




Neuromuscular Disorders
Viral and retroviral myositis
Jun. 16, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Malignant hyperthermia is a pharmacogenetic disease that typically manifests during or immediately following general anesthesia. The authors discuss the disease and its diagnosis, including genetic testing. The primary mode of inheritance is autosomal dominant, although malignant hyperthermia does not occur with each anesthetic exposure. Non-anesthesia related cases of severe rhabdomyolysis linked to hereditary neuromuscular disorders or RYR1 or CACNA1S mutations are reported in the literature with increasing frequency.
Without early identification and treatment, mortality is 70+%; thus, early diagnosis and treatment are lifesaving. However, early signs can be mistaken for inadequate anesthesia or for a febrile reaction of any cause. Thus, treatment must be started before the definitive diagnosis; in retrospect, many patients who are treated may not have malignant hyperthermia. Diagnosis of malignant hyperthermia for suspicious episodes may be confirmed only by the caffeine halothane contracture test, which is performed at a very limited number of centers in North America and requires fresh muscle tissue. Genetic testing is available at several designated centers. Despite ongoing advances, molecular genetic testing has limited sensitivity—it is used to confirm susceptibility to malignant hyperthermia rather than to clear patients of the diagnosis. Patients who are susceptible to malignant hyperthermia may receive general or regional anesthesia, but the anesthetic must be trigger-free, consisting of regional and local anesthetics or intravenous general anesthesia with or without nitrous oxide. The muscle relaxant succinylcholine and all potent inhalational agents carry a risk of triggering malignant hyperthermia in these patients. Reports of nonanesthetic-associated rhabdomyolysis, with or without hyperthermia, have been associated with malignant hyperthermia susceptible contracture tests or malignant hyperthermia–associated RYR1 mutations (27; 98; 130; 69; 145). Patients presenting in this manner represent a smaller subset of individuals susceptible to malignant hyperthermia given that most patients with anesthetic-induced malignant hyperthermia do not develop life-threatening hyperthermia or rhabdomyolysis with exercise or heat exposure.
|
• Malignant hyperthermia is a hypermetabolic response to potent inhalational agents or succinylcholine that can lead to death. | |
|
• Most patients have no signs or symptoms prior to a malignant hyperthermia crisis. | |
|
• Carriers may be at risk of nonanesthetic-related malignant hyperthermia or rhabdomyolysis triggered by heat, exertion, febrile illness, or a combination of these conditions. | |
|
• Dominant inheritance is mostly of RYR1 mutations. | |
|
• RYR1 variants of unknown significance are not uncommon in an unselected population (ie, no history of malignant hyperthermia or myopathy). | |
|
• Early recognition, discontinuing triggers, treatment with dantrolene, and symptomatic therapy are critical. | |
|
• Adequate temperature monitoring has been associated with preventing death in analysis of malignant hyperthermia cases. | |
|
• For the known malignant hyperthermia-susceptible patient, general anesthesia is safe as long as known triggering agents are avoided. | |
|
• Family members must be identified to alert them about the potential risk. |
Malignant hyperthermia is a rare but potentially lethal complication of anesthesia. This syndrome was first identified in Australia in 1960. The proband had survived an anesthetic-induced syndrome characterized by hypotension, cyanosis, tachycardia, hyperthermia, and postoperative difficulty with movement. A family history of anesthesia-related deaths (10 of 24 relatives receiving anesthetics) suggested that this syndrome was an inherited disorder (25). An analogous syndrome occurs in various strains of pigs and is referred to as malignant hyperthermia, porcine stress syndrome, or pale, soft, exudative pork syndrome. Similar disorders have also been described in horses, dogs, and cats. Mice with knock-in of abnormal RyR1 have been produced and allow a better understanding of the syndrome (141; 87; 31). Malignant hyperthermia is associated with exposure to triggering agents—volatile anesthetics or succinylcholine, a depolarizing neuromuscular blocker—in otherwise normal patients (114; 144). The observation that an in vitro contracture occurred in a muscle bundle in response to low doses of caffeine or halothane in individuals susceptible to malignant hyperthermia formed the basis for a diagnostic test and suggested that skeletal muscle was the target tissue. Genetic studies have demonstrated that malignant hyperthermia is a heterogeneous disorder (112).
Dantrolene was first synthesized in 1967 and accidentally found to have antispasmodic properties. Extensive work by Keith Ellis, a research scientist with a strong background in skeletal muscle physiology, confirmed that it caused dose-dependent muscle relaxation without significant side effects or cardiopulmonary depression. Ellis systematically narrowed down the site of action of dantrolene to involve intracellular calcium release. When he read about porcine stress syndrome and recognized that dantrolene was a potential treatment, he forwarded the drug to several researchers of malignant hyperthermia and, thus, pioneered its use as the life-saving treatment for malignant hyperthermia crises (108). Since then it has been established that dantrolene reduces calcium release or leak from RYR1-dependent intracellular stores; it also suppresses increased extracellular calcium entry and store overload-induced calcium release (31; 21).
Susceptibility to malignant hyperthermia is impossible to predict in the absence of a challenge with triggering agents, confirmation by diagnostic testing, or a positive family history. In the latter case, there is typically a 50% chance of inheritance of the malignant hyperthermia gene from either parent. The earliest signs of a malignant hyperthermia episode include tachycardia and increased end-tidal carbon dioxide. Muscle rigidity may be an early sign but is not always present. Hyperthermia may appear as soon as 15 minutes after onset and is one of the first to third signs in the majority of patients (72; 70). Muscle injury during a malignant hyperthermia episode may produce myoglobinuria with elevated plasma potassium and creatine kinase. The most sensitive indicator of an acute episode of malignant hyperthermia is an unanticipated increase in end-tidal carbon dioxide. Rigidity of the entire body following succinylcholine or potent inhalational anesthetics is a less sensitive sign than end-tidal carbon dioxide, but it is more specific for malignant hyperthermia if myotonia and voluntary or reflex muscle activity have been ruled out. If generalized rigidity is due to malignant hyperthermia, it will not respond to conventional paralytic drugs such as vecuronium. Muscle rigidity may be limited to masseter spasm, most frequently observed after administration of succinylcholine (53). Malignant hyperthermia has a wide range of presentations, from indolent onset over hours with mild signs to evolution of severe acidosis and hyperthermia in minutes. The syndrome can be fatal if not arrested early in its course, before cardiovascular collapse or extreme temperature elevation result in multiorgan failure. With effective intervention, complete recovery occurs, and most of the muscle damage is reversible. However, weakness may last for months after an acute episode of malignant hyperthermia (91).
Although it has long been assumed that most patients with malignant hyperthermia susceptibility have no obvious signs or symptoms of their predisposition to malignant hyperthermia until they are exposed to a trigger, a prospective study by Bersselaar and colleagues found that patients with a history of RYR1-related exertional rhabdomyolysis or malignant hyperthermia susceptibility are more likely to suffer from cramps, myalgia, and exertional myalgia than healthy controls (05). In addition, those patients were found to more frequently consult healthcare professionals for apparent neuromuscular symptoms and to have a higher likelihood of developing muscle weakness later in life.
With rapid recognition and treatment, the prognosis of an acute episode is good. Whereas mortality was as high as 70% prior to the availability of dantrolene, it is now estimated to be between 6% and 10% (116; 70). The syndrome usually resolves rapidly. However, both acute and delayed recrudescence have been reported. In North America, recrudescence is common and present in about 20% of patients treated with dantrolene, leading to the suggestion that the dosing interval be shortened from 6 to 4 hours (14). Complications during and after the episode include hyperkalemia, disseminated intravascular coagulation, brain injury, compartment syndrome, and myoglobinuric renal failure.
Forrest and colleagues reported a previously healthy 14-year-old patient who had a severe episode of malignant hyperthermia during appendectomy and suffered a cardiac arrest, with resuscitation including extracorporeal membrane oxygenation (ECMO) and treatment with dantrolene (37). Despite successful weaning from ECMO, the patient developed progressive cerebellar edema with subsequent herniation and brain death. The patient had a confirmed malignant hyperthermia causal mutation, and the authors noted that RYR1 is present in both hippocampal neurons and cerebellar Purkinje cells. The authors hypothesized that, in addition to ischemic injury, RYR1-mediated intracellular calcium elevation may explain the atypical cerebellar edema (37).
In 2014, Larach and associates reported 84 cases of very likely or almost certain malignant hyperthermia episodes submitted to the North American Malignant Hyperthermia Registry between 2007 and 2012 (70). Eight of 84 patients died. The mean peak temperature of those who died was 41.6° C, and absence of temperature monitoring or monitoring only with skin temperature (ie, not core temperature) were strongly associated with mortality. This firmly suggested that absence of core temperature monitoring was associated with delayed recognition and initiation of treatment of acute malignant hyperthermia (70), highlighting the importance of accurate temperature monitoring for preventing morbidity and mortality.
A 32-year-old man sustained an open fracture of the proximal humerus in an automobile accident. He was healthy and gave no history of neuromuscular diseases; family history was negative for anesthetic problems. Blood pressure was 140/75. He was taken to the operating room for debridement and open fixation of the fracture. Anesthesia was induced with propofol, fentanyl, and succinylcholine, and he was intubated without problem. Inhalational anesthesia was started with 60:40 nitrous oxide:oxygen and sevoflurane 1.5%. Ventilation was controlled with a tidal volume of 850 mL at a rate of eight per minute. End-tidal carbon dioxide was 34 mmHg; peak inspiratory pressure was 20 cm H2O. Blood pressure and heart rate were normal. Temperature in the distal esophagus was monitored and started at 35.9. Over the next hour the end-tidal carbon dioxide increased in spite of increasing respiratory rate to 15. The patient was overbreathing the ventilator until paralyzed with rocuronium. Tachycardia reappeared despite addition of 250 µg of fentanyl (total 500 µg) and increasing the sevoflurane to 2%. Some rigidity in the arm was noted, despite absence of twitch response to electrical stimulation of the facial nerve. The temperature, which had earlier started to fall, was now 37.2. An arterial blood gas was obtained, and the pH measured 7.10, partial pressure of carbon dioxide was 60 mm Hg, partial pressure of oxygen was 70 mm Hg, and base deficit was 9. The diagnosis of malignant hyperthermia was presumed, the sevoflurane was stopped, ventilation increased, and an infusion of propofol was started at 150 µg/kg per minute. A total of 240 mg of dantrolene (twelve 20 mg bottles dissolved with sterile water) was given as quickly as possible, and the surgery was rapidly finished. The intravenous fluid rate was increased. The Malignant Hyperthermia Association of the United States hotline was contacted, an arterial line was started, and the patient was cooled. Further blood was obtained for potassium, creatine kinase, and myoglobin. Urine was sent for to see if dipstick was positive for blood. Shortly after administration of dantrolene, the end-tidal carbon dioxide fell, the tachycardia and rigidity resolved, and minute ventilation could be reduced to starting values with normal end-tidal carbon dioxide. The potassium was 5.2 mEq/L in the first specimen but 4.2 mEq/L in the second specimen.
The patient was then allowed to wake up. In this case there was no slow awakening, and the patient was extubated after full recovery from neuromuscular blockade. The patient complained of some muscle weakness and aching. Dantrolene was continued intravenously at 1 mg/kg Q6 hours for 24 hours; the patient was observed in the ICU and then sent to the regular floor for a further 24 hours. Repeated blood gases showed a return to normal, although there was one possible recrudescence of malignant hyperthermia treated with an additional dose of dantrolene.
Creatine kinase peaked at 22,000 IU/L 18 hours postoperatively, the urine dipstick was positive for blood, and there was myoglobin in the blood and urine, though the myoglobin results did not come back for over 1 week.
Six months later, an in vitro caffeine halothane contracture test was performed; responses to both halothane and caffeine confirmed malignant hyperthermia susceptibility. At the time of the contracture test, the creatine kinase was 280 IU/L, with a normal range up to 180. Histology and histochemistry of a muscle biopsy failed to show any specific abnormality.
The patient and all his blood relatives were advised of their risk and that the in vitro caffeine halothane contracture test could be used to discriminate non-susceptible family members from those at risk. Presently, we would advise the patient to undergo molecular genetic testing prior to scheduling a caffeine halothane contracture test; if a malignant hyperthermia causal mutation was found, this could be used to confirm malignant hyperthermia susceptibility in family members.
The cause of human malignant hyperthermia is not entirely understood, but mutations in different genes have been identified. Anesthesia-induced or, in some patients, heat- or exertion-induced loss of intracellular calcium regulation and consequent grossly elevated myoplasmic calcium are believed to be the turning point in the progression of the syndrome. Control of intracellular calcium is complex (138; 22; 21). Five proteins have been shown as necessary for conformational coupling of Cav 1.1 to the RYR1 encoded channel in excitation-contraction coupling. Freeze-fracture electron microscopy indicates the junctional proteins β1α, Stac3, and junctophilin 2 are also required for skeletal muscle contraction (105).
The functions of several different proteins and modulators may be altered in human malignant hyperthermia, including the RYR1 encoded calcium release channel (101; 89), Cav1.1, the principal subunit of the L-type calcium channel also known as the dihydropyridine receptor and encoded by CACNA1S (89; 30; 40; 03), Stac3 (52), and nonspecific sarcolemmal cation channels (31). In a study by Endo and colleagues, a pathogenic heterozygous variant in ASPH, a gene encoding junctin, was identified after performing genomic sequencing on a cohort of patients with exertional heat illness or malignant hyperthermia and/or abnormal caffeine halothane contracture testing (32). Junctin is one of several proteins involved in the regulation of excitation-contraction coupling in muscle function. Results of this study point to yet another possible cause for malignant hyperthermia susceptibility.
There is evidence that deranged intracellular calcium regulation in malignant hyperthermia involves impaired inhibition of RYR1 by intracellular calcium and magnesium as well as excessive extracellular calcium entry and store overload-induced calcium release (51; 31; 43; 21). In a knock-in mouse model of malignant hyperthermia, Canato and colleagues demonstrated excessive calcium accumulation in mitochondria, associated with greatly increased production of reactive oxygen species (ROS); this was associated with increased sarcoplasmic reticulum calcium release with either caffeine or electrical stimulation. In the knock-in mouse, mitochondrial calcium uniporter silencing decreased calcium entry into the mitochondrial matrix, resulting in reduced reactive oxygen species accumulation and normalization of sarcoplasmic reticulum calcium release, similar to the wild type (16).
In a study using metabolomics to analyze malignant hyperthermia-susceptible and malignant hyperthermia nonsusceptible muscle fibers, impairment of skeletal muscle metabolism could be seen in multiple pathways (08). Measurable changes included increased oxidative stress, increased levels of histidine pathway metabolites, and a shift from carbohydrates to lipids for energy production. These metabolic derangements would add evidence to the hypothesis that mitochondrial dysfunction is responsible alongside the abnormal Ca2+ regulation (64).
Fletcher and colleagues hypothesized that fatty acids may play a modulatory role in malignant hyperthermia, as they markedly enhance halothane-induced calcium release from the sarcoplasmic reticulum (36). Concentrations of free fatty acids were also shown to be elevated in the muscles of patients with malignant hyperthermia (20). A metabolomics study by Bojko and colleagues expands on these findings by demonstrating increased levels of polyunsaturated fatty acids and hypothesizing that this increase in free fatty acids in the malignant hyperthermia-susceptible group indicates an increased, though impaired, utilization of lipids for energy production (08). Because beta-oxidization of fatty acids occurs in the mitochondria, these findings further strengthen the implication that defects in mitochondrial function play a significant role in the pathophysiology of malignant hyperthermia. This research complements work by Lopez and colleagues on TRCP3/6 mitochondrial ion channels that are activated by one of the increased lipid metabolites, diacylglycerol (85).
Chang and colleagues investigated the association between malignant hyperthermia susceptibility and the expression of genes associated with oxidative phosphorylation and fatty acid metabolism (19). Their aim was to shed more light on the purported role of mitochondrial bioenergetics on the phenotypic expression of malignant hyperthermia. Using human muscle samples and subjecting them to contracture testing, bulk RNA sequencing was employed to compare variable gene expression in malignant hyperthermia susceptible and non-susceptible groups. Overall, the study supports a link between malignant hyperthermia susceptibility and dysregulated expression of genes associated with mitochondrial bioenergetics.
The European Malignant Hyperthermia Group currently lists 72 RYR1 variants as pathogenic or likely pathogenic as well as two CACNA1S variants as pathogenic. (See www.emhg.org/ for the accepted malignant hyperthermia causative mutations). A Clinical Genome Resource expert panel used criteria from the American College of Medical Genetics and the Association of Molecular Pathologists in a pilot study of 84 variants of RYR1 and added an additional 251 variants to their review in 2022 (59). As a result, the panel of experts now classifies 29 variants as pathogenic, 57 as likely pathogenic, 219 as variants of unknown significance, 17 as likely benign, and 13 as benign.
In most families, malignant hyperthermia susceptibility is linked to the RYR1 gene on chromosome 19q13.1, whereas in other families this is not the case (33; 112; 99; 68; 128). In North America, approximately 25% of malignant hyperthermia-susceptible people have identified mutations in the three hot spots of RYR1 tested (124; 10). In 2018, Miller and colleagues reported molecular genetic testing results in 770 independent families with a history of malignant hyperthermia in the United Kingdom. They found that 546 out of 722 families had an RYR1 variant that was known or potentially pathogenic, seven families had a CACNA1S variant that was known or potentially pathogenic, and five families had both RYR1 and CACNA1S variants that were known or potentially pathogenic (96). Nearly 10% of malignant hyperthermia-susceptible families had at least two RYR1 variants classified as likely pathogenic or pathogenic; it is important not to limit examination to only one exon or variant. It is estimated that 50% to 75% of all malignant hyperthermia cases may eventually be associated with RYR1. Conversely, Miller and colleagues estimate that pathogenic variants in RYR1, CACNA1S, or Stac3 will not be found in 14% to 23% of malignant hyperthermia-susceptible families.
Epigenetic silencing may play a role in the variable penetrance of the disorder. It has been identified in RYR1 and shown to be tissue specific (143; 111).
In 2013, five families affected by Native American myopathy, an autosomal recessive disorder associated with malignant hyperthermia, were found to have a missense mutation in the Stac3 gene on chromosome 12 that encodes a t-tubule protein essential for excitation-contraction coupling. In a zebrafish model, the mutant protein resulted in decreased Ca+2 transients in fast twitch muscle (52). Linsley and colleagues confirmed that the Stac3 protein is essential for this coupling by regulating Ca2+ channels in muscle cells (80); the NAM mutation resulted in increased caffeine-induced Ca2+ release across a wide range of concentrations as well as increased Ca2+ in internal stores, consistent with increased SR luminal Ca2+. Malignant hyperthermia episodes have not been reported in heterozygous carriers of the mutation. Native American myopathy has been reported in other ethnic groups; the p.Trp284Ser Stac3 variant is present in 0.12% of the African ExAC population (129; 96).
Several additional rare variants of genes involved in skeletal muscle Ca2+ regulation were identified in a study of malignant hyperthermia susceptible patients (Bjorksten and Gillies 2016). Among them, the DHPR subunit β1α is known to modulate RYR-1 function and regulates the DHPR α1S subunit. A study expressing the identified DHPR β1α mutation V156A in mouse cultured myotubes resulted in disrupted Ca2+ homeostasis similar to that seen in malignant hyperthermia-linked mutations of the DHPR α1S subunit (104).
Additional functional studies will help classify variants of unknown significance as pathogenic or benign. White and colleagues performed a functional analysis of RYR1 variants in 21 patients with a history of a clinical malignant hyperthermia episode along with a positive contracture test (137). Variants were tested in HEK293 cells and exposed to 4-chloro-m-cresol and analyzed in silico using the EMHG scoring matrix as well as ClinGen expert panel guidelines. Three additional variants were classified as disease associated.
In the absence of triggering anesthetics, there are usually no identifiable signs or symptoms of this disorder. No muscle abnormalities are consistently observed in people susceptible to malignant hyperthermia. Small subpopulations of subjects with malignant hyperthermia, identified either by clinical presentations or by diagnostic testing, have evidence of muscle disorders, including central core disease, multiminicore disease, King-Denborough syndrome, centronuclear myopathy, congenital fiber type disproportion, Native American myopathy, and late-onset myopathies (114; 28; 29; 86; 61). Central core disease is known to be a disorder of calcium release channels (12; 89), although malignant hyperthermia and central core disease are not always concordant (93). Patients with central core disease may have asymptomatic family members who are malignant hyperthermia susceptible; the family member may have a dominant malignant hyperthermia RYR1 mutation (66). Recessive inheritance of malignant hyperthermia-associated RYR1 mutations has been described in myopathic patients, and these mutations may also be present in asymptomatic family members. Malignant hyperthermia susceptible causal RYR1 mutations and novel RYR1 variants have also been demonstrated in unrelated patients with King-Denborough syndrome; some patients also had significant decreases in the amount of RYR1 protein expressed (28). In a single-center 25-year retrospective analysis of children with severe reactions to anesthesia, one third of patients with confirmed malignant hyperthermia had an underlying neuromuscular disorder. Patients with CK levels greater than 8000, those who were given dantrolene, or those who had a diagnosis of malignant hyperthermia were included (02).
Using the USA Nationwide Inpatient Sample, approximately 1 in 100,000 patients were discharged with the diagnosis of malignant hyperthermia between 2000 and 2005 in the United States. A study of 47 patients with a discharge diagnosis of malignant hyperthermia showed that less than 25% had an episode of malignant hyperthermia during admission, around 25% had high fever without any evidence of malignant hyperthermia, and nearly 50% had a personal or family history of malignant hyperthermia (106). Mortality has fallen in the same period from 16% to 6% (116). The New York State database for the same period indicates a similar incidence in New York State hospitals (09). Data from New York State ambulatory surgery centers estimate the prevalence of malignant hyperthermia as 0.18 in 100,000 between 2002 and 2011 (88). The Kids’ Inpatient Database (KID) pediatric inpatient database showed an incidence of malignant hyperthermia diagnosis in 3 per 100,000 discharges, with males having a 3-fold greater incidence (79). Similar incidence has been described in other countries (127). Malignant hyperthermia has been identified in both sexes, with a male-to-female predominance of clinical episodes of about 3 to 1 (09). Male preponderance is seen with in vitro testing as well (57; 110). In American women undergoing caesarean section, the prevalence of malignant hyperthermia discharge diagnosis is 1 out of 125,000; this is similar to patients undergoing nonobstetric surgery (Guglielminotti 2020). Malignant hyperthermia occurs in all races and areas of the globe. The incidence of clinical episodes is greater in children than adults (102). The incidence varies from 1 in 10,000 (13) to 1 in 250,000 (103) and varies with the concentration of malignant hyperthermia families in a given geographic area. Because many subjects who are genetically predisposed to malignant hyperthermia do not express the syndrome on some or all exposures (48; 04), the true prevalence is not known. An analysis of the French population predicted a prevalence, possibly as high as 1 in 4000 (99), and more recent analyses of population genomics data estimate the prevalence as no less than 1 in 1500 (100) to as high as 1 in 300. The incomplete penetrance and variable gene expression result in far fewer cases of malignant hyperthermia than anticipated by mutation prevalence (54; 126). Shaw and colleagues suggest a threshold non-Mendelian model where “weaker” RYR1 variants inherited with non-RYR1 variants combine to confer malignant hyperthermia susceptibility. De novo mutations in RYR1 have been reported, explaining some of the inconsistencies in genetics (117).
Preoperative screening for family history of malignant hyperthermia or for signs of the syndrome in previous surgeries is the most important and still the most easily available tool for preventing a malignant hyperthermia episode. Immediate response by the anesthesiologist or nurse anesthetist to the first signs of the malignant hyperthermia episode and the availability of dantrolene in the operating room are the two most important means of preventing mortality. Given that studies provide strong evidence that succinylcholine, in the absence of potent inhalational anesthetics, may trigger malignant hyperthermia (110; 134), having dantrolene available in anesthetizing locations that do not use volatile anesthetics, but have succinylcholine available for emergencies, is vital.
Because the disorder is typically inherited in an autosomal dominant pattern, members of families in which malignant hyperthermia has been identified are at high risk. In addition, nonmyopathic family members of myopathic patients with either homozygous recessive or compound heterozygous malignant hyperthermia-associated RYR1 mutations may be at risk (125). Thus, patients and their family members with central core disease, multi minicore disease, King-Denborough syndrome, and some other myopathies are at risk. Signs of the syndrome during a previous anesthetic, including masseter muscle or whole-body rigidity, elevated creatine kinase values, and myoglobinuria, are also indicative of high risk (102).
Patients with muscular dystrophy conditions, such as Becker, Duchenne, and Brody myopathies, may be at increased risk for malignant hyperthermia, and precautions need to be taken. These disorders will be discussed in the section on associated disorders.
Whether statin drugs are a risk for patients known to be malignant hyperthermia susceptible (or if patients with muscle-related problems with statin drugs are likely to be malignant hyperthermia susceptible) is under investigation. A report that malignant hyperthermia-susceptible porcine muscle but not normal porcine muscle produced an in vitro contracture on exposure to clinically relevant concentrations of statins suggests that there may be a relationship (95). A study using a knock-in mouse heterozygous for a known malignant hyperthermia-associated RYR1 mutation, Y524S, demonstrated a hypermetabolic reaction in mice injected intraperitoneally with 60 to 80 mg/kg simvastatin, as well as increased in vitro sarcoplasmic reticulum calcium leak in muscle fibers exposed to 500 µM or 1 mM simvastatin (67). Given the very high in vivo doses and in vitro concentrations (many orders of magnitude higher than therapeutic plasma levels, eg, < 0.1 µM), it is uncertain if this finding has relevance to the association of malignant hyperthermia susceptibility with statin-induced myopathy. Two studies have examined the incidence of RYR1 mutations in patients with severe statin myopathy (135; 56). Patients in the earlier study were also screened for CPT2 deficiency, McArdle disease, and myoadenylate deaminase deficiency, whereas Isackson’s study employed whole-exome sequencing. Isackson and colleagues found that 10 of 76 patients with severe statin-associated muscle symptoms had pathogenic or probably pathogenic variants in RYR1 or CACNA1S. The authors concluded that severe statin myopathy is frequently associated with probably pathogenic variants in RYR1 or CACNA1S (135; 56). On the other hand, Dr Hopkins of the Leeds, United Kingdom, Malignant Hyperthermia Testing Center noted that
|
“many patients attending our unit for malignant hyperthermia testing have been taking a statin with no evidence of adverse clinical or biochemical responses associated with malignant hyperthermia status. It would seem sensible, though, when initiating statin treatment in a known malignant hyperthermia patient to take a pre-treatment creatine kinase level and instruct the patient to report muscle symptoms or dark urine” (51). |
Malignant hyperthermia must be differentiated from other causes of increased end-tidal carbon dioxide, tachycardia, fever, and rhabdomyolysis. A common clinical problem is differentiation of malignant hyperthermia from sepsis in the intraoperative and postoperative period. Drug reactions may produce high fever and associated signs suggesting malignant hyperthermia. The syndrome triggered by certain antipsychotic agents, termed neuroleptic malignant syndrome, is similar to malignant hyperthermia in some ways but a distinct entity.
Exposure to other drugs that alter dopaminergic, serotonergic, or catecholaminergic function can produce signs similar to those of malignant hyperthermia, including severe rhabdomyolysis. Endocrine disease such as pheochromocytoma and thyroid storm may produce increased oxygen consumption, fever, and cardiovascular collapse. The increasing frequency of laparoscopic surgery has led to the recognition that some patients, especially when carbon dioxide has dissected subcutaneously, may rapidly develop high end-tidal carbon dioxide concentrations or require high minute ventilation to maintain normocarbia. In the above-mentioned cases mimicking a malignant hyperthermia reaction, the response to in vitro contracture testing for malignant hyperthermia susceptibility as well as genetic work-up is usually negative, but the problem of how to treat the acutely ill patient remains. Treatment for the underlying disease process should not be delayed. Administration of dantrolene is not expected to alter the course of these diseases. However, as dantrolene is an excellent nonspecific antipyretic, it may be given to abate a hyperthermic crisis in the absence of a diagnosis. A decrease in temperature after dantrolene administration is not diagnostic of malignant hyperthermia. Dantrolene can be given while evidence is sought to narrow the differential diagnosis.
One clinical problem related to malignant hyperthermia is masseter muscle rigidity (110; 134; 53). This is more often observed in children, particularly after halothane induction and succinylcholine administration, but it can occur in adults as well. Although incomplete relaxation of the jaw in children given halothane and succinylcholine is not uncommon, masseter muscle tension to the point of compromising intubation is rare (49; 76). Approximately 50% of patients with masseter muscle rigidity have tested positive for malignant hyperthermia, but development of clinical signs and symptoms of malignant hyperthermia occurred in only about 15% of patients (102; 115; 110). Rhabdomyolysis and arrhythmias during anesthesia can be due to causes other than malignant hyperthermia. Ruling out the diagnosis of malignant hyperthermia susceptibility following masseter rigidity unaccompanied by hypermetabolism requires in vitro contracture testing (62). In a study by Hudig and colleagues, no clinical characteristics separated malignant hyperthermia susceptible patients who had experienced masseter muscle rigidity from those who were not malignant hyperthermia-susceptible, as determined by genetic analysis and contracture testing (53). Furthermore, the degree of severity of the masseter muscle rigidity could not be linked to malignant hyperthermia susceptibility.
It has been suggested that patients with Duchenne muscular dystrophy are at an increased risk of malignant hyperthermia. Frequently, the presentation in patients with dystrophinopathies consists of rhabdomyolysis and cardiovascular collapse rather than hypermetabolism (the sine qua non of malignant hyperthermia). Anesthetized patients with Duchenne or the related Becker muscular dystrophy are also at risk for dramatic hyperkalemia and cardiac arrest, especially after succinylcholine administration (74), though also well-documented after potent inhalational anesthesia without succinylcholine. A clinical series from the Mayo Clinic did not observe any episodes of malignant hyperthermia in 47 patients with Duchenne or Becker muscular dystrophy who underwent 117 general anesthetics, 66 of which included a volatile anesthetic (123). One patient with undiagnosed Becker dystrophy had succinylcholine-induced hyperkalemic cardiac arrest. Most complications were associated with underlying respiratory or cardiac disease or major blood loss. Still, it is strongly recommended that the physicians caring for patients with dystrophinopathies or suspected to be susceptible to malignant hyperthermia avoid volatile anesthetics and succinylcholine.
Brody myopathy, an autosomal recessive myopathy due to mutations in the gene for SERCA1 (sarcoplasmic reticulum Ca2+ ATPase type 1), has been associated with postanesthetic rigidity and rhabdomyolysis in abnormal caffeine-halothane contracture testing in an adult patient and his siblings (119). Affected family members had a history of childhood-onset exercise intolerance. Typical malignant hyperthermia (ie, intraoperative signs of hypermetabolism) was not observed and has never been reported in a patient with Brody disease. A review recommended that succinylcholine and potent inhalational anesthetics be avoided in patients with Brody disease (97).
Patients with idiopathic hyperCKemia may also be at increased risk of anesthetic-induced malignant hyperthermia (63; 128).
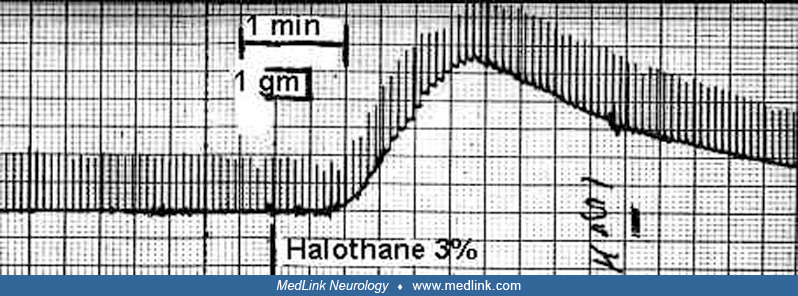
The only highly sensitive diagnostic test for malignant hyperthermia susceptibility is the caffeine and halothane contracture test of biopsied skeletal muscle.
The patient must be referred to a testing center for the diagnosis of malignant hyperthermia because the tissue is not viable long enough to allow shipment and because it is crucial that the surgeon be familiar with the procedure. Muscle is usually removed under regional anesthetic, but nontriggering general anesthesia can be used. There are two different protocols for the in vitro contracture test. These are the European Malignant Hyperthermia Group protocol and the North American Malignant Hyperthermia Group protocol. Both protocols employ halothane or caffeine addition to tissue baths containing fiber bundles and determine the resulting contracture response (increase in resting tension).
The sensitivity and specificity of the North American Malignant Hyperthermia Group Protocol has been estimated in humans to be about 97% and 78%, respectively, using a 2-component test (01). Results of the North American and European tests are in close agreement (35). In patients who have had normal caffeine halothane contracture tests (ie, malignant hyperthermia non-susceptible), there are no reported cases of malignant hyperthermia during subsequent general anesthetics that include triggering agents (107). The worldwide experience includes 232 malignant hyperthermia non-susceptible patients who have been administered 429 triggering anesthetics. In Pollock’s diagnostic center, no patients with a causal RYR1 mutation had a non-susceptible (ie, false-negative) contracture test. This contrasts with the United Kingdom experience, where 16 out of 280 families with malignant hyperthermia-causal mutations had some members who were genotype positive/phenotype negative (96).
A review of the diagnosis of malignant hyperthermia has been published (81). Use of ryanodine or 4-chloro-m-cresol may increase the specificity of contracture testing. Because of concern regarding the continued availability of halothane for contracture testing, Metterlein and colleagues compared it with currently used potent inhalational anesthetics (94). Using surplus muscle from human patients, he found that halothane was superior, producing the strongest contractures in malignant hyperthermia susceptible patients, as well as the best discrimination between malignant hyperthermia susceptible and malignant hyperthermia non-susceptible individuals.
It is common to remove additional small specimens to examine for muscle pathology. Many laboratories now routinely draw blood specimens during preadmission testing for use in future molecular genetics analyses, allowing a more definitive diagnosis without the need for the patient to return to the testing center. The European group has published guidelines for diagnosis based on genetic testing in well-characterized families. Although they agree to label someone as susceptible to malignant hyperthermia based on genetic testing, they are unwilling to call a family member malignant hyperthermia nonsusceptible unless confirmed by in vitro contracture testing (133; 41). Family members who have abnormal RYR1 can be assumed to be malignant hyperthermia susceptible and avoid muscle biopsy (82).
Although genetic testing is rapidly evolving, diagnosis of malignant hyperthermia via genetic analysis alone remains problematic due to great genetic and phenotypic heterogeneity and the high prevalence of variants of unknown significance in RYR1 and CACNA1S. Levano and colleagues developed a resequencing array that allows for screening of the entire coding sequences of RYR1, CACNA1S, as well as BCHE genes (78). They identified 16 rare RYR1 and CACNA1S variants, four of them novel, in samples from malignant hyperthermia susceptible patients with no known pathogenic variants. This high-throughput technique may help elucidate why 30% to 50% of malignant hyperthermia susceptible patients have none of the known causative variants identified via traditional sequencing of exons encoding functionally critical domains (78; 109).
Sadhivasam and colleagues employed Bayesian modeling to predict malignant hyperthermia susceptibility and pathogenicity of variants in RYR1, CACNA1S, and Stac3 in 57 North American subjects (118). Posterior probability for a variant was determined by application of four conditions:
|
1. A strongly positive caffeine-halothane contracture test (CHCT) (caffeine contracture > 0.6 g; halothane contracture > 1.1 g) | |
|
2. Identification of all variants in RYR1, CACNA1S, and Stac3 by sequencing | |
|
3. No known pathogenic variants are present in RYR1, CACNA1S, or Stac3 (though this condition discounts the probability of coinheriting a second pathogenic variant) | |
|
4. All but one of the variants in those three genes are known to be benign or meet strong criteria (eg, allele frequency for a benign variant) |
The authors found four novel variants, three in RYR1 and one in Stac3, with a high posterior probability in malignant hyperthermia susceptible subjects. They point out that an abnormal caffeine-halothane contracture test does not confirm pathogenicity of a single variant, as multiple variants may be present in a malignant hyperthermia susceptible individual. In addition, more than 25% of malignant hyperthermia susceptible subjects had either no variants or benign/likely benign variants in these three genes, suggesting either pathogenic variants in other genes or possible false-positive contracture tests (118).
There are three centers in North America that perform caffeine-halothane contracture testing and an increasing number of laboratories that perform molecular genetic testing for malignant hyperthermia. Information regarding contacting diagnostic centers can be obtained in the United States from the Malignant Hyperthermia Association of the United States (www.mhaus.org; 607-674-7901) or from the British Malignant Hyperthermia Association (0113 206 5270).
Elevated creatine kinase values in a relative of someone known to be susceptible to malignant hyperthermia significantly increase the likelihood of malignant hyperthermia susceptibility, although a normal creatine kinase does not rule out the diagnosis.
Because of the variability of clinical signs of malignant hyperthermia and the similarity of these signs to those of other perioperative complications, diagnosis of an episode has been subjective. In an attempt to make diagnosis more objective, a malignant hyperthermia clinical grading scale has been developed by agreement among a panel of experts (73). This scale uses commonly seen signs and gives them a numerical value. However, use of this scale cannot replace muscle biopsy and contracture testing.
A full-blown malignant hyperthermia episode may be avoided by immediate discontinuation of triggering anesthesia at the first signs (102). In addition to increasing the flow of fresh oxygen to 10 L/min, placing activated charcoal filters in the breathing circuit will rapidly reduce the concentration of potent inhalational anesthetic to less than 5 ppm (06). The patient should be hyperventilated (two to four times normal minute ventilation) with 100% oxygen at high fresh gas flow rates. To date, dantrolene is the only effective treatment to abate a malignant hyperthermia episode. The formulations of dantrolene that are currently sold in the United States include Dantrium® IV, from JHP Pharmaceuticals, Revonto®, from US World Meds, and Ryanodex®, from Eagle Pharmaceuticals. Dantrium® or Revonto® require mixing each ampoule of 20 mg with 60 mL of water for injection; one should, therefore, call for help to prepare and give the starting treatment dose, 2.5 mg/kg, as rapidly as possible. In 2014, the FDA approved Ryanodex® (Eagle Pharmaceuticals), a nanocrystalline formulation of dantrolene sodium, for clinical use. Ryanodex® is vastly more water soluble than Dantrium® or Revonto® – 250 mg of Ryanodex powder (one vial) is dissolved in 5 mL of sterile water. Each vial of Ryanodex® contains only 125 mg of mannitol, compared with 3000 mg of mannitol per 20 mg bottle of Dantrium® or Revonto®. The initial intravenous dose should be at least 2.5 mg/kg, and this should be repeated as needed while monitoring heart rate, body temperature, the partial pressure of end-tidal and arterial carbon dioxide, and muscle tone. Although a maximum of 10 mg/kg is recommended, higher doses may be deemed necessary in extreme cases. Central mixed venous or femoral venous blood gas determinations may be useful to assess acidosis. Significant metabolic acidosis should be treated with 2 to 4 mEq/kg sodium bicarbonate while increasing ventilation. Monitoring exhaled carbon dioxide with continuous capnography is highly recommended. Avoid verapamil or diltiazem, as they can induce hyperkalemia in the presence of dantrolene. If a patient already taking oral verapamil or diltiazem is suspected of having an acute malignant hyperthermia crisis, one should not hesitate to give dantrolene and should not reduce the initial treatment dose or avoid giving additional dantrolene if there are persistent signs of hypermetabolism or rigidity.
To reduce body temperature, rapid intravenous infusion of cold 0.9% saline is the fastest way to lower core temperature. If skilled personnel are available, intraperitoneal lavage with cool isotonic crystalloid is also a rapidly effective technique (26). One may also employ surface cooling by placing blankets that circulate cold water (or air) over and under the patient, use of Arctic Sun®, or applying external ice packs to the axillae and groin (42). Stop cooling when core temperature is less than 38.0°C to avoid “overshoot” hypothermia. Hyperkalemia is managed in the usual fashion, including intravenous calcium, NaHCO3, insulin, and glucose followed by frequently monitoring the potassium level. Inhaled albuterol in doses 2- to 4-fold those used for bronchodilation may be used in addition to insulin to lower the potassium level. Potassium replacement should be performed only when essential. Monitor the patient in the intensive care unit for at least 24 hours. Give the patient dantrolene for at least 24 hours. Due to the possibility of disseminated intravascular coagulation, coagulation studies should be carried out during and following an episode. The presence of myoglobinuria dictates therapy with fluid loading, to maintain urine output greater than 2 mL/kg/hour. Mannitol 3 gm is present in each 20 mg vial of Dantrium® or Revonto®, so for every 2 mg/kg of dantrolene the patient gets 0.3 g/kg of mannitol; extra mannitol is usually not necessary. Fluid overload due to aggressive fluid administration or hypovolemia due to diuresis, uptake into injured skeletal muscle, or bleeding necessitates careful assessment of volume status (11).
Hotlines are available for assistance. In the United States contact the Malignant Hyperthermia Hotline of the United States (800-MH-HYPER). The same hotline can be reached from outside the United States (001-1-315-464-7079). In Great Britain call 07947 609 601. Many European countries as well as Australia, New Zealand, South Africa, and Brazil have malignant hyperthermia testing and research centers; however, not all list emergency numbers online. The North American Malignant Hyperthermia Registry was formed as a repository for patient information regarding acute episodes and results of contracture testing. The registry has merged with the Malignant Hyperthermia Association of the United States and is hosted by the University of Florida Department of Anesthesiology. Information may be obtained by e-mailing Amy Gunnett RN CCRC at agunnett@anest.ufl.edu or calling 888-274-7899. Study of the registry provides evidence-based recommendations for preparation and management of patients (72).
It is estimated that more than 15 million patients per year undergo surgery at freestanding ambulatory surgery centers. If a malignant hyperthermia episode occurs at an ambulatory surgery center, treatment of the episode, including dantrolene, should be initiated prior to transfer to a hospital with critical care expertise. An interdisciplinary collaboration resulted in publication of a transfer-of-care protocol that should be individualized by each ambulatory surgery center that uses malignant hyperthermia triggering agents (71). Adaptation of the protocol must take into account the following (71):
|
• The availability of an adequate number of personnel at the ambulatory surgery center who have rehearsed how to respond to a malignant hyperthermia crisis | |
|
• Presence of appropriate personnel during transport who will continue treatment of the crisis and complications | |
|
• Clear communication between the anesthesiologist or nurse anesthetist at the ambulatory surgery center with emergency medical technicians as well as with physicians at the receiving hospital. |
Safe anesthetic alternatives exist for known or suspected cases. The use of reassuring counseling and non-triggering anesthesia as well as careful monitoring of heart rate, end-tidal carbon dioxide, muscle tone, and temperature should result in an uneventful anesthetic. Although some physicians previously recommended the prophylactic use of 2.5 mg/kg of dantrolene for patients who have previously experienced a malignant hyperthermia episode, dantrolene prophylaxis is usually unnecessary and no longer recommended.
In many cases, the anesthetic of choice for completion of surgery or transport of intubated patients after discontinuation of triggering inhaled anesthetics is propofol. A study by Joseph and colleagues elucidated propofol binding sites on the RYR1 receptor via affinity labeling and inhibition of RYR1 channel opening (60). The authors proposed that propofol may not only be safely used but also be protective in malignant hyperthermia susceptible patients.
Family members of patients who suffered a malignant hyperthermia event must be notified to alert them of their potential for increased risk of anesthesia-induced malignant hyperthermia. Patients scheduled for surgery should be told to contact their anesthesia provider with information about their status as early as possible. If surgery is elective, this should be done at least 24 hours in advance.
In most cases, there is no reason for a patient known to be susceptible to malignant hyperthermia to alter his or her lifestyle. Exceptions are patients who have experienced serious “awake” episodes, such as exertional heat illness or rhabdomyolysis. Management of awake muscular symptoms in malignant hyperthermia-susceptible patients has been achieved with oral dantrolene therapy. Case reports and a retrospective study show a common starting dose of 25 mg to significantly improve symptoms, but no consensus recommendation exists to date (130; 15; 120). A 25-year retrospective cohort study on the tolerability and adverse effects of oral dantrolene in the management of myalgias, fatigue, and rhabdomyolysis in malignant hyperthermia susceptibility patients with positive contracture tests showed that oral dantrolene was generally well tolerated and had no serious adverse effects at doses of 25 to 400 mg per day, with a median dose of 50 mg in patients responding to treatment (55).
Malignant hyperthermia-susceptible patients are advised to wear a medic alert bracelet, and patient support services are available in the United States from the Malignant Hyperthermia Association of the United States (11 East State St., PO Box 1069, Sherburne, NY 13460-1069).
In 2011, Brandom and colleagues reported the incidence of complications associated with dantrolene treatment of malignant hyperthermia episodes in 368 patients (11). These cases had been submitted to the North American Malignant Hyperthermia Registry over a 20-year period of time. The most frequently observed complications were muscle weakness (nearly 15%), phlebitis (9.2%), and gastrointestinal upset (4.3%). Fourteen patients had acute respiratory failure, of whom six had pulmonary edema. Cases of respiratory failure were associated with either underlying cardiac or pulmonary disease or the severity of the malignant hyperthermia episode. Use of furosemide was associated with higher total doses of dantrolene as well as a decreased incidence of dantrolene complications. Use of monitors to better assess intravascular volume status (eg, CVP, PA catheter, or echocardiography) was also associated with a decreased incidence of complications (11).
Although prompt detection and treatment of acute malignant hyperthermia usually results in complete recovery, deaths are still reported. Sequelae may include persistent weakness or neurologic deficits associated with either brain injury or compartment syndrome. Outcomes have been shown to be worse with inadequate temperature monitoring (70).
Malignant hyperthermia is rare during pregnancy. The normal precautions regarding malignant hyperthermia should be used for the obstetric patient. Paternal as well as maternal malignant hyperthermia susceptibility may be a risk for the fetus; thus, the mother should not be exposed to triggering agents if the father is susceptible to malignant hyperthermia. Dantrolene does have a fetal to maternal blood concentration ratio of 0.4. Although dantrolene has not been suggested to cause any significant problems in the fetus or newborn, few data are available. The prophylactic use of dantrolene is generally not recommended (92).
The main triggering agents that should be avoided are the volatile anesthetics (including halothane, isoflurane, desflurane, and sevoflurane) and the depolarizing neuromuscular blocking agent succinylcholine. For tracing family histories of anesthetic problems, enflurane, methoxyflurane, cyclopropane, fluroxene, and ethers are also regarded as triggering agents.
Drugs that appear safe for use in patients susceptible to malignant hyperthermia include antibiotics, anticholinergics, anticholinesterases or sugammadex (as reversal agents for the non-depolarizing muscle relaxants), antihistamines, antipyretics, barbiturates, benzodiazepines, nondepolarizing muscle relaxants, droperidol, etomidate, ketamine, local anesthetics, opioids, nitrous oxide, propofol, propranolol, and vasoactive drugs (113).
The preoperative interview should collect detailed information about past anesthetic experiences of the patient and family members, including uneventful anesthetics. The patient should be reassured that death from malignant hyperthermia is very unlikely. A bracelet that identifies the diagnosis or a card describing the diagnosis should be carried at all times. Anxiolytic agents are suggested. Safe premedications include benzodiazepines or anticholinergics, antihistamines, or opioids when indicated. Careful monitoring of exhaled carbon dioxide, heart rate, muscle tone, and temperature in addition to immediately available dantrolene and iced solutions should be sufficient precaution. The anesthesia machine should be prepared by preventing use of the vaporizers and flushing the machine with 10 L/minute air or oxygen for 10 to 20 minutes. High flow of fresh gas during anesthesia has been recommended to prevent a rebound increase in volatile anesthetic concentration. The breathing circuit and carbon dioxide absorbent should be changed prior to use. Newer anesthesia machines may take much longer (eg, more than 1 hour) to prepare as the anesthetic takes longer to leach out of the system. The use of an activated charcoal filter will significantly shorten the preparation time (47; 06) of newer anesthesia machines and may permit lower fresh gas flow rates during maintenance of anesthesia.
Capnography for monitoring exhaled carbon dioxide is a required standard monitor; increased end-tidal carbon dioxide is an early sign of the syndrome. Blood gas and electrolyte measurement should be available. Increased heart rate is also an early but nonspecific sign of malignant hyperthermia. Body temperature should be monitored by nasopharyngeal, esophageal, or bladder probe for all patients and procedures under general anesthesia. Peripheral (eg, skin) temperature measurement is not recommended as the only means of monitoring (70).
Regional, local, or major conduction anesthetics with either amide or ester local anesthetic are advised when appropriate. In other cases, intravenous induction followed by nitrous oxide and oxygen and nondepolarizing neuromuscular blockade and opioid supplementation may be appropriate, or total intravenous anesthesia (TIVA) may be employed. Anticholinesterases and anticholinergics are safe for use.
RYR1 variants and malignant hyperthermia causal RYR1 mutations as well as caffeine halothane contracture tests indicating malignant hyperthermia susceptibility have been found in families with members suffering from recurrent or otherwise unexplained exertional myalgias or rhabdomyolysis (17; 139). These individuals did not appear myopathic and, with rare exceptions, did not have a personal or family history of anesthetic-induced malignant hyperthermia (131; 130; 109). Rhabdomyolysis was commonly triggered by exercise and heat and, less frequently, viral infections, alcohol, or drugs (27; 130). A study of patients who survived an episode of exertional heat illness employed next-generation sequencing of the entire coding region of RYR1 and CACNA1S. Three novel RYR1 variants were found, as well as two RYR1 variants previously reported in association with malignant hyperthermia and two variants in association with centronuclear myopathy. Three variants in CACNA1S were also found. The clinical importance of most of the discovered variants remains unclear and requires additional study (34). Retrospective cohort analyses of Canadian and European patients with exercise-induced rhabdomyolysis or myalgia lend additional support for the association between exertional rhabdomyolysis, RYR1, or CACNA1S variants, and malignant hyperthermia susceptibility. Functional effects of numerous variants of unknown significance have yet to be confirmed. Meanwhile, one might consider a person with a history of recurrent exertional rhabdomyolysis to be malignant hyperthermia-susceptible unless exercise intolerance can be attributed to a disorder of muscle metabolism or to an endocrine, inflammatory, or drug-induced process (139; 69). A study of 64 subjects who had served in the military and experienced exertional heat illness or exertional rhabdomyolysis, described in vitro contracture test results and variants in 38 genes (38). Thirty-five percent were malignant hyperthermia susceptible; 27% had RYR1 variants. In 38 patients, the authors found 51 potentially pathogenic variants in 20 different genes, including genes for fatty acid or carbohydrate metabolism or the sarcolemmal chloride or sodium channels, as well as subjects with more than one variant in either the same or different genes. This supports the idea that susceptibility to exertional heat illness is oligogenic. The authors suggest that prerecruitment genetic screening may identify individuals at increased risk of complications from heat stress.
Using knock-in mouse models with human RYR1 malignant hyperthermia mutations, exercise per se does not show an increase of rhabdomyolysis, but if temperature is allowed to increase, these animals die of malignant hyperthermia (24; 142). A knock-in mouse model where the homozygote is viable demonstrates that gene dose (homozygote vs. heterozygote), male gender, and elevated environmental temperature increase lethal expression of susceptibility (142). This threshold genetic model is supported by a study by Lopez and colleagues in which knock-in mice with the RYR1 variant G2435R showed gene dose-dependent responses to both pharmacological and environmental stressors, which are parallel to those seen in patients with the human variant G2434R, the most common malignant hyperthermia-causative variant in the United Kingdom (84). This once again highlights the likely dependence of clinical penetrance of a mutation on other genetic and nongenetic factors. Chang and colleagues provided evidence of metabolic defects at trigger-free conditions by investigating muscle fiber bioenergetics in the same knock-in mice (RYR1 G2435R variant) (18). Functional analysis, protein expression, as well as transcriptomics showed increases in oxidative phosphorylation and reactive oxygen species pointing towards mitochondrial damage from chronic oxidative stress exposure in the muscle fibers of these knock-in mice.
An identical de novo RYR1 mutation was described in two unrelated children who had fatal nonanesthetic episodes of malignant hyperthermia; one child had signs of congenital myopathy, and the other child also had a second maternally inherited RYR1 missense variant (45). A third myopathic child with the same RYR1 mutation, p.Arg3983His, had an anesthetic-induced malignant hyperthermia episode at the age of 3 and a lethal awake episode associated with acute gastroenteritis at the age of 5 (39). A fourth case of lethal awake malignant hyperthermia was reported in a 6-year-old male who in retrospect had delayed motor development; a novel RYR1 variant was demonstrated in the decedent and in a parent who underwent caffeine halothane contracture testing that confirmed malignant hyperthermia susceptibility as well as central core disease on histology (75). An editorial suggested that awake malignant hyperthermia episodes may be associated with either homozygous recessive or compound heterozygote RYR1 mutations, or a single heterozygous RYR1 mutation in the setting of a myopathic individual (eg, with weakness/muscle spasms, hypotonia, or external ophthalmoplegia) (77). Additional possible explanations would include epigenetic silencing of the wild-type allele or an intronic mutation resulting in altered splicing of transcripts. Lehmann-Horn and colleagues also questioned the sensitivity of the caffeine halothane contracture test to detect those at risk of nonanesthetic triggered malignant hyperthermia episodes, though without documented false-negative tests in this setting. To place this in perspective, MacLennan and Zvaritch noted that even those with homozygous recessive malignant hyperthermia mutations may appear normal and asymptomatic, and that humans at risk of nonanesthetic-induced malignant hyperthermia are a small subset of malignant hyperthermia susceptible patients (90). Zvaritch and colleagues reported fatal nonanesthetic malignant hyperthermia deaths of two young athletic males in a family with a history of malignant hyperthermia susceptibility (145). The lethal episodes were triggered by physical exertion coupled with febrile illness. A likely pathogenic RYR1 variant was found that segregates with malignant hyperthermia susceptibility in that family.
An increasing number of case reports of malignant hyperthermia without anesthetic triggers demonstrates the wide spectrum of RYR1-related pathology and the need for further exploring this aspect of malignant hyperthermia. Symptoms similar to those of a malignant hyperthermia crisis, such as hyperthermia, rhabdomyolysis, and muscle rigidity, can be seen in patients exposed to high environmental temperatures, exertion, or febrile illness. In one retrospective study, 64% of patients who had nonanesthesia-related persistently elevated CK, post-exercise rhabdomyolysis, post-viral chronic fatigue, or muscle weakness of unknown cause tested positive on caffeine-halothane contracture testing (130). The Malignant Hyperthermia Association of the United States addresses the issue of malignant hyperthermia and malignant hyperthermia-like illness in its consensus statement (83). Although it endorses education of patients, families, physicians, and athletic coaches to recognize signs and symptoms of malignant hyperthermia-like events, it does not conclude that exercise restriction is indicated for all malignant hyperthermia-susceptible individuals because a solid risk-benefit analysis is currently not possible. Neither could the expert panel definitively recommend that all individuals who have suffered from heat-related or exertional rhabdomyolysis be considered susceptible to malignant hyperthermia, though certain clinical characteristics were identified that place such patients at higher risk for malignant hyperthermia susceptibility. Unfortunately, noninvasive tests do not exist at this time that would accurately confirm or exclude malignant hyperthermia susceptibility in relatives of malignant hyperthermia susceptible patients who do not have a known malignant hyperthermia-causative mutation. MHAUS recommends that individuals or parents of children who have experienced awake symptoms “restrict their activity based on their own experience and consult with a malignant hyperthermia expert, expert neurologist, or sports medicine physician familiar with both malignant hyperthermia and the adverse effects of heat and exercise.”
Watt and colleagues addressed the apparent lack of familiarity of youth sports coaches as well as first responders with malignant hyperthermia and the associated risk for a subgroup of athletes exercising in hot environments in one study (136). It proposes educating personnel about the condition to speed up appropriate treatment and to save lives. The lead author of the study also devised questions to be asked in the U.S. Olympic and Paralympic Committee’s existing athlete intake survey in order to identify individuals at risk for malignant hyperthermia or exertional heat illness. This will likely be an ever increasingly important topic as future locales for summer Olympic Games are likely to expose athletes to higher ambient temperatures than ever before.
Febrile illness has been linked to rhabdomyolysis and excessive, uncontrollable temperature in multiple case reports. Patients in these reports present as if experiencing a malignant hyperthermia episode, including increases in CK, myoglobin, uncontrollable temperatures, and associated sequelae, even if they do not have malignant hyperthermia or are malignant hyperthermia susceptible. Rhabdomyolysis during infection may point towards a heritable predisposing condition. Signs that may distinguish infection with fever and rhabdomyolysis from nonanesthetic-induced malignant hyperthermia include: (1) absence of hypercarbia; (2) shivering while feeling chilled; and (3) absence of sweating. Lack of culture identification of a specific pathogen does not confirm susceptibility to anesthetic-induced malignant hyperthermia.
With the advent of the COVID-19 pandemic, although rare, additional cases in which patients present with malignant hyperthermia-like symptoms after high fevers and respond to dantrolene have been documented. The authors of one case report suggest that dantrolene may prevent the release of IL-6 and, thus, contribute to the recovery of COVID-19 patients via antiinflammatory effects in addition to resolving rhabdomyolysis (23).
Because dantrolene has been shown to reduce hypoxia/ischemia, mitochondrial damage, oxidative stresses, inflammation, impairment of autophagy, and apoptosis, among other effects, it is currently under investigation as a treatment for COVID-19 infection (58); however, there is no evidence of improved outcomes to date.
To date, it is not possible to predict whether a malignant hyperthermia episode will occur, and pretreatment with dantrolene is not recommended in patients deemed susceptible to malignant hyperthermia and undergoing anesthesia. However, the increasing understanding, not only of the responsible genetic mutations but also their intracellular mechanistic implications, has led to the investigation of possible preventive therapies.
The use of mutant allele-specific gene silencing in malignant hyperthermia-susceptible mouse model resulted in normalization (ie, similar to wild-type) of SR calcium release in response to caffeine as well as prevention of the increase in resting calcium level with elevated temperature in flexor digitorum brevis muscle fibers. Although application was limited to the hind footpad, this work lays the foundation for systemic administration of mutant-allele-specific siRNA for assessment of the effect on different muscle groups as well as potential prevention of anesthetic-induced malignant hyperthermia (87).
The use of genomic DNA capture, next-generation sequencing, or resequencing arrays may lead to identifying associations of malignant hyperthermia susceptibility with other genes involved in excitation-contraction coupling or calcium homeostasis (65; 121). These techniques may reveal candidate RYR1 mutations that have been missed by “hot spot” screening or Sanger sequencing (44; 65; 78). As pointed out by Kim and colleagues, the finding of an RYR1 missense variant is insufficient to demonstrate causality. In a control population of 5379 individuals, more than one third had at least one protein-coding change in RYR1. As Kim and colleagues state, “Given the low estimated incidence of malignant hyperthermia, it follows that the majority of rare, missense RYR1 variants in the population are not causal for malignant hyperthermia.” Gonsalves and colleagues concluded that “some RYR1 and CACNA1S variants may have been misclassified as pathogenic without adequate genetic (eg, co-segregation) or functional data.”
Multiple bioinformatics programs aim to predict pathogenicity of gene variants. A comparison of eight such programs revealed that their sensitivity and specificity to designate RYR1 and CACNA1S variants as damaging versus benign, was highly variable. Thus, the authors of the comparison study warn that prediction results should be treated with caution and only used in combination with additional data, as none of the programs tested predicted all the variants correctly (122). In a study of patients with severe statin-associated myopathy, Isackson and colleagues employed the REVEL predictive tool to assess pathogenicity of RYR1 and CACNA1S variants, noting that REVEL had superior sensitivity to SIFT in correctly assigning pathogenicity to known malignant hyperthermia-causal mutations (56).
Hoppe and colleagues compared three freely available pathogenicity prediction tools and tested their results against those of blood samples and muscle biopsies of patients with a history of clinical malignant hyperthermia or their close relatives (50). Pathogenic variants as well as malignant hyperthermia susceptible variants were all correctly identified by the prediction tools. Despite high sensitivity, bioinformatic tools are still of limited value in the diagnosis of malignant hyperthermia susceptible variants due to a significant discordance between genotype and phenotype.
Individual RYR1 mutations continue to be evaluated for their functional impact on intracellular calcium control. Although nearly 700 RYR1 variants have been identified to date, the European Malignant Hyperthermia Group currently lists only 66 diagnostic mutations of RYR1 as pathogenic or likely pathogenic, as well as two mutations of CACNA1S as pathogenic.
As Gonsalves and colleagues further state: “It is important to stress that in addition to robust genetic analysis, there is a critical need for a robust and noninvasive functional test for MHS, which together with genetic data could allow accurate determination of the prevalence and penetrance of this trait” (44). In an article Riazi and colleagues suggest that each RYR1 variant considered as potentially malignant hyperthermia-causative should be assayed in one of the recombinant in vitro expression systems to measure the variant’s effects on channel properties. The European Malignant Hyperthermia Group guidelines in turn no longer require description of a variant in more than one family if it has been well characterized by genetic manipulation (109). Ongoing studies continue to investigate variants in genes other than RYR1 and CACNA1S in the search for alternative genes causative for malignant hyperthermia. An example is a study of a cohort of 30 Australian malignant hyperthermia susceptible patients in whom analysis of RYR1 and CACNAS1 did not yield any functional mutations. Four rare variants in four different genes encoding proteins involved in skeletal muscle calcium regulation (CACNB1, CASQ1, SERCA1 and CASQ2) were identified (Bjorksten and Gillies 2016). Although their significance with respect to malignant hyperthermia susceptibility remains unclear at this point, their discovery highlights the need to continue the search for causative mutations other than within the RYR1 and CACNA1S genes.
Use of cryoelectron microscopy and x-ray crystallography continues to elucidate the structure of the RYR1-encoded channel and its domains. High-resolution imaging and 3-D modeling of the channel and its associated proteins promise better understanding of the functional characteristics of the many identified variants (105). Todd and colleagues published a comprehensive discussion of the relationship of RYR1 variants and their position in the channel’s 3-dimensional protein structure with associated RYR1-related disorders (132). Yamazawa and colleagues employed live cell Ca+2 imaging and molecular dynamic simulation to describe the behavior of RYR1 variants associated with loss of function (eg, Y523S associated with CCD/malignant hyperthermia) or gain of function (R402C associated with malignant hyperthermia) (140). Although molecular dynamics simulation has been employed for decades to investigate proteins, combining this with live Ca+2 imaging allows for a deeper understanding of malignant hyperthermia pathophysiology by directly linking functional changes to structural changes of specific RYR1 mutations.
In an effort to better understand sarcoplasmic reticulum leak and increased reactive oxygen species during malignant hyperthermia crises, Canato and colleagues investigated knock-in mice and demonstrated increased mitochondrial Ca+2 uptake (16). Inhibition of this increased uptake by silencing the mitochondrial calcium uniporter may be a potential area for developing new pharmacological means of halting the malignant hyperthermia process.
In research by Lopez and colleagues, focus was placed on sarcolemmal transient receptor potential cation channels 3 and 6 (TRCP3/6), which play an important role in skeletal muscle calcium and sodium homeostasis (85). In a knock-in mouse model, malignant hyperthermia susceptible muscle showed a significantly increased response to extracellular Ca2+ as well. In blocking these receptors, or with nonconducting TRP6 channels, it was demonstrated again that the dysregulation of Ca2+ homeostasis was not only due to increased release of Ca2+ from sarcoplasmic reticulum stores but also from extracellular calcium entry. Increased influx from the extracellular space could be prevented by TRCP3/6 blockers. Overexpression of a nonconducting TRP6 channel increased the length of survival after halothane exposure but did not prevent death of knock-in mice. These findings nonetheless open new avenues for possible new pharmacologic treatment of malignant hyperthermia.
Multiple metabolomics studies continue to investigate the role of impaired biochemical pathways in the development of malignant hyperthermia and promise to provide greater insight into the connection between genotype and phenotypic expression of the illness.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Harvey K Rosenbaum MD
Dr. Rosenbaum of the David Geffen School of Medicine at the University of California, Los Angeles has no relevant financial relationships to disclose.
See ProfileDorothea Hall MD
Dr. Hall of the UCLA Department of Anesthesiology has no relevant financial relationships to disclose.
See Profile
Aravindhan Veerapandiyan MD
Dr. Veerapandiyan of University of Arkansas for Medical Sciences has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuromuscular Disorders
Jun. 16, 2026
Neuromuscular Disorders
May. 27, 2026
Neuromuscular Disorders
May. 21, 2026
Developmental Malformations
May. 08, 2026
Neuroimmunology
Apr. 26, 2026
Neuromuscular Disorders
Apr. 23, 2026
Neuromuscular Disorders
Apr. 23, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026