Epilepsy & Seizures
Driving and epilepsy
Jul. 08, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Lafora disease is a severe form of progressive myoclonus epilepsy characterized by the onset of seizures or cognitive decline during late childhood or adolescence with a progressive course, usually ending in death 5 to 10 years after the initial symptoms. Lafora disease is caused by mutations in the EPM2A or EPM2B/NHLRC1 genes, encoding the laforin phosphatase and the malin ubiquitin ligase, respectively, two cytoplasmically active enzymes that regulate glycogen construction. Pathologically, the disease is characterized by the presence of polyglucosan intracellular inclusion bodies called Lafora bodies in neurons and other tissues. Despite improved patient diagnosis, prenatal diagnosis, and genetic counseling for this devastating disease, treatment is primarily palliative, aiming at seizure reduction. In this article, the author summarizes the clinical manifestations and rapidly expanding information on the genetic basis and metabolic derangements of Lafora disease and some promising results from the treatment of animal models.
|
• Lafora disease is an autosomal recessive progressive myoclonus epilepsy characterized by the accumulation of insoluble abnormal glycogen deposits in the brain and peripheral tissues. Clinically it manifests with cortical myoclonus, other types of epileptic seizures, progressive intellectual decline, ataxia, and early death. | |
|
• Onset of symptoms is in the second decade of life, typically between the ages of 11 and 18 years. Death usually occurs 2 to 10 years after onset (mean 5 years). | |
|
• The pathological hallmark of Lafora disease is the presence of cytoplasmic polyglucosan inclusions, the Lafora bodies. | |
|
• The disease is caused by mutations in either the EPM2A gene, encoding the protein phosphatase laforin, or the EPM2B gene, encoding the ubiquitin ligase malin. | |
|
• The MRI shows subtle or no abnormalities, and the EEG is grossly abnormal even in early stages. Biopsy or genetic studies are required for a definitive diagnosis. | |
|
• Management is only supportive and palliative, limited to symptomatic management of the epileptic seizures, myoclonus, and intercurrent complications. There are promising therapies in development in preclinical stages that soon may proceed to clinical trials. |
The neuropathological features typically found in Lafora disease were first described by Gonzalo Rodríguez Lafora in 1911 (40; 40).
It was soon established that the presence of typical Lafora inclusion bodies in the pathological material was associated with a progressive neurologic disorder with myoclonus, different types of seizures, and dementia. This triad of symptoms characterizes a group of diseases categorized as progressive myoclonic epilepsies (07; 30; 63; 52). Lafora disease is recognized as one of the major causes of progressive myoclonus epilepsy with well-defined clinical and pathological characteristics. In 2021, the Commission on the Classification and Terminology of the International League Against Epilepsy (ILAE) classified Lafora disease in the group of epilepsy syndromes with developmental and/or epileptic encephalopathy, or with progressive neurologic deterioration (57).
Lafora disease is characterized by symptoms common to the other progressive myoclonus epilepsies: severe myoclonus, major generalized convulsive seizures, and dementia (07; 30; 63; 31; 48; 52; 68; 37). Focal occipital seizures occur in about half of the patients.
Onset of symptoms is in the second decade of life, typically between the ages of 11 and 18 years.
A few cases of the adult-onset form of the disease have been described in patients with typical Lafora inclusion bodies and a more benign course (38; 66). Slowly progressive forms are most commonly due to EPM2B mutations (33). Also, an early-onset form of Lafora disease with dyslexia and learning disorder followed by epilepsy and neurologic deterioration has been described (26). This form is associated mainly with mutations in exon 1.
The natural course of Lafora disease can be divided into three phases (16). Initially, most cases start with convulsive tonic-clonic seizures and myoclonic jerks, and some cases can mimic juvenile myoclonic epilepsy; the jerks are massive and synchronous bilateral myoclonic jerks of brief duration, usually limited to one or more muscle groups. The myoclonic jerks are not associated with loss of consciousness and are usually triggered by visual stimulation or proprioceptive impulses (07; 30; 63; 31; 48; 52). Epileptic visual seizures with elementary visual hallucinations may be an early sign of the disease. Behavioral changes or decline of school performance (35), hepatic disease (34), and, occasionally, parkinsonism (56) may also be the starting symptoms.
With progression, myoclonus increases in intensity and becomes severe and multifocal, precipitated by posture or action. At this point, intellectual deterioration occurs rapidly. An advanced stage characterized by severe dementia, spastic quadriparesis, and almost constant myoclonic seizures and frequent generalized tonic-clonic seizures is reached within 2 to 10 years after the onset of symptoms in classic Lafora disease.
Lafora disease has a poor prognosis. In the final stages, severe dementia is always a major feature. Death usually occurs 2 to 10 years after onset (mean 5 years). The mean age at death is 20 years (30).
A milder phenotype has been described in those with NHLRC1 mutations, where a later age of onset, slower disease progression, and longer life expectancy was observed (24; 12). However, a “mild” or slowly progressive course may also be seen in patients with EPM2A mutation (personal observation).
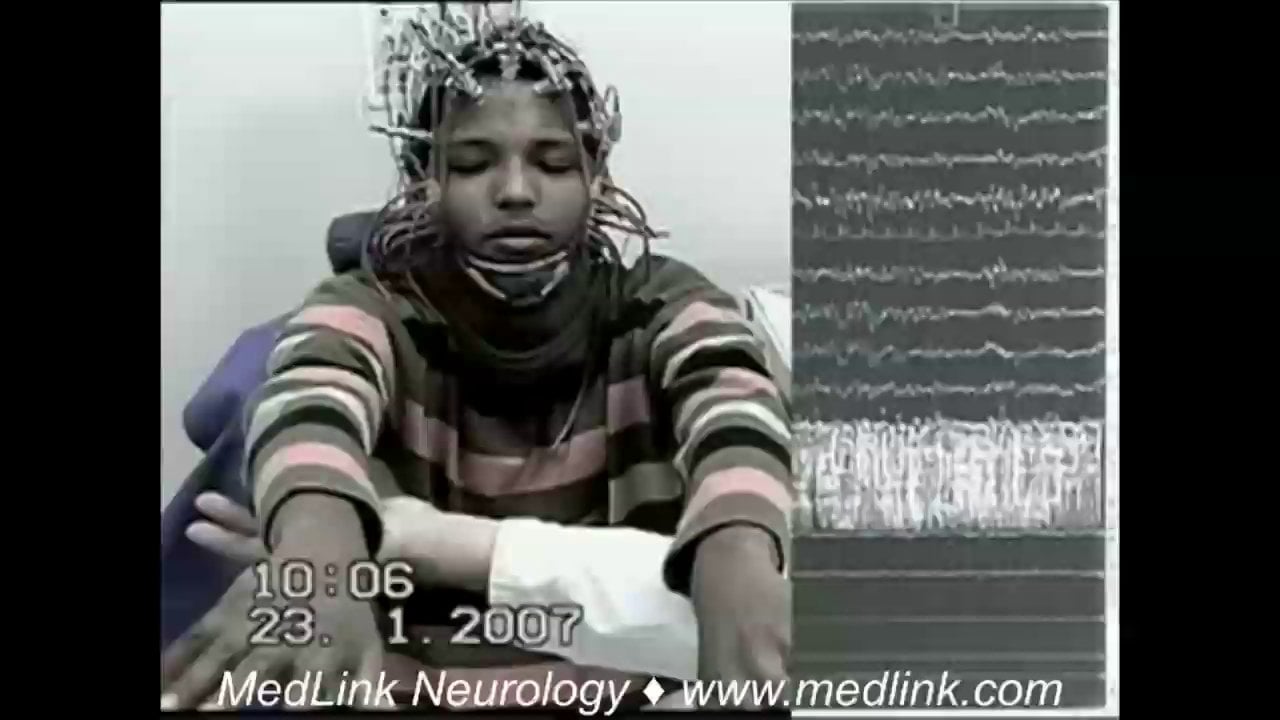
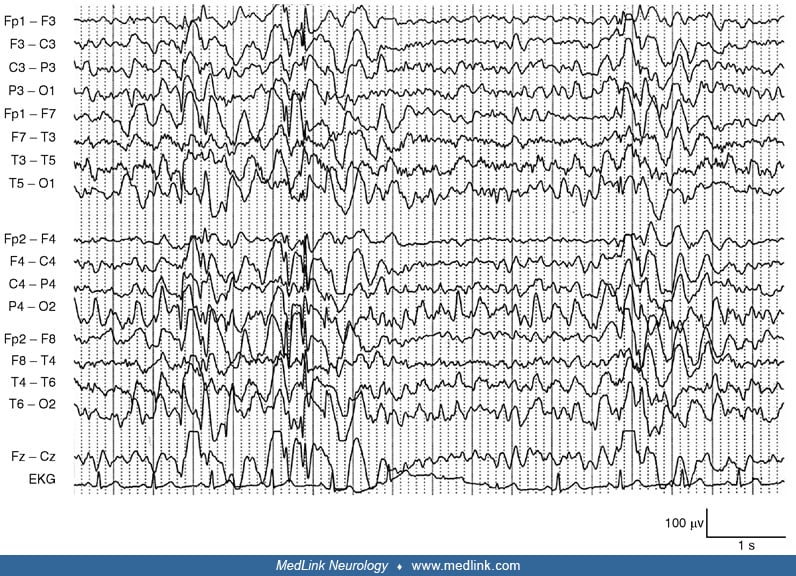
A 19-year-old girl started at age 14 years with myoclonic jerks followed early in the evolution by generalized tonic-clonic seizures and elementary visual hallucinations. She was misdiagnosed with juvenile myoclonic epilepsy and started on antiepileptic drugs without achieving seizure control. One year later, she developed gait unsteadiness and cognitive and speech impairment causing school failure. Her neurologist suspected a progressive myoclonus epilepsy, and the genetic test revealed a homozygous mutation in EPM2B. At age 19 years, she had almost continuous myoclonus, severe cognitive deterioration, and frequent generalized tonic-clonic and myoclonic seizures despite having tried different antiepileptic drugs. The EEG showed a slow and disorganized background activity with very frequent polyspike and spike-waves discharges that decreased during sleep.
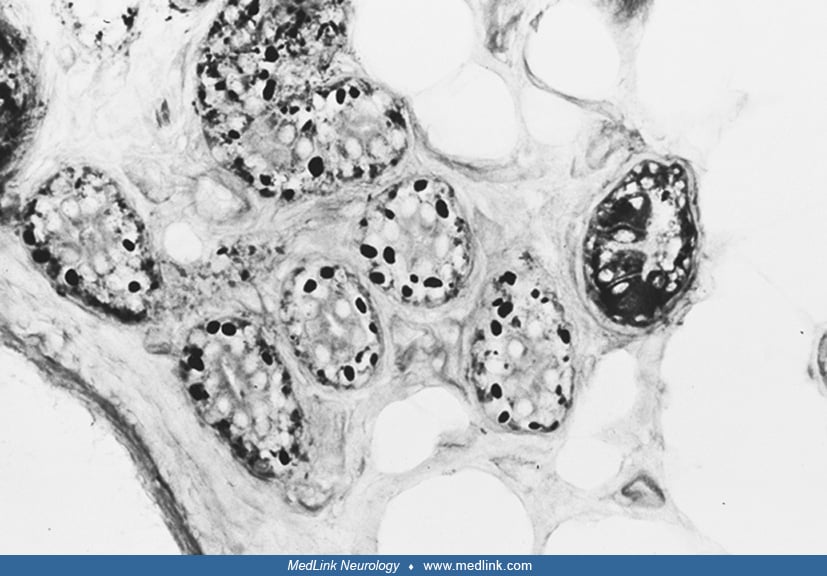
The pathologic picture of Lafora disease is unique (40). PAS-positive inclusions (Lafora bodies) are present throughout the nervous system, particularly in the dentate nucleus, red nucleus, substantia nigra, and hippocampus. Lafora bodies are also seen in various tissues, including liver, skeletal and cardiac muscle, and skin (14). The inclusions consist largely of polyglucosans that are poorly branched and insoluble forms of glycogen, highly phosphorylated and with small variable sulfate groups (59; 14; 69). Lafora bodies occupy vast numbers of neuronal dendrites and perikarya in Lafora disease in time-dependent fashion, leading to intractable and fatal progressive myoclonus epilepsy. Studies of different tissues have revealed that the storage material has some histochemical and morphological differences, varying from organ to organ. Lafora bodies, as seen by electron microscopy, appear to be free in the cytoplasm of neurons and in eccrine sweat-gland duct cells. In hepatic cells, the storage material displaces various organelles to the periphery, and the center of the cytoplasm is occupied by hypertrophic smooth endoplasmic reticulum containing the PAS-positive material. In skeletal muscle, the inclusions are definitively surrounded by membranes (68).
Lafora disease is an autosomal recessive disorder caused by loss-of-function mutations in the EPM2A/NHLRC1 gene, which encodes a protein tyrosine phosphatase (laforin), or in the EPM2B gene, which encodes an E3 ubiquitin ligase (malin) that ubiquitinates and promotes the degradation of laforin (46). It is characterized by the presence of abnormally branched water-insoluble glycogen inclusions known as Lafora bodies in the neurons and other tissues (62; 15; 36; 27; 29; 43; 18; 65; 64; 31; 48; 39; 68; 51; 53; 37; 10). Laforin's principal function is to control glycogen chain lengths in a malin-dependent fashion, and loss of this control underlies Lafora disease. Mouse models of Lafora disease, developed by targeted disruption of the EPM2A or EPM2B genes, recapitulated most of the symptoms and pathological features as seen in humans and offered insight into the pathomechanisms (28). Evidence from these models indicates that Lafora bodies are a principal driver of neurodegeneration and neurologic disease.
Over 100 distinct mutations in EPM2A or EPM2B/NHLRC1 have been discovered in over 200 independent families affected by Lafora disease; nearly half of them are missense mutations, one quarter are deletion mutations (64), and novel mutations are common (42; 19; 61; 25; 02; 58). In a homogeneous environment and gene pool, the same Lafora epilepsy mutation resulted in highly uniform ages of onset (14 years) and death (21 years) (67).
Experimental work has shown that glycogen synthesis is requisite for Lafora body formation and that Lafora bodies are pathogenic. Elimination of brain glycogen synthesis in Lafora disease mice prevents Lafora body formation, neurodegeneration, and seizure susceptibility (54; 20).
The myoclonus of Lafora disease is epileptic cortical myoclonus. However, with progression of the disease, the degenerative process affects both cortical and subcortical structures; therefore, epileptic and nonepileptic myoclonus may coexist.
Cognitive impairment in Lafora disease is predominantly due to metabolic damage in the frontal cortex (55). Areas of decreased glucose metabolism in Lafora disease are extensive, often involving multiple cortical and subcortical regions, with the thalamus and the temporal, frontal, and parietal lobes being the most severely affected (49).
Lafora disease is an ultra-rare disease and is seen worldwide but is more common in geographic isolates and areas with a high degree of inbreeding. In Western countries, prevalence is estimated to be well below 1 in 1,000,000. The disease probably accounts for 10% of patients with progressive myoclonus epilepsy. In one study, 12 of 84 (14.2%) patients with progressive myoclonic epilepsy had Lafora disease (04).
Lafora disease is a frequent cause of progressive myoclonus epilepsy in populations of Mediterranean origin, whereas it is rare or absent in Scandinavian countries, particularly Finland, where Unverricht-Lundborg disease is the predominant form. A cluster of families has been described in the south of India (01). There are several reports of cases in Japan (71) and China, and a few cases have been reported in Great Britain (22).
Due to its genetic nature, genetic counseling should be offered to families with confirmed cases of the disorder (37). No environmental factors are believed to play a role in causing the disease.
Prenatal testing. If EPM2A or EPM2B pathogenic variants have been identified in an affected family member, prenatal testing for pregnancies at increased risk may be offered.
Preimplantation genetic diagnosis. This may be an option for some parents in whom EPM2A or EPM2B pathogenic variants have been identified.
Recognition of Lafora disease in its fully developed form is not difficult, though in its initial stages the disease can resemble juvenile myoclonic epilepsy or occipital epilepsy. The presence of: (1) tonic-clonic and focal visual seizures to bilateral tonic-clonic seizures, followed by myoclonic jerks (not exclusively on awakening), associated with myoclonus that is more often postural and triggered by movements; (2) EEG background slowing with superimposed diffuse epileptiform abnormalities with occipital epileptiform discharges; and (3) drug-resistant epilepsy and cognitive impairment during the evolution of the disease gives rise to the suspicion of a progressive disease, such as Lafora disease (17).
Another important distinction has to be made from the other major causes of progressive myoclonic epilepsy: a hallmark of this group of epilepsies is progressive myoclonus that is triggered by action, is multifocal, and becomes refractory (12). The group of progressive myoclonic epilepsies include Unverricht-Lundborg disease, neuronal ceroid lipofuscinosis, sialidosis, and mitochondrial encephalomyopathy (06; 30; 52). Rare causes of progressive myoclonus epilepsy include dentatorubral-pallidoluysian atrophy, noninfantile neuronopathic Gaucher disease, late-infantile and juvenile forms of GM2 gangliosidosis, the juvenile form of neuroaxonal dystrophy, Hallervorden-Spatz syndrome, and action myoclonus-renal failure syndrome. Most of the above disorders can be differentiated from Lafora disease by their typical ages at onset of symptoms and by the absence of Lafora bodies. The presence of the typical pathological findings is important in patients in whom the disease presents as a predominantly dementing illness with relatively infrequent seizures or when it mimics nonspecific secondary generalized epilepsy with mild myoclonus. Nonepileptic myoclonus disorders can also be differentiated based on clinical and pathological characteristics.
The EEG shows progressive slowing of the background rhythm and generalized epileptiform discharges. Focal or multifocal, often posterior, epileptiform abnormalities are also seen. Photosensitivity is common. Mid and even early stages of the disease can reveal a grossly abnormal recording with severe slowing and very frequent epileptiform discharges that may almost disappear during sleep. EEG patterns through the disease course can also change. At onset of epilepsy, the EEG may only show findings of a generalized genetic epilepsy, which then progresses to slowing of the background, multifocal epileptiform discharges, and occipital abnormalities. In the final stage of the disease, which is characterized by severe dementia and refractory epilepsy, the EEG background may slow further with frequent fast multifocal spikes (16). Neurophysiological study of the myoclonus is consistent with cortical myoclonus (52).
1H-magnetic resonance spectroscopy is more sensitive than structural MRI to detect brain involvement. The greatest metabolic changes are seen in the frontal cortex, cerebellum, and basal ganglia (70). In a study, a statistically significant difference was found between NAA/Cho ratio in the cerebellum in patients with Lafora disease compared with normal controls (P= 0.04) (03). In addition, both myoclonus and ataxia scores showed significant correlation with NAA/Cho ratios in the pons (P= 0.03, P= 0.04) and in the cerebellum (P= 0.04, P= 0.01), respectively (03).
A study by d'Orsi and colleagues looked at biomarkers FDG-PET, CSF Aβ42, CSF p-tau181, and t-tauAg to correlate with severity of disease (16). The group found that in the mid to final stages of the disease, CSF patterns were similar to non-Alzheimer dementia. PET hypometabolism was more often seen in the parietal, precuneus, posterior cingulate, and frontal regions. As the disease progressed, there was involvement of the thalamus and lateral temporal lobes.
The definitive diagnosis of Lafora disease depends on the detection of the characteristic PAS-positive inclusions, but this approach has both false negatives and false positives (43; 23). Skin biopsy of the axilla is best site because of the typical presence of Lafora bodies in the sweat glands and ducts. To offer adequate samples, the skin biopsy should be deep enough to include entire sweat gland ducts and glands. Cryostat sections stained with PAS are best for demonstrating the inclusions. Electron microscopic confirmation is advisable (14).
As the biopsy may be negative due to sampling error, molecular genetic testing of the EPM2A and EPM2B gene is now the diagnostic procedure of choice. Genetic testing results in the finding of mutations in EPM2A or EPM2B.
Management of Lafora disease is only supportive and palliative (31; 48; 52).
Clonazepam and valproate are the main classic antiepileptic drugs with antimyoclonic efficacy. Of the newer antiepileptic drugs, levetiracetam (09) and zonisamide (72) are considered the most effective. Clobazam and phenobarbital are second options. Fast-acting benzodiazepines are used for temporary relief of myoclonus during social events and the treatment of myoclonic status epilepticus.
Lamotrigine may worsen myoclonus. Other contraindicated antiepileptic drugs include carbamazepine, oxcarbazepine, eslicarbazepine acetate, phenytoin, gabapentin, pregabalin, tiagabine, and vigabatrin—all of which often worsen myoclonus.
Perampanel adjunctive therapy appears to confer particular benefit on seizures and myoclonus in Lafora disease (32).
Vagal nerve stimulation improved seizures in a case with Lafora disease (45). The ketogenic diet did not prevent the deterioration in five patients with Lafora disease (13).
A mouse model of Lafora disease (Epm2b-/-) was subjected to different pharmacological interventions aimed to alleviate protein clearance and endoplasmic reticulum stress. Metformin, an AMPK activator, administration decreased the accumulation of Lafora bodies and polyubiquitin protein aggregates, diminished neurodegeneration (neuronal loss and reactive gliosis), and ameliorated neuropsychological tests (08). Burgos and colleagues looked at outcomes of early versus late metformin treatment (after neurologic symptoms appeared) in both mouse models (Epm2a-/- and Epm2b-/-) and human patients (11). They found that patients treated with metformin (both early or late) had a slower progression of disease, specifically in preserving speech and multiple activities of daily living. Metformin has been granted orphan drug designation by the United States Food and Drug Administration and the European Medicines Agency and is being used in clinical practice using standard dosing for diabetes management (1000–2000 mg per day).
In a study, fingolimod was administered in a mouse model to target neuroinflammation. The study found that fingolimod decreased reactive astroglial neuroinflammation and T lymphocytes with improved behavioral performance (60).
In a study of EPM2B deletion and wild type mice, cannabidiol showed some improvement in cognitive function but no effect on seizure control (05; 44). In another knockout mice model of EPM2B -/-, propranolol and epigallocatechin gallate (green tea antioxidant extract) showed improvement in some cognitive domains, and propranolol decreased astrogliosis and microgliosis in mice (44; 47).
New approaches to the treatment of Lafora disease have been discussed in the sixth International Lafora Epilepsy workshop. In brief, there are several potential lines of therapy: (1) small molecule inhibitors of glycogen synthase, (2) enzyme therapy to digest Lafora bodies, (3) antisense oligonucleotides (ASOs) to block glycogen synthase, and (4) repurposed drugs such as metformin. Gene therapy replacing laforin or malin is probably the most promising therapeutic modality but is still in its infancy (44). A mouse model of EPM2a -/- injected with an adenovirus vector to for the expression of EPM2a showed a decrease Lafora body synthesis and neuroinflammation (73). There is a current study of VAL-122, a recombinant glucosidase plus antibody, which may degrade cerebral polyglucans and delay disease progression (50). Another study is looking into the use of empagliflozin, a medication used for type 2 diabetes and chronic kidney disease, to increase glucose excretion (21).
Prenatal testing. If EPM2A or EPM2B pathogenic variants have been identified in an affected offspring, prenatal testing may be available for further pregnancies.
Preimplantation genetic diagnosis. This may be an option for some families in which EPM2A or EPM2B pathogenic variants have been identified in the parents in a carrier state.
There is not enough information on the effect of pregnancy on the disorder.
This article is dedicated to the memory of C P Panayiotopoulos MD PhD FRCP, in whose previous outstanding version this update is based.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Aparna Polavarapu MD
Dr. Polavarapu of Albert Einstein College of Medicine and Montefiore Medical Center has no relevant financial relationships to disclose.
See Profile
Solomon L Moshé MD
Dr. Moshé of Albert Einstein College of Medicine has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Epilepsy & Seizures
Jul. 08, 2026
Neurobehavioral & Cognitive Disorders
Jun. 17, 2026
Epilepsy & Seizures
Jun. 02, 2026
Neurobehavioral & Cognitive Disorders
May. 20, 2026
Movement Disorders
May. 18, 2026
Neuropharmacology & Neurotherapeutics
May. 11, 2026
Epilepsy & Seizures
May. 08, 2026
Infectious Disorders
May. 01, 2026