/kg/day for adults), which relieves most of the clinical manifestations (34). Clonazepam, a GABA-A receptor agonist, may compensate for the defective glycine-gated chloride channel by enhancing the GABA-gated chloride channel function.
Small doses of clobazam are also effective and well tolerated and may be added for clonazepam-resistant cases (24). Valproic acid has been found to be effective as well, but the risk of hepatotoxicity in neonates and infants exists. Trihexyphenidyl has been described as effective in a clonazepam-resistant case (31). Other attempted therapies include carbamazepine, phenytoin, diazepam, sodium oxybate, and phenobarbital (34).
A case of neonatal hyperekplexia resistant to clonazepam subsequently responded to levetiracetam (15). A 5.5-year-old initially treated with clonazepam experienced a recurrence of symptoms after self-discontinuation but had subsequent resolution of symptoms with escitalopram alone (06). Patients report improvement in symptoms with alcohol, likely owing to its effect on glycine receptors.
The startle response stops when the baby is flexed, that is, made to curl up by pressing the head towards the knees. This simple Vigevano maneuver of forced flexion may be lifesaving when prolonged stiffness impedes respiration (43). If a frequent exaggerated startle is not controlled, nutrition may need adjustment for increased caloric needs.
Dehydroxylcannabidiol (DH-CBD), a synthetic nonpsychoactive cannabinoid, is being studied as a potential candidate drug to treat hyperekplexia (46).
With clonazepam, the gait becomes normal, the falling attacks are abolished, the increased tone reverts to normal, and the spontaneous prolonged tonic attacks are almost, but often not completely, abolished. Developmental delay may be present, but overall outcomes are good with normal cognition. Clonazepam is not as effective for infantile hypertonicity as it is for exaggerated startle, but the stiffness often resolves by 2 to 5 years of age (34).
The stiffness is apparent in the neonatal period in some affected children. Episodes of stiffening felt by the mother before birth may have occurred in some instances. Close supervision of affected children with apnea and treatment after oxygen monitors during the perinatal period are important as failure to act on prolonged stiffening may result in death.
Anesthesia should be used with extreme caution as the effects of neuromuscular blockade in hyperekplexia is not entirely known. Stimuli that could trigger the reflex should be avoided. Clonazepam and diazepam are used to prevent and control the spasms. Propofol and other agents with the ability to potentiate both GABAergic and glycinergic transmission may be appropriate choices for anesthesia (34). Reaction to neuromuscular blockers may be unpredictable.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Daniel N Lax MD
Dr. Lax of Albert Einstein College of Medicine received research support from Biohaven and a consultant honorarium from Theranica.
See Profile
Solomon L Moshé MD
Dr. Moshé of Albert Einstein College of Medicine has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Epilepsy & Seizures
Jun. 02, 2026
Neuropharmacology & Neurotherapeutics
May. 11, 2026
Epilepsy & Seizures
May. 08, 2026
Epilepsy & Seizures
May. 01, 2026
Epilepsy & Seizures
Apr. 30, 2026
Epilepsy & Seizures
Apr. 17, 2026
Epilepsy & Seizures
Apr. 13, 2026
Epilepsy & Seizures
Apr. 08, 2026