
Neurobehavioral & Cognitive Disorders
Mental status examination
Jun. 17, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
The mucopolysaccharidoses are a group of seven disorders caused by a deficiency of any of 11 lysosomal enzymes catalyzing the degradation of glycosaminoglycans (mucopolysaccharides). Depending on the enzyme deficiency, the catabolism of dermatan sulfate, heparan sulfate, keratan sulfate, chondroitin sulfate, or hyaluronan may be blocked in isolation or in combination. A disorder that looks like mucopolysaccharidosis has been identified. Unlike classic mucopolysaccharidoses, it is not due to a missing lysosomal enzyme but instead results from changes in the VPS33A gene, which disrupts lysosomal function. In all mucopolysaccharidoses, accumulation of glycosaminoglycans results in cell, tissue, or organ dysfunction. Severe intellectual disability is characteristic of mucopolysaccharidoses I H (Hurler syndrome), the severe form of mucopolysaccharidoses II (Hunter syndrome), and all subtypes of mucopolysaccharidoses III (Sanfilippo syndrome), including acquired behaviors related to autism, whereas average intellect may be retained in other mucopolysaccharidoses. Secondary neurologic complications may occur, such as hearing loss, hydrocephalus, or spinal cord compression that may develop at any stage of the disease and independent of therapy. Specific therapy with enzyme replacement has been approved for mucopolysaccharidoses I, II, IVA, VI, and VII. Novel brain-penetrating enzyme replacement therapy is being evaluated in patients with mucopolysaccharidoses I and II.
|
• The mucopolysaccharidoses are a group of storage disorders caused by the deficiency of enzymes catalyzing the degradation of glycosaminoglycans (mucopolysaccharides). | |
|
• Most patients with mucopolysaccharidosis I, II, and III develop variable degrees of intellectual disability, often with features of autism. | |
|
• Specific therapy consisting of enzyme replacement, systemic brain-penetrating or intrathecal, and gene therapy is being developed for several of the mucopolysaccharidoses. | |
|
• Symptomatic therapy, such as early ventricular-peritoneal shunt of obstructive hydrocephalus, is important as well. | |
|
• Signs of spinal cord compression should prospectively be monitored, including after initiation of enzyme replacement therapy. |
The mucopolysaccharidoses are a group of inherited single-gene metabolic disorders that result from deficiencies in the lysosomal enzymes required for the degradation of mucopolysaccharides. Hunter and Hurler provided the first reports of patients with the typical constellation of clinical features seen in the mucopolysaccharidoses: coarse facies, short stature, corneal clouding (in Hurler, but not Hunter Syndrome), skeletal anomalies, and hepatosplenomegaly (90; 91). Further in-depth historical accounting of the mucopolysaccharidoses, since Hunter's and Hurler's first reports, can be reviewed elsewhere (122; 84).
Brante identified the material stored in these patients as mucopolysaccharides and, thus, proposed the term "mucopolysaccharidoses" to describe these disorders (16). Dorfman and Lorincz identified heparan sulfate and dermatan sulfate in the urine of a patient with Hurler syndrome (47). Van Hoof and Hers surmised that excessive mucopolysaccharide accumulated in these patients secondary to deficiencies of specific lysosomal hydrolases involved in the degradation of mucopolysaccharides, and that the storage of mucopolysaccharides occurred in the lysosome (204). Hers defined the characteristics of lysosomal storage diseases (79). McKusick proposed a system of classification of the mucopolysaccharidoses into six separate entities, based on phenotype, family patterns, and pattern of specific mucopolysaccharides in the urine (123).
In classic experiments utilizing cultured fibroblasts, Dr. Elizabeth Neufeld and her colleagues demonstrated a deficiency of "corrective factors" in individual mucopolysaccharidoses (52; 08; 07). These corrective factors were purified over the next few years, and they were determined to be the various enzymes involved in the stepwise degradation of mucopolysaccharides. Dr. Neufeld's studies led to further classification of the mucopolysaccharidoses because two separate defects were found to cause the same phenotype (Sanfilippo syndrome type A and B), and it was also determined that Hurler and Scheie syndromes resulted from deficiency of the same factor, despite their widely different phenotypes.
There are currently 11 known enzyme deficiencies that give rise to seven distinct mucopolysaccharidoses. A new lysosomal storage disease state was described in 1996 by Natowicz and colleagues. The disease state has since been identified as a new mucopolysaccharidosis, MPS IX. They identified two mutations in the HYAL1 allele. They also believe this finding predicts the existence of other hyaluronidase deficiencies (139; 196). It is now apparent that, like most genetic disorders, there is a continuous spectrum of phenotypes from the severe to the mildly affected. It may now be preferable to consider speaking of these disorders in terms of specific enzyme deficiency (Table 1) rather than an eponym (221). Since the 1990s, the techniques of molecular genetics have been employed in the study of mucopolysaccharidoses to further enhance classification, diagnosis, and carrier detection and to develop novel approaches to treatment through gene therapy (77). A novel disorder with impaired glycosaminoglycan metabolism caused by a mutation in the VPS33A gene in the Yakut population in the eastern Siberian region called Sakha has been described (104). Although the detailed mechanism and disease pathophysiology, as well as the domain-specific function of VPS33A remain to be elucidated, the author found that VPS33A regulates lysosomal acidification and glycosaminoglycan metabolism rather than the expected regulation of endocytosis or autophagy (104). Clinical phenotypes of this disease are like conventional mucopolysaccharidosis caused by enzymatic deficiencies, although there were in some patients other abnormalities such as cerebral hypomyelination, hypertrophic cardiomyopathy, bone marrow dysfunction, and kidney disease. Nevertheless, this disease should be considered as a differential diagnosis for mucopolysaccharidosis (104).
• In general, the mucopolysaccharidoses are progressive disorders that involve multiple organ systems. | |
• A single enzyme deficiency can give rise to multiple phenotypes: mucopolysaccharidosis I (Hurler), mucopolysaccharidosis II (Hunter), mucopolysaccharidosis VI (Maroteaux-Lamy), mucopolysaccharidosis VII (Sly). | |
• In contrast, different enzyme deficiencies can give rise to the same phenotype: mucopolysaccharidosis III A-D (Sanfilippo), and mucopolysaccharidosis IV A and B (Morquio). | |
• The mucopolysaccharidoses are grouped according to their clinical manifestations and specific enzyme deficiencies (Tables 1 and 2). | |
• All the mucopolysaccharidoses are autosomal recessive disorders except for mucopolysaccharidosis II (Hunter), which is X-linked recessive. | |
• Somatic manifestations are seen in all mucopolysaccharidoses (Table 2), although somatic features may be mild as in mucopolysaccharidosis III (Sanfilippo syndrome) and mucopolysaccharidosis IX. |
Characteristic features for all the mucopolysaccharidoses:
• Coarse facies (less pronounced in MPS III) |
Similar phenotypes may be seen with different enzyme deficiencies (mucopolysaccharidosis III and IV). In contrast, a single enzyme deficiency can give rise to multiple phenotypes (mucopolysaccharidosis I, II, VI, and VII). These multiple phenotypes result from different mutant alleles at the same genetic locus. Thus, within a group, the severity of enzyme deficiency and clinical picture can be different. For example, all the mucopolysaccharidosis I disorders result from defects in the enzyme alpha-L-iduronidase. However, the spectrum of clinical manifestations runs from mild in mucopolysaccharidosis I S (Scheie) to severe in mucopolysaccharidosis I H. The clinical phenotypes resulting from the same enzyme deficiency are so distinct that mucopolysaccharidosis I S was initially classified as a separate disorder (mucopolysaccharidosis V) until it was later recognized that both conditions involve the same enzyme deficiency. Differences in phenotypes of lysosomal storage diseases, including mucopolysaccharidoses, may be due to epigenetic factors influencing differentiation of glycosidase properties not associated with a primary hereditary defect (96). In mucopolysaccharidosis I, measuring two trisaccharides in fibroblast extracts enabled stratification of patients based on the presence or absence of central nervous system disease (57).
Visual impairment. Visual impairment may result from somatic changes (corneal clouding, glaucoma) or from direct neurologic involvement (retinal pigmentary degeneration). Although corneal clouding is common in the mucopolysaccharidoses, retinal disease is present most commonly in mucopolysaccharidosis I (177). Studies in animal models of mucopolysaccharidoses suggest that corneal clouding may result from disorganization of the collagen fibril architecture in the corneal stroma (05). Alroy demonstrated that although mucopolysaccharidosis I, III A, and III B all demonstrate increased collagen fibril diameters, only mucopolysaccharidosis I had alterations in the fibril spacing and increased fibril packing (05). Comparable findings were noted between control and mucopolysaccharidosis I cat models. Histopathologic and ultrastructural examination of the cornea from a patient with mucopolysaccharidosis VI B revealed accumulation of membrane-bound vacuoles containing fibrillogranular and lamellated material in keratocytes and endothelial cells, along with thinning of Descemet’s membrane and the presence of excrescences (114). Donor corneal transplants have been helpful for some patients with mucopolysaccharidosis, although the reaccumulation of mucopolysaccharides in the grafts varies in timing and extent (205). One can follow changes in corneal opacification over time using iris camera corneal opacification measurement scores (97).
Voice abnormalities. Many patients have alterations in voice related to abnormalities of the larynx and pharynx (137). A hoarse voice is common. In addition, incomplete glottal closure, dysphonia, tense voice, and development of vocal cord nodules are reported (183).
Hearing loss. Auditory impairment in patients with mucopolysaccharidoses can result from somatic changes or, alternatively, from direct neurologic involvement. Hearing loss is usually of mixed conductive and sensorineural type. The conductive component, caused by somatic abnormalities, results from chronic eustachian tube obstruction, thickened middle ear secretions, possible deformity of the ossicles, and temporal bone pathology (103). Middle ear pressure-equalization tubes are often required. The sensorineural component of the hearing loss results from damaged cells of the spiral and vestibular ganglia.
Central nervous system involvement. The neurologic manifestations (Table 2) of the mucopolysaccharidoses may result from direct involvement of nervous system structures or may be secondary to somatic abnormalities. The central nervous system is primarily affected in mucopolysaccharidosis I H, II (severe), and III. In these syndromes, intellectual disability with progressive neurodevelopmental regression occurs secondary to the direct toxic effect of accumulated lysosomal storage on neurons. In more mildly affected patients, this progressive neurodegeneration may present initially with subtle behavioral changes, including hyperactivity, which coexists with dysmorphic features. However, in more severely affected patients, progressive psychomotor deterioration is observed. Many mild cases of mucopolysaccharidosis III may go undiagnosed due to the absence of overt somatic features seen in the other mucopolysaccharidoses. Thus, these patients often have nonspecific intellectual disability. A careful neurodevelopmental history is, therefore, essential to document the progressive nature of the neurobehavioral impairments (198; 174). Even in the mucopolysaccharidoses that show definite coarse facial features, such as mucopolysaccharidosis II, the most common initial complaints are delayed developmental milestones and delayed speech (180).
Spinal cord compression can occur in many mucopolysaccharidoses subtypes due to atlanto-axial instability, hypertrophy of the dura and ligamentum flavum, reactive hypertrophy of the cartilage around the odontoid process, as well as soft-tissue thickening. Resultant myelopathy with quadriparesis is well described in mucopolysaccharidoses IV and mucopolysaccharidoses VI (88; 176). All patients with mucopolysaccharidoses should have regular neurologic examinations to assess for signs of spinal cord disease, including hyperreflexia, loss of proprioception, abnormal gait, and bowel and bladder dysfunction. Spinal cord MRI is recommended at baseline and every 2 to 3 years, or more often if clinically indicated. Some authors recommend prophylactic decompression with fusion of the occipital cervical junction in patients with mucopolysaccharidoses IV (218).
Communicating hydrocephalus can develop in patients with mucopolysaccharidoses I, II, III, VI, and VII. The pathophysiology is not well understood. Because glycosaminoglycans are deposited throughout the meninges, it is believed that hydrocephalus occurs due to impaired CSF absorption through the arachnoid granulations. In addition, bone dysostosis at the skull base may contribute to reduced venous outflow and subsequent chronic dural venous hypertension. In general, hydrocephalus is found to be slowly progressive and, thus, may be difficult to distinguish from ventricular dilation owing to brain atrophy. Communicating hydrocephalus is found more often in MPS Hurler syndrome (37).
Peripheral nervous system involvement. Peripheral nerve entrapment syndromes, such as carpal tunnel syndrome, also frequently occur (213). Carpal tunnel syndrome is seen in virtually all patients with mucopolysaccharidosis type II (96%) (111). Electrodiagnostic studies should be performed promptly and regularly in these patients and initiated relatively early to allow intervention before irreversible damage occurs (36).
Visceral features. Other somatic problems seen in various mucopolysaccharidoses include noncirrhotic portal hypertension and nodular regenerative hyperplasia of the liver. This has been studied in dogs with mucopolysaccharidosis I. Such studies suggest the need for monitoring in humans (121). Increased upper airway resistance is seen in mucopolysaccharidoses and is due to the multilevel skeletal, oral, adenotonsillar, laryngeal, and tracheal involvement, which often causes sleep disorders (146).
Skeletal problems. Skeletal abnormalities are an early and prominent feature of most mucopolysaccharidoses.
Dysostosis multiplex is a constellation of congenital chondrodystrophic skeletal changes manifesting as abnormally shaped vertebrae and ribs, enlarged skull, hypoplastic epiphyses, thickened diaphyses, and bullet-shaped metacarpals. Mucopolysaccharidoses IV present with a unique type of skeletal dysplasia known as “Morquio-like dysostosis multiplex.” These patients have severe short stature with a disproportionally short trunk, kyphoscoliosis, pigeon chest, short neck, large-appearing hypermobile joints, platyspondyly, vertebral beaking and shortening, and dysplasia of the long bones.
Thoracolumbar kyphosis or gibbus deformity occurs due to poor growth of the anterior-superior aspect of the vertebrae, usually in the upper lumbar region. Neurologic complications secondary to the gibbus are not common; however, some patients require surgical stabilization to halt the progression of the deformity.
Odontoid hypoplasia, another somatic feature seen in mucopolysaccharidoses (most notably mucopolysaccharidosis IV), may result in neurologic manifestations via atlantoaxial subluxation. One review of brain and cervical spine MRI in 11 patients with mucopolysaccharidosis IV demonstrated that all had abnormal odontoid pegs with cord compression due to an anterior soft tissue mass and indentation by the posterior arch of the atlas. In each case, the cord compression was more marked than suggested by the symptoms and signs, leading to the recommendation that MRI of the cervical spine in all patients with mucopolysaccharidosis IV be performed before the development of symptoms in order to optimize surgical intervention (88). A review of the relatively common form of mucopolysaccharidosis type IIIA showed a very large cohort of patients with a wide variety of symptoms, but a clear progression that depended on the specific genotype and residual enzyme activity (200). A review of brain and cervical spine MRIs of patients with the mild variant of mucopolysaccharidosis II showed thickening of the soft tissue posterior to the odontoid peg with associated canal stenosis (147). Central nervous system dysfunction secondary to storage disease is implicated as the cause (30). Paradoxically, the increased joint flexibility that develops with enzyme replacement therapy in young children with type VI mucopolysaccharidosis can promote the development of spinal cord compression (85). Early monitoring using MRI combined with motor and sensory evoked potentials in treated patients is recommended (85; 176).
Joint disease. Joint stiffness and contractures are found in all the mucopolysaccharidoses, except for mucopolysaccharidoses IV and IX. Limited shoulder flexion can make activities of daily living, such as showering, grooming, and dressing, a challenge. Furthermore, hand function can be impaired due to stiffness and contractures in all the phalangeal joints, resulting in a claw hand deformity. Ankle stiffness can present with toe-walking.
Joint stiffness can occur proximally in mucopolysaccharidoses IV (Morquio syndrome), but the majority of mucopolysaccharidoses IV patients have joint hypermobility.
Genu valgum is seen in all children with mucopolysaccharidoses IV and in many patients with mucopolysaccharidoses I, even those with post-hematopoietic stem cell transplant. Many of these patients will have a deformity severe enough to warrant surgery.
Hip dysplasia is present in most of the mucopolysaccharidoses and is not prevented by hematopoietic stem cell transplantation. Common features include acetabular dysplasia, epiphyseal growth failure of the femoral head, valgus deformity, osteonecrosis, and subsequent degeneration and dislocation. Patients with symptoms of pain and limited mobility should be referred to orthopedic surgery (217).
Skin. A 1996 case report of an initial presentation of grouped papules on the extensor surfaces of the upper arms and legs in a patient who went on to develop progressive flexion contractures and mild developmental delay and who was then found to have mucopolysaccharidosis I H/S, is the only report of its kind so far (169). However, skin pebbling beginning on the upper back and spreading has been seen in mucopolysaccharidosis II (188). Extensive dermal melanocytosis has also been associated with mucopolysaccharidosis II (165).
MPS group designation | Enzyme deficiency | Gene | Genetics | Glycosaminoglycan in urine |
MPS I H | Alpha-L iduronidase | IDUA | AR | DS, HS |
MPS I H/S | Alpha-L-iduronidase | IDUA | AR | DS, HS |
MPS I S | Alpha-L-iduronidase | IDUA | AR | DS, HS |
MPS II (severe) | Iduronate sulfatase | IDS | X-linked recessive | DS, HS |
MPS II (attenuated) | Iduronate sulfatase | IDS | X-linked recessive | DS, HS |
MPS III A | N-sulfoglucosamine sulfohydrolase (Heparan-N-sulfatase) | SGSH | AR | HS |
MPS III B | N-alpha-acetylglucosaminidase (Alpha-N-acetyl-glucosaminidase) | NAGLU | AR | HS |
MPS III C | Heparan acetyl-CoA: | HGSNAT | AR | HS |
MPS III D | N-acetylglucosamine-6-sulfatase (N-acetylglucosamine-6-sulfate sulfatase); Glucosamine-6-sulfatase | GNS | AR | HS |
MPS IV A | Galactosamine-6-sulfate sulfatase (N-acetyl-galactosamine-6-sulfatase) | GALNS | AR | KS, CS |
MPS IV B | Beta-galactosidase 1 | GLB1 | AR | KS, CS |
MPS VI | Arylsulfatase B (N-acetyl-galactosamine-4-sulfatase) | ARSB | AR | CS |
MPS VII (Sly) | Beta-glucuronidase | GUSB | AR | DS, HS, CS |
MPS IX | Hyaluronoglucosaminidase 1 (Hyaluronidase) | HYAL1 | AR | None |
MPS I H | ||
Somatic features | ||
• coarse facial features | ||
Neurodevelopmental features | ||
• progressive neurodevelopmental degeneration (onset age 1 to 2 years). | ||
Neurological features | ||
• hearing loss (conductive and sensorineural) | ||
MPS I H/S | ||
Somatic features | ||
• intermediate between MPS I H and MPS I S syndromes | ||
Neurodevelopmental features | ||
• cognition mildly impaired or unaffected | ||
Neurologic features | ||
• hearing loss | ||
MPS II (severe) | ||
Somatic features | ||
• coarse facial features | ||
Neurodevelopmental features | ||
• progressive neurodevelopmental degeneration (onset 2 to 4 years) | ||
Neurologic features | ||
• hearing loss (conductive and sensorineural) | ||
MPS II (attenuated) | ||
Somatic features | ||
• similar to MPS II (severe), but reduced rate of progression | ||
Neurodevelopmental features | ||
• cognition mildly impaired or unaffected | ||
Neurologic features | ||
• hearing loss | ||
MPS III A, B, C, and D | ||
Somatic features | ||
• minimal dysostosis multiplex | ||
Neurodevelopmental features | ||
• progressive neurodevelopmental degeneration (onset 2 to 10 years, possibly later onset in milder forms) | ||
MPS IV A and B | ||
Somatic features | ||
• coarse facial features | ||
Neurodevelopmental features | ||
• hearing loss | ||
MPS VI | ||
Somatic features | ||
• coarse facial features | ||
Neurodevelopmental features | ||
• hearing loss | ||
MPS VII | ||
Somatic features | ||
• coarse facial features | ||
Neurodevelopmental features | ||
• wide range of severity from severe neurodegeneration beginning neonatally to unaffected cognition | ||
MPS IX | ||
Somatic features | ||
• coarse facial features | ||
Neurodevelopmental features | ||
• presumed unaffected cognition | ||
The prognosis of patients with mucopolysaccharidoses depends on the mucopolysaccharidosis group, as well as on the specific allelic variant of the disease that the patient possesses. All the mucopolysaccharidoses are progressive disorders; however, the spectrum of clinical manifestations varies from mild to severe presentations. Prognosis is determined by the presence of neurologic involvement and the severity of cardiorespiratory complications, with mucopolysaccharidosis I H representing the most severe form. Mucopolysaccharidosis I H represents the most severe course of the mucopolysaccharidoses. Developmental delay is usually apparent by 12 to 24 months of age, and the maximum functional age obtained is 2 to 4 years developmental age. This is followed by progressive neurologic deterioration. Death usually occurs by the age of 10 years, usually due to cardiorespiratory complications. On the other hand, patients with the mucopolysaccharidoses that do not involve neurodegeneration may have a normal life span. Their prognosis is related to the degree of cardiorespiratory difficulties that they encounter.
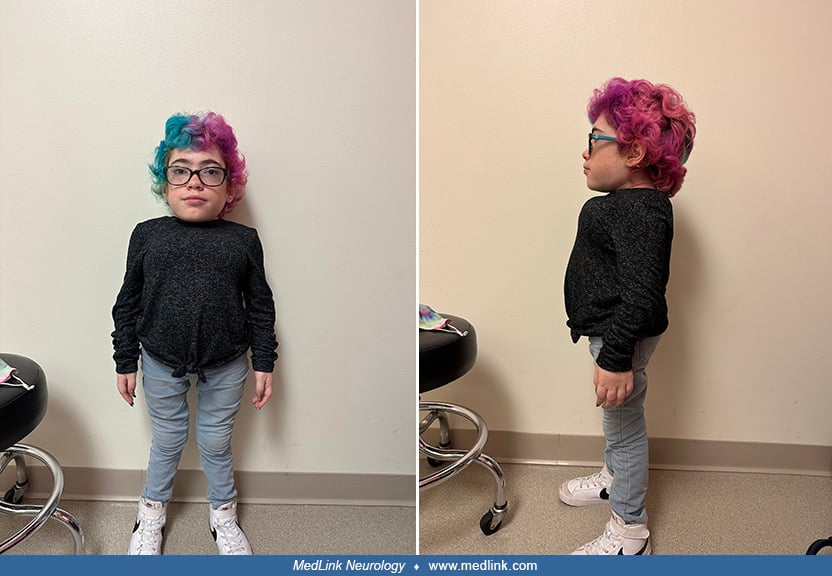
A 16-month-old male was referred to a tertiary medical center for evaluation of “balance” difficulty related to macrocephaly and increasing joint stiffness.
The patient was born at full-term, following an uncomplicated pregnancy, labor, and delivery. Birth weight was 3.9 kg (75th percentile), and fronto-occipital circumference at birth was 36.5 cm (75th to 90th percentile). Corneal clouding was noted at 4 to 6 months of age. An inguinal hernia was noted at 4 months of age and repaired surgically at 10 months of age. An umbilical hernia was noted at 14 months of age. Progressive macrocephaly was noted over time. Computerized tomography scanning of the patient’s brain revealed ventriculomegaly. In addition, progressive joint stiffening, with restriction of movement at the elbows and knees, was also reported. Thoracic kyphosis had been noted by 6 months of age. Additionally, the child was noted to have chronic nasal congestion and a large tongue with tongue thrusting since infancy.
Developmental history. Rolling occurred at 4 to 5 months, but head control with pull-to-sit was not present until 6 to 7 months, presumably secondary to macrocephaly. Sitting occurred at 9 months with a reciprocal hand-knee crawl (with head in contact with the floor) by 11 months. The patient pulled to stand at 11 months, and independent ambulation was present by 15 months, with persistent problems with both head control and “balance.” Squat and recover was equivocally present at evaluation, with forward toppling apparently due to head size and weight. Spoon and cup use were age-appropriate. The patient was smiling by three months and babbling consonant strings by 7 months. He used a specific "mama and papa" by 8 months. At the time of this examination, he communicated primarily through gestures. During the evaluation, he was able to follow verbal commands as expected for his chronological age of 16 months.
Family history. The parents were first cousins. None of the patient’s five siblings manifested similar signs and symptoms, nor was there any other family history of similar symptoms.
Physical examination. Height was 83 cm (5%), weight was 11.5 kg (50% to 75%), and fronto-occipital circumference was 52.5 cm (greater than 98%, 50% for a 7-year-old). Blood pressure was 131 over 67. The patient was a well-developed, well-nourished, mildly dysmorphic appearing male with macrocephaly and thoracic kyphosis. The general physical exam was notable for the following: macrocephaly with frontal bossing; coarse facial features with thickened eyebrows and prominent cheeks; diffuse corneal haze extending from limbus to limbus in each eye, with normal anterior segments, normal pupillary light reflex, and normal discs; normally shaped and placed ears; broad nasal bridge with copious nasal discharge; and gingival hyperplasia with normal teeth and prominent alveolar ridges. Spine examination showed that thoracic kyphosis was present. The abdominal exam was notable for a reducible umbilical hernia. There was no hepatosplenomegaly. Extremity examination showed symmetrical restriction of passive and active range of motion at the elbows and knees bilaterally. The remainder of the physical and neurologic examinations was normal.
Audiometric evaluation. Auditory brainstem evoked response showed moderate sensorineural hearing loss in both the low (500 Hz) and high (800 Hz) frequency regions. Acoustic immittance measures indicated a significant increase in stiffness of middle ear mechanisms.
Laboratory evaluation. The laboratory evaluation is summarized in Table 3.
|
Urine metabolic screen |
+ mucopolysaccharides (151/14) |
|
Arylsulfatase A (from white blood cells) |
2.11 nmol/min per mg protein (normal) |
|
• Control 1 |
1.64 |
|
Arylsulfatase B (from white blood cells) |
0.00 nmol/min per mg protein (low) |
|
• Control 1 |
1.98 |
|
Alpha-L-iduronidase (from white blood cells) |
214 pmol/min per mg protein (normal) |
|
• Control 1 |
314 |
|
Beta-D-glucuronidase (from plasma) |
0.98 nmol/min per mL plasma (normal) |
|
• Control 1 |
5.63 |
|
Glycosaminoglycan quantitation | |
|
• mg uronic acid (carbazole reaction)/g creatinine |
191 |
|
| |
Summary. The patient was a 16-month-old boy with macrocephaly, coarse facial features, thoracic kyphosis, short stature, joint stiffness, corneal clouding, umbilical and inguinal hernias, chronic nasal congestion, hearing loss, and average intelligence. This clinical phenotype, combined with the laboratory finding of deficient arylsulfatase B, indicates the patient has mucopolysaccharidosis VI (Maroteaux-Lamy).
Mucopolysaccharidoses are caused by genetically inherited deficiencies of specific lysosomal enzymes involved in the stepwise degradation of mucopolysaccharides. The genes encoding the enzymes deficient in most of the mucopolysaccharidoses have been localized to specific chromosomes (145). A superfamily of metalloenzymes with similar metal-binding sites that contain a set of conserved amino acid residues likely to be required for enzymatic activity has been identified. Mutational changes in the vicinity of the set of these conserved residues in several sulfatases cause mucopolysaccharidoses II, III, IV, and VI as well as metachromatic leukodystrophy (60).
Mutation analysis at the DNA level has provided greater insight into the causes of the variability observed in the mucopolysaccharide disorders (67; 208; 14; 54; 68; 95; 125; 191; 152). For example, large deletions of the gene encoding iduronate 2-sulfatase (IDS), found in 8% of patients, result in severe mucopolysaccharidosis II (Hunter) (22; 83; 152). The atypical presentation of seizures in patients with this disorder is associated with the proximal continuation of the deletion of the iduronate-2-sulfatase locus to include deletion at the FMR2 locus (189). On the other hand, in mucopolysaccharidosis II, mutational analysis has identified more than 333 alterations (available at: Human Gene Mutation Database: IDS), accounting for the wide spectrum of clinical phenotypes (35). In a study, Li and colleagues identified 17 variants in 18 unrelated patients, nine of which were novel (116). Variants occurring at the same codon can lead to significantly differing phenotypes, as is seen with the p.Arg468Trp variant, producing a mild variant of mucopolysaccharidosis II (Hunter), whereas p.Arg468Glu is associated with a severe form of the disease (62). Two nonsense variants are associated with severe mucopolysaccharidosis II (Hunter) (26). One study of a gene alteration in a severely affected patient with mucopolysaccharidosis II (Hunter) suggested that an iduronate-2-sulfatase gene-pseudogene exchange was responsible for that patient’s disease (11). Major rearrangements due to gene-pseudogene recombination have also been observed elsewhere (21). Skewed X chromosome inactivation of the paternal gene and a point mutation in the maternal gene are the basis for mucopolysaccharidosis II (Hunter) in one female (181). A 1998 variant analysis of 57 unrelated patients with mucopolysaccharidosis II indicates that many variants have arisen (199).
Milder forms of mucopolysaccharidosis I (Scheie syndrome) result from allelic variants that permit some residual enzyme activity (62). This residual activity of the deficient enzyme determines the neuropathic versus nonneuropathic course of the disease (33).
Mutation analysis at the DNA level has identified new variants accounting for mucopolysaccharidosis III A (Sanfilippo A) (12; 46; 128; 216). Now that the gene for the deficient enzyme in the mucopolysaccharidosis III B (Sanfilippo B) has been isolated, the number of identified causative variants has grown (09; 170; 20; 215). In a study of 22 patients, 21 different mutations were identified, with 18 of these being novel mutations. There was a vast genetic heterogeneity in this population (20). Another study of 40 patients demonstrated 31 different mutations, with 25 of these being novel. Twenty-two and a half percent of IDS mutations are de novo (152).
In mucopolysaccharidosis IV A (Morquio A), point mutations associated with differing phenotypes have been identified (62; 197; 225), as have three other novel small deletions (56; 55).
Since 1994, when six unique arylsulfatase B (ARSB) gene alleles causing variable mucopolysaccharidosis VI (Maroteaux-Lamy) phenotypes were documented (92), there have been more point mutations documented, with approximately 40 currently being documented (211). Again, the milder variants retain some residual enzymatic function (62; 211). A 1996 case report of a patient with severe mucopolysaccharidosis VI (Maroteaux-Lamy), shows two novel frameshift mutations as the cause of the patient’s problem (93).
Variants associated with mucopolysaccharidosis VII (Sly) continue to be found (62; 208; 210). Some of these variants result in hydrops fetalis (62; 202; 208). Two variant mutations that individually cause minor enzyme deficiencies appear to act together, resulting in a significant enzyme deficiency that leads to mucopolysaccharidosis VII (Sly) (94). Neither of these mutations appears to exist independently as a benign polymorphism (155).
Correlation of phenotype to genotype based on direct DNA analysis or the distinct biology of the mutated enzyme (214) aids in genetic counseling of families regarding prognosis and potential treatment strategies.
In MPS IX, Triggs-Raine and colleagues proposed that additional hyaluronidase deficiency mutations likely exist beyond the one they reported (196).
Storage substance. Mucopolysaccharides or glycosaminoglycans, such as dermatan sulfate, heparan sulfate, keratan sulfate, and chondroitin sulfate, are heteropolysaccharides usually containing two types of monosaccharide units, of which at least one has an acidic group. They occur as complexes with specific proteins called mucins or mucoproteins to form glycoproteins or proteoglycans. In this class of glycoproteins, the polysaccharide makes up most of the weight. Proteoglycans are major structural components of cartilage, bone, cornea, and other connective tissue structures. Glycosaminoglycans are degradation products derived by proteolytic cleavage of proteoglycans, the macromolecular form in which glycosaminoglycans exist in the extracellular matrix. Glycosaminoglycans are jelly-like, sticky, or slippery substances that function to provide lubrication and serve as intercellular cement and extracellular ground substance. As such, glycosaminoglycans are present in all tissues, including synovial fluid and the vitreous humor of the eye. Thus, unlike other metabolic disorders with single enzyme defects, in mucopolysaccharidoses, single enzyme defects result in diffuse organ involvement and dysmorphic features.
Metabolism of stored substrates. Glycosaminoglycans are normally metabolized in lysosomes. Lysosomes are spherical, membrane-bound vesicles, 0.2 to 0.5 µ in diameter, that contain a large variety of lytic enzymes. Lysosomal storage diseases result when specific lysosomal enzymes are absent or inactive. In these instances, the metabolism of certain substrates (mucopolysaccharides, cerebrosides, gangliosides, etc.) does not occur. As a result, these substances accumulate in the lysosome, and the swollen lysosomes interfere with normal cellular function.
The enzymes involved in each of the mucopolysaccharidoses cleave terminal sugars from the polysaccharide chains that are complexed with mucoproteins. When there is a defect in the removal of a terminal sugar, the remainder of the polysaccharide chain cannot be further degraded; thus, these chains accumulate within the lysosome in various tissues and organs of the body. From a microscopic perspective, affected cells are distended and show clearing of cytoplasm to create the so-called "balloon cells." The clear cytoplasm contains numerous vacuoles that, on electron microscopy, have been shown to be swollen lysosomes filled with a finely granular periodic acid-Schiff–positive material that can be identified as glycosaminoglycan biochemically. This accumulation of glycosaminoglycan alters normal cellular metabolism, leading to the severe somatic and neurologic changes seen in patients with mucopolysaccharidoses. In addition, dermatan sulfate normally plays a role in the extracellular organization of collagen, and it has been hypothesized that an excess of dermatan sulfate may induce abnormally extensive collagen deposition (124). This secondary collagenosis may be responsible in part for such symptoms as joint stiffness, nerve entrapment syndromes, pachymeningitis cervicalis, and communicating hydrocephalus.
Pathogenesis of CNS dysfunction. In the mucopolysaccharidoses, as with other lysosomal storage disorders, accumulation of indigestible substrates may result in brain dysfunction. This brain dysfunction can result from direct cytotoxic events as the swollen lysosomes mechanically displace organelles required for normal metabolic activity. In addition, the accumulation of glycosaminoglycan in neurons results in distortion in neuronal geometry (154). It has been shown that large neural processes referred to as "meganeurites" develop as storage sites for accumulated indigestible materials. These meganeurites tend to develop between the base of the perikaryon and the initial portion of the axon. The meganeurites further possess spines, some of which are contacted by presynaptic processes forming abnormal synapses. In addition, as the meganeurites preferentially develop adjacent to the initial axon segment, they also affect the electrical properties of the neuron. Thus, the accumulated indigestible substrates may have a direct toxic effect on the neurons, or may induce the elaboration of meganeurites. The meganeurites, and the meganeurite synapses, in turn contribute to the onset and progression of neuronal dysfunction by altering electrical properties of the neuron and by modifying integrative operations of the dendritic synaptic inputs.
In the mucopolysaccharidoses with neurologic manifestations, in addition to the swollen lysosomes filled with periodic acid-Schiff-positive material, some of the lysosomes in neurons are replaced by lamellated "zebra bodies" like those seen in Tay-Sachs disease, other gangliosidoses, and sphingolipidoses. Thus, it has been hypothesized that glycosaminoglycan accumulation in the lysosome interferes with the functioning of other lysosomal enzymes involved in sphingolipid degradation, and it is, in fact, the sphingolipid accumulation that results in the neurodevelopmental sequelae of patients with mucopolysaccharidoses. Constantopoulos and colleagues have shown that the aberration of sphingolipid metabolism that results from glycosaminoglycan accumulation parallels the presence of intellectual disability in patients with mucopolysaccharidoses (32). In their study, neurons of patients with severe intellectual disability showed substantial increases in glycosaminoglycan concentration as well as in sphingolipid concentration. In patients with moderate intellectual disability, a moderate amount of glycosaminoglycan accumulated in neurons, with a smaller increase in sphingolipid concentration. In patients with mucopolysaccharidosis l S with average intelligence, there was a normal concentration of glycosaminoglycan in neurons, and this was associated with normal sphingolipid accumulation. It is possible that high residual activity or high activity of various endoglycosidases could enhance the elimination of undegraded glycosaminoglycan from the neurons of patients with mucopolysaccharidosis I S, thus, sparing them from the accumulation of gangliosides or sphingolipids. Oxidative damage and cytotoxic cell involvement has been found in the brains of a mucopolysaccharidosis III B mouse model in the early phases of the disease (212).
Pathologic studies of neurons from patients with mucopolysaccharidosis IV (Morquio syndrome) provide further support that is indeed for the accumulation of gangliosides or sphingolipids that results in the neurodevelopmental sequelae of patients with mucopolysaccharidoses, rather than a direct effect of accumulated glycosaminoglycan (106). Patients with mucopolysaccharidosis IV usually do not show any neurologic signs or symptoms except for those secondary to their significant skeletal abnormalities, and intelligence is usually in the average range. Their neurons do show accumulation of glycosaminoglycan; however, no zebra bodies or accumulation of sphingolipids have been documented in these patients.
An increased level of dolichol in the CNS had previously been regarded as pathognomonic for neuronal ceroid-lipofuscinosis. However, increased CNS dolichol has now been detected in mucopolysaccharidosis II (severe), mucopolysaccharidosis III B, and in GM1 gangliosidosis as well. This suggests that an increased level of dolichol in the cerebral cortex is a nonspecific phenomenon related to some generalized lysosomal dysfunction.
The concept that neurologic dysfunction in mucopolysaccharidoses reflects a global lysosomal disturbance, rather than direct neurotoxicity from glycosaminoglycan accumulation, is indirectly supported by observations of increased dolichol levels in the cerebral cortex of some patients. Lysosomal beta-hexosaminidase consists of an alpha subunit and a beta subunit. When only alpha subunit activity is lacking, Tay-Sachs disease occurs. When only beta subunit activity is lacking, Sandhoff disease occurs. Neither disease has a mucopolysaccharidosis phenotype. The visceral organs of these mice lacking both alpha and beta subunit activity show an accumulation of glycosaminoglycans. Cultured fibroblasts from these mice show a defect in the degradation of glycosaminoglycans. Thus, beta-hexosaminidase appears to play an essential role in the degradation of glycosaminoglycans, and the lack of glycosaminoglycan storage in Tay-Sachs and Sandhoff diseases is probably due to functional redundancy in the beta-hexosaminidase enzyme system (164; 182). A novel method of analyzing neuronal dysfunction in vitro is to model the disease using patient-derived induced pluripotent stem (iPS) cells that are secondarily differentiated to neurons (115). This tool should be used in the future to facilitate a true understanding of the mechanism of disease in the various mucopolysaccharidoses.
The mucopolysaccharidoses are rare disorders. They occur in all ethnic groups and have a combined incidence of 1 in 30,000 to 1 in 150,000 (129). However, patients with mucopolysaccharidoses exhibit a spectrum of clinical phenotypes from mild to severe, and the total incidence may be as high as 1 in 10,000 if there are undetected patients with clinically mild phenotypes (84). This potential underreporting of the incidence of mucopolysaccharidoses might well be the case with milder variants of Sanfilippo syndrome, where somatic changes can be mild, and urine screening tests often yield false-negative results.
The only means of preventing the mucopolysaccharidoses is through prenatal diagnosis and carrier detection. All families with a family history of any of the mucopolysaccharidoses should be provided with appropriate genetic counseling. For families who have a child seeming to have a sporadic case of mucopolysaccharidosis II (Hunter), genetic counseling should include consideration of germline mosaicism in the proband’s mother (53). Both mucopolysaccharidosis IV A and VII have been identified in the group of lysosomal storage diseases leading to nonimmune hydrops fetalis. In prenatal diagnosis of nonimmune hydrops fetalis, in utero testing for prenatal diagnosis of mucopolysaccharidosis VII reference ranges for fetal serum levels of lysosomal enzymes are available (179; 69). Postnatally, much effort should be made to detect subtle features of mucopolysaccharidoses because such a diagnosis would have implications for genetic counseling. Electron microscopy has been used as part of a structured necroscopy protocol to diagnose mucopolysaccharidoses in such cases (127). If an infant with hydrops fetalis has early bone maturation accompanying intrauterine growth acceleration, the infant probably has mucopolysaccharidosis VII, as other etiologies for hydrops fetalis are not known to cause the early bone maturation-IGA association (192).
Prenatal diagnosis is possible for all the mucopolysaccharidoses. The diagnosis can be accomplished through amniocentesis and culture of amniotic fluid cells or via chorionic villus biopsy (203; 108). The diagnosis is based on simple enzyme assays that measure enzyme activity in the cultured cells. Assays are being developed to improve the speed and accuracy of such prenatal diagnoses (101). Mucopolysaccharidosis II (Hunter syndrome) presents a difficulty in prenatal diagnosis. As a result of nonrandom inactivation of the X chromosome, iduronate sulfatase activity in cells obtained from female fetuses may occasionally be almost as low as that from affected male fetuses (34). Thus, prenatal diagnosis of Hunter syndrome should always include the determination of sex. Newborn screening has been initiated in a number of countries and states in the United States. For example, in North Carolina, one true patient with mucopolysaccharidosis I was found in 62,734 newborns that were screened (186). As of 2025, 44 of 50 states in the United States, as well as the District of Columbia, have mucopolysaccharidoses I on the newborn screen. Twelve states screen for mucopolysaccharidosis type II (78).
Identification of heterozygote carriers of the mucopolysaccharidoses using enzyme measurement in leukocytes or serum has been difficult (142). The results of such carrier detection based on enzyme activity are most reliable when the enzyme activity is in the low range, as this would positively identify a carrier of the mucopolysaccharidosis. However, these results are least reliable when enzyme activity is in the normal range, as there is a significant degree of overlap between normal and heterozygote ranges of enzyme activity. With advances in molecular genetics, carrier detection of heterozygotes that carry variants for inherited metabolic disorders should be facilitated by direct DNA analysis. Amplification of DNA using the technique of polymerase chain reaction has allowed for direct DNA analysis, and, thus, provides a vehicle for specific genetic diagnosis even when insufficient fetal material is obtained for enzyme assay. Amplification of the canine beta-glucuronidase gene by polymerase chain reaction to determine the genotype of the mucopolysaccharidosis VII locus by two independent methods was helpful in distinguishing phenotypically normal mucopolysaccharidosis VII-carrier dogs from homozygous normal dogs (157). Direct dye primer sequencing of polymerase chain reaction products has been used to identify mixed bases in an obligate carrier of mucopolysaccharidosis II (190). Carrier detection in mucopolysaccharidosis II using cDNA probes has been reported (58; 23). Use of restriction fragment length polymorphisms to detect haplotypes in order to delineate the molecular heterogeneity of mucopolysaccharidosis IV A haplotypes and make prenatal diagnosis and carrier detection possible has been done (158). These molecular genetic techniques also offer opportunities for preimplantation diagnosis of early embryos after in vitro fertilization (50).
Clinical manifestations of the mucopolysaccharidoses are rather heterogeneous, both between mucopolysaccharidosis groups and within each mucopolysaccharidosis group. Thus, the major differential diagnosis appears to be distinguishing between the various mucopolysaccharidoses themselves.
Mucopolysaccharidoses are relatively easy to distinguish from other disorders based on their progressive nature and clinical symptoms that include distinctive skeletal changes (dysostosis multiplex), coarse facies, corneal opacities, short stature, stiff joints, and hepatosplenomegaly. The mucopolysaccharidoses are heterogeneous in regard to expression of these signs and symptoms; however, some milder variants of the mucopolysaccharidoses may be confused with other disorders. The mucopolysaccharidoses with neurologic sequelae especially need to be distinguished from other neurodegenerative disorders, including other lysosomal storage diseases. Other lysosomal storage diseases share similar patterns with the mucopolysaccharidoses. Deficiency of a single lysosomal enzyme results in the accumulation of substrates ordinarily degraded by that enzyme and results in a progressive clinical course. In mucopolysaccharidoses and other lysosomal storage diseases, a considerable variation of phenotype may also exist within a single disorder.
There are several lysosomal storage diseases that may present with coarse facial features, dysostosis multiplex, hepatosplenomegaly, and a varying degree of progressive neurologic deterioration. These disorders include disorders of glycoprotein degradation (mannosidosis, fucosidosis, sialidosis, aspartylglycosaminuria) and mucolipidosis II (I-cell disease), mucolipidosis III (pseudo-Hurler polydystrophy), mucolipidosis IV with mucolipidosis IV, and sialidosis I lacking facial and skeletal dysmorphisms (102). The gangliosidoses, particularly GM1 gangliosidosis, can also present with coarse facial features, dysostosis multiplex, and a variable degree of neurologic deterioration. Immunoprecipitation and immunostaining have been used to demonstrate the different pathophysiologies surrounding mutant beta-galactosidase in GM1 gangliosidosis and mucopolysaccharidosis IV B (Morquio-B), although both diseases have a beta-galactosidase deficiency (184). In addition, in the mucopolysaccharidoses with neurologic sequelae, various sphingolipids (including gangliosides) accumulate, and it has been suggested that the accumulation of sphingolipids could be responsible for some of the neurologic manifestations seen in the mucopolysaccharidoses (32). Electron microscopy of CNS neurons, in both the gangliosidoses and mucopolysaccharidoses, shows zebra bodies that represent lysosomal accumulation of sphingolipids among the seven lysosomal storage diseases presenting as nonimmune hydrops fetalis, in addition to mucopolysaccharidoses IV A and VII--type 2 Gaucher disease, sialidosis, Farber disease, infantile free sialic acid storage disease, and mucolipidosis II (I-cell disease).
A rare form of metachromatic leukodystrophy (multiple sulfatase deficiency) may present with coarse facial features, hepatosplenomegaly, and progressive neurologic deterioration (119). Fabry disease, an X-linked inborn error of glycosphingolipid catabolism, may present with pain and paresthesia in the extremities, mimicking the carpal tunnel syndrome seen in mucopolysaccharidosis II (which also shows X-linked inheritance). Finally, the mucopolysaccharidoses need to be distinguished from neurodegenerative diseases that originate from defects other than those in lysosomal storage. For example, the peroxisomal disorders such as Zellweger syndrome and neonatal adrenoleukodystrophy can present with facial dysmorphism, hepatosplenomegaly, and progressive neurologic deterioration (18). Double knockout mice (Hexa and Hexb) show a new phenotype of both mucopolysaccharidosis and gangliosidosis (163).
Mucopolysaccharidosis III (Sanfilippo) presents a challenge in differential diagnosis. These patients may have mild somatic symptomatology; thus, in the absence of coarse facial features and dysostosis multiplex, the diagnosis of a mucopolysaccharidosis may be missed. Even when this diagnosis is suspected, patients with mucopolysaccharidosis III have a higher rate of false negatives in urine screening tests (156). Thus, mild cases of mucopolysaccharidosis III may present as intellectual disability with accompanying behavioral disturbance (223). Because behavioral difficulties are common in children with intellectual disability, a diagnosis of mucopolysaccharidosis III can easily be missed. They are often misdiagnosed as idiopathic developmental delay, attention deficit/hyperactivity disorder, and autism spectrum disorders (15; 220; 162). Mucopolysaccharidosis IIIA and IIIC may even present as a Kluver-Bucy syndrome (153; 86). However, a careful neurodevelopmental history of any child with intellectual disability and behavior problems should elicit a pattern of neurologic, intellectual, or behavioral regression. Such a history of regression would indicate the need for further workup for neurodegenerative disorders, including mucopolysaccharidosis III. A history of corneal opacity and congenital glaucoma in a hypotonic infant with no dysmorphic features; no hirsutism; no hepatosplenomegaly with normal toxoplasmosis, rubella, cytomegalovirus, and herpes simples; and amino acid studies, is an indication for a mucopolysaccharidosis III workup (117).
The mucopolysaccharidoses result from the accumulation of undegraded glycosaminoglycans. These glycosaminoglycans are highly soluble in water, and they are in part excreted in the urine. Thus, the diagnosis of the mucopolysaccharidoses is based on clinical suspicion and can be supported by evidence of abnormal glycosaminoglycan excretion in the urine (179). Qualitative urine spot tests have been developed that employ binding of an appropriate dye, or flocculating agent, to the acidic glycosaminoglycan macromolecules to give a visual reaction. However, these qualitative urine spot tests, such as the Ames and Berry tests, result in a large number of false-negative and false-positive results. Thus, they are not reliable in diagnosing the mucopolysaccharidoses. They are particularly poor in diagnosing mucopolysaccharidosis III (Sanfilippo syndrome) (156). This is unfortunate because Sanfilippo syndrome is the most difficult of the mucopolysaccharidoses to diagnose clinically, due to its lack of striking somatic features; therefore, screening measures would be most valuable for the diagnosis of mucopolysaccharidosis III. Quantitative urine screening tests have been developed (dimethylmethylene blue test and cetylpyridinium chloride turbidity test), and these have been shown to have improved sensitivity in diagnosing mucopolysaccharidoses (41). The diethylene blue test is the test of choice for evaluating urine with less distinctly increased glycosaminoglycan content, namely in patients suspected of having mucopolysaccharidosis I S (Scheie) or mucopolysaccharidosis III (Sanfilippo) (42).
Analysis of fluorophore-labeled urinary glycosaminoglycans using a high-resolution electrophoresis technique has been useful in screening for mucopolysaccharidoses types I-VII. This technique has identified patients not identified by conventional chromatography methods used for mucopolysaccharidoses screening (63). Methods of newborn screening based on mass spectrometry have been developed for mucopolysaccharidoses I, II, and IIIA (227; 166).
Definitive diagnosis of the mucopolysaccharidoses requires direct demonstration of reduced activity of the specific lysosomal enzyme in each of the disorders. Enzyme assays can be performed using cultured fibroblasts, leukocytes, or serum (for mucopolysaccharidoses I, II, III B, VII, and IX) (142). A fluorometric enzyme assay for the diagnosis of mucopolysaccharidosis III A has also been developed (100). As above, direct DNA mutational analysis is also now available for many mucopolysaccharidoses, and whole exome sequencing can reveal novel disorders associated with mucopolysaccharidosis phenotype (104).
By using quantification of urinary glycosaminoglycans, electrophoresis, and correlation with enzymatic activity in leukocytes, investigators have been able to show subtle biochemical differences within the same mucopolysaccharidosis type and between the various types of mucopolysaccharidoses (29; 59; 19; 209; 167). Heparitinase II has been used to distinguish between the nonreducing end of heparan sulfate excreted in urine by a person with mucopolysaccharidosis II and the nonreducing end of heparan sulfate excreted in urine by a person with mucopolysaccharidosis III (194). A combination of anion-exchange chromatography and 30% to 40% gradient polyacrylamide gel electrophoresis has been useful to purify and characterize urinary glycosaminoglycans from various mucopolysaccharidoses (25). Electron microscopy of skin, conjunctiva, and peripheral nerve biopsies is useful for diagnosis when urinary excretion of mucopolysaccharides is normal (06; 195; 161).
The management of a patient with a mucopolysaccharidosis usually requires a multidisciplinary team to address the neurologic, cardiovascular, respiratory, and musculoskeletal complications of the disease (130). Because the mucopolysaccharidoses were discovered to result from single lysosomal enzyme deficiencies, attempts at designing therapeutic regimens have focused on enzyme replacement. Historically, unsuccessful attempts at enzyme replacement have involved direct infusion of plasma (45) and plasma exchange transfusion (17). Transplantation of cells that produce the deficient enzyme has been attempted with fibroblasts (40) and amniotic epithelial cells (01). The main difficulties with transplantation were the low number of transplanted cells that produced an insufficient amount of enzyme, and a requirement for repeated transplants because survival of the cellular grafts was only short-term.
Animal models of the mucopolysaccharidoses have provided new methods to study the safety, toxicity, and efficacy of the various proposed treatments, enabling researchers to become less hesitant to design and implement clinical trials. Models of mucopolysaccharidoses that have been used include mouse, dog, and cat for both enzyme replacement therapy and gene therapy.
Bone marrow transplantation. Bone marrow transplantation has demonstrated the ability to provide some enzymatic correction in patients with mucopolysaccharidoses (226). Bone marrow transplants serve to provide donor cells to degrade the glycosaminoglycans present in host tissues; they could also transfer their lysosomal enzymes to deficient host cells. The plasma infusions, fibroblast transplants, and amniotic cell transplants would have been expected to improve only the somatic effects of lysosomal enzyme deficiency and not the central nervous effects, as the replacement enzymes would be unable to cross the blood-brain barrier. On the other hand, it has been hoped that bone marrow transplants could provide enzymes that would cross the blood-brain barrier and correct lysosomal enzyme deficiencies in the central nervous system. This is based on the determination that perivascular glial cells and microglial cells within the brain are derived from bone marrow precursors (81). Bone marrow transplants have markedly prolonged survival (160), improved somatic symptomatology by increasing joint mobility, decreasing hepatosplenomegaly and corneal clouding, increasing linear growth, improving facial appearance, improving respiratory and cardiac function, and resolving communicating hydrocephalus (219; 87; 80). However, bone marrow transplantation does not appear to affect the skeletal changes of the mucopolysaccharidoses (120; 185; 206; 71; 73; 118). There is poor penetration of the musculoskeletal tissues by the enzyme derived from transplanted leukocytes (51; 175). Long-term follow-up of patients with mucopolysaccharidoses who have had successful bone marrow transplants suggest that ocular disease continues to progress (201).
Despite the hope that bone marrow transplantation would be able to allow replacement enzyme to cross the blood-brain barrier, neurologic and intelligence outcomes following bone marrow transplants have shown varying results (142; 71; 118). This varying neurologic outcome may be partly explained by the fact that some of the neurologic improvement in these children could be due to somatic improvements in hearing, vision, joint mobility, and resolution of communicating hydrocephalus, rather than improvements resulting from direct effects on the CNS. However, Hugh-Jones has reported no neurologic regression in several patients with mucopolysaccharidoses for at least two years after bone marrow transplant (89). Bergstrom also has reported sustained subjective and objective improvements in symptoms for more than three years following bone marrow transplant in a patient with mucopolysaccharidosis II (Hunter) (10). Vellodi and colleagues have reported that of 13 patients with mucopolysaccharidosis I who underwent bone marrow transplant and survived for at least five years post transplantation, most have shown an arrest or slowing down of psychomotor regression (206).
Whitley prospectively followed nine surviving patients with Hurler syndrome 3.8 to 8.9 years after transplantation. The four patients with pretransplantation standard scores above 80 (on various developmental instruments) maintained scores in a comparable range. The five patients with initial standard scores below 80 demonstrated a further lowering of scores on subsequent testing. Thus, these authors advocate for earlier diagnosis and transplantation, whereas intelligence indicators remain in a low normal range (219). Similarly, Shapiro (173) and Krivit (107) looked at the outcomes of children with various mucopolysaccharidoses. In mucopolysaccharidosis I (Hurler), they found that transplantation in 14 children under 24 months of age resulted in stabilization of cognition in a low normal range, whereas transplantation in seven children between 24 months and 36 months resulted in a decline in cognitive function. Subsequent data on long-term monitoring of patients with mucopolysaccharidosis led to the conclusion that mucopolysaccharidosis I patients, particularly those less than 24 months old and having a baseline Mental Developmental Index of greater than 70 at the time of bone transplantation, can achieve a favorable long-term outcome with continuing cognitive development (149). Boelens and colleagues reviewed 146 MPS I patients and found that cord blood gives the best likelihood of full long-term engraftment with the highest enzyme level (13).
In mucopolysaccharidosis II (Hunter), all the children in the Shapiro and Krivit studies were beyond 24 months of age, and all but one child had a Mental Developmental Index on a Bayley Infant Intelligence Scale of less than 50 on follow-up. An analysis of 146 patients with mucopolysaccharidosis II, including 27 newly transplanted patients and 119 published patients were compared with 51 patients on enzyme replacement therapy and 15 untreated patients (109). Results showed that hematopoietic cell transplantation is more effective than enzyme replacement therapy for mucopolysaccharidosis II in a wide range of disease manifestations (109). Finally, in mucopolysaccharidosis III (Sanfilippo), patients after bone marrow transplant identified no cases in which bone marrow transplantation altered the progressive neurodegenerative course of the disease (107; 173). Vellodi has reported on the results of 10 bone marrow transplant patients with mucopolysaccharidosis II, with only three surviving for up to 7 years. The high mortality is attributed to poor donor selection. Of the three survivors, only one has demonstrated a favorable outcome on intellectual development without a significant physical handicap (207).
As above, many studies suggest that if bone marrow transplantation is to be considered, it should be considered at younger ages, as younger patients have lower incidences of transplant-related complications, and their underlying disease will also be less advanced.
Although bone marrow transplantation has been associated with significant morbidity and mortality, results based on 2005 European guidelines for mucopolysaccharidoses have shown promising results. A study of 56 patients with MPS I, II, III, and IVA showed an overall survival of 95% and even free survival of 90.3% (03). To achieve maximal engrafted survival rates, a noncarrier matched sibling, fully matched unrelated cord blood, or adult unrelated donor remains the highly preferred donor in mucopolysaccharidoses patients (03). These results were better than the ones obtained in the past.
Long-term cognitive outcomes for MPS 1H patients who received hematopoietic cell transplantation appear stable and in the mildly impaired range. In a study of 47 individuals who were 9 years post-transplant on average, the average full-scale IQ was 67.1 (normal, 85 to 115), but there was a significant difference in patients who had a known severe mutation and those with other genotypes (110). Long-term cognitive outcomes posttransplant for other mucopolysaccharidoses are mixed, and studies are limited by a large variation in age of transplant (187).
Given the above, hematopoietic stem cell transplantation is considered the standard-of-care for young children with the most severe form of MPS I, Hurler syndrome. For more attenuated MPS I and the other mucopolysaccharidoses, hematopoietic stem cell transplantation is considered on a case-by-case basis.
Enzyme replacement therapy. Enzyme replacement currently exists for mucopolysaccharidoses I, II, IVA, VI, and VII (64; 76).
Enzyme replacement therapy for MPS I. Effective treatment for MPS I has been achieved using mannose-targeted delivery of enzymes to the lysosomes with impressive clinical results in patients with this disease.
A study aimed to evaluate the usefulness and efficacy of recombinant human alpha-L-iduronidase (laronidase) and its impact on functional parameters (222). Patients treated with laronidase demonstrated improvements in the 6-minute walk test, better sleep quality in those with sleep apnea, and significant reductions in liver volume (8.9%) and glycosaminoglycan excretion (54.1%), changes that were not observed in the placebo group. The results of this study confirmed that laronidase reduced clinical GAG accumulation and excretion and that this was translated into clinically important improvements in respiratory function and in physical capacity. The approved 0.58 mg/kg/week laronidase dose regimen provided near-maximal reductions in glycosaminoglycan storage and the best benefit-to-risk ratio (66). When initiated in adults, enzyme replacement therapy lowers glycosaminoglycans and liver volume and possibly improves the 6-minute walk test but shows no other clinical benefit (148). It should be stressed, however, that these intravenous enzyme infusions have no effect on the neurologic aspects of the disease. For secondary abnormalities such as spinal cord compression, intrathecal enzyme therapy may be helpful (135), but possibly also for the neurologic aspects of the disease (141; 28). A combination of bone marrow transplantation with enzyme replacement may have substantial advantages. Severe cardiac disease in an infant with MPS I significantly improved with enzyme replacement therapy followed by bone marrow transplantation (82). The experience with a larger group of patients suggested that enzyme infusion prior to bone marrow transplantation is beneficial and is not associated with an increased rate of rejection (193; 224). Furthermore, all MPS I patients, including those who have not been transplanted or whose graft has failed, may benefit significantly from enzyme replacement; it should be started at diagnosis and may be of value in patients awaiting transplantation (43).
Enzyme replacement therapy for MPS II. Recombinant human iduronate 2-sulfatase is approved for the treatment of MPS II (134). A systematic review of enzyme replacement therapy for MPS II found that although the randomized clinical trial identified was of good quality, it failed to describe important outcomes. It has been demonstrated that enzyme replacement therapy with idursulfase is effective in relation to functional capacity (distance walked in 6 minutes) and forced vital capacity (liver and spleen volumes and urine glycosaminoglycan excretion) when compared with placebo in patients with MPS II. There is no available evidence in the included study or in the literature on outcomes such as improvement in growth, sleep apnea, cardiac function, quality of life, and mortality (39). However, using the Hunter Outcome Survey registry, it is shown that mostly in children, idursulfase has a positive effect on urinary glycosaminoglycan levels, 6-minute walk test results, left ventricular mass index, forced vital capacity, absolute forced expiratory volume in 1 second, and hepatosplenomegaly after three years of treatment (131). Similar effects have been observed with enzyme replacement therapy in MPS VI (75). A salutary effect on secondary neurologic complications has been described (105). Surprisingly, iduronate 2-sulfatase produced by an adeno-associated virus 2 vector was able to substantially cross the blood-brain barrier in an animal model of mucopolysaccharidosis II (151). Generally, the effects of “specific” therapy in patients, such as enzyme replacement or bone marrow transplantation, are assessed by looking at various biomarkers such as organ volume, heart and lung function, and joint movement. Novel biomarkers have been suggested, such as heparin cofactor II-thrombin complex and dermatan sulphate:chondroitin sulfate ratio; however, their real usefulness remains to be established (112).
The safety and efficacy of intrathecal enzyme administration is being studied (44; 132; 133). The efficacy is studied in terms of spinal stenosis and improving neurologic outcome. Modified enzyme replacement consisting of a fusion protein that includes a ligand to cerebral vascular endothelial cells for better brain delivery of systemic therapy is being tried in patients with mucopolysaccharidoses I and II (65; 74). A drug that fuses human iduronate sulfatase with human transferrin receptor antibody has been approved in Japan for the treatment of neuronopathic mucopolysaccharidoses II (144).
Enzyme replacement therapy now exists for mucopolysaccharidoses VII, which the FDA approved after a one-arm trial with a novel method (76).
Gene therapy. There are currently no approved gene therapies for the mucopolysaccharidoses; however, there are active clinical trials for gene therapy in mucopolysaccharidoses I, II, III, and VI. Gene therapy techniques involve in vivo or ex vivo methods. In vivo therapy involves treating the patient directly with the vector that delivers the correct gene to the cells. Ex vivo therapy involves treating the patient’s cells with the vector carrying the correct gene and then delivering the corrected cells back to the patient. Vectors used have included retroviruses, lentiviruses, and adeno-associated viruses. Retrovirus vectors were used in the earliest attempts at gene therapy for mucopolysaccharidosis. Current trials involving a lentiviral vector utilize ex vivo methods to transduce the patients’ hematopoietic stem cells with the correct gene for mucopolysaccharidoses I, II, and MPS IIIa (NCT03488394; NCT05665166, NCT04201405). Current trials involving an adeno-associated virus (AAV) vector use in vivo methods. One advantage of AAV vectors is that they do not integrate with the patients’ nuclear DNA; instead, they replicate as episomes inside the nucleus. There are active in vivo gene therapy trials using AAV vectors in mucopolysaccharidoses I, II, and IIIa (NCT03580083; NCT03566043; NCT03612869; NCT02716246).
Supportive treatment for patients with advanced diseases or mucopolysaccharidoses without available enzyme replacement therapy. Supportive management and close monitoring of the progression of somatic disease and neurodevelopmental status are required in these patients. These patients need to be closely monitored for the development of communicating hydrocephalus, cervical spinal cord compression, atlantoaxial instability, nerve compression syndromes, visual disturbance, hearing impairment, as well as multiple other respiratory, cardiac, and otolaryngologic difficulties (38; 171; 02). These children require a strong primary care medical provider to serve as a case manager to make timely referrals for appropriate neurosurgical, orthopedic, and otolaryngologic procedures to improve quality of life. Such procedures as coronary valve replacement, coronary artery bypass grafting, ventriculoperitoneal shunting for communicating hydrocephalus, occipital cervical fusion, surgical decompression of the spinal cord, peripheral nerve decompression, middle ear pressure equalization tubes, and tracheostomy have all been shown to improve the quality of life for patients with mucopolysaccharidoses (126; 159; 228). Screening for the presence of hydrocephalus and even preventive shunting before bone marrow transplantation are now recommended (04). Special procedures, such as an endoscopic adenoidectomy for patients with symptomatic adenoid hypertrophy to avoid complications associated with abnormal cervical vertebrae, have been used (140). Implication of a laryngeal mask as a cause of airway obstruction in a male with mucopolysaccharidosis II is part of a databank of problems seen with patients having a mucopolysaccharidosis that could be used in development management plans to avoid future problems in these patients (24).
Children with the neurologic and orthopedic manifestations of the mucopolysaccharidoses require appropriate special education services as well as direct and consultative physical therapy, occupational therapy, and speech and language therapy services as indicated. Parents of these children benefit greatly from respite services provided by local agencies, as well as from participation in parent support groups, which foster increased understanding, awareness, and confidence in caring for their children. The primary care medical provider can best serve patients with mucopolysaccharidoses and their families by assisting them in obtaining the physical, psychological, and educational services they need (49; 143). Controlling the behavioral manifestations of mucopolysaccharidosis III is a particular challenge. The mainstay of recommended practices includes understanding the behavior in the context of the cognitive skill level and creating predictable routines in a safe home and school environment (136). There are limited studies for the use of behavior-modifying medications in the mucopolysaccharidosis IIIA population. A study of twelve mucopolysaccharidoses IIIA patients showed some benefit for hyperactivity and oppositional behavior (98), but other studies have shown that antipsychotic medication can worsen hyperactivity and aggression (48). In general, limited medication use targeting specific symptoms at the lowest effective doses with careful monitoring for side effects is recommended. For procedures such as MRI, dexmedetomidine may be safer than propofol for sedation of patients with mucopolysaccharidoses (99).
Anakinra, a recombinant human interleukin 1 receptor antagonist, was administered to patients with MPS III (Sanfilippo syndrome) to address the inflammation that underlies many of the challenging neurobehavioral and systemic symptoms, such as hyperactivity, agitation, poor sleep, and abnormal stooling. This phase I/II trial demonstrated that anakinra was safe and associated with clinically meaningful improvements in neurobehavioral and quality-of-life outcomes (150). A phase III trial is anticipated.
The availability of data regarding long-term treatment outcomes, of course, depends on how long enzyme replacement has been available for each of the different mucopolysaccharidoses. Laronidase for mucopolysaccharidoses I was FDA-approved in 2003, idursulfase for mucopolysaccharidoses II in 2006, elosulfase alfa for mucopolysaccharidoses IVA in 2014, galsulfase for mucopolysaccharidoses VI in 2005, and vestronidase alfa for mucopolysaccharidoses VII in 2017. In mucopolysaccharidoses I, long-term outcome data are divided into patients with Hurler syndrome who received hematopoietic stem cell therapy and patients with more attenuated mucopolysaccharidoses I who received enzyme replacement therapy only. Treatment with both HSCT and ERT has extended lifespan, improved cardiorespiratory tolerance, reduced hepatosplenomegaly, and improved growth. Both HSCT and ERT have limitations on addressing bone and joint symptoms, heart valve disease, and corneal opacity, with resultant loss of mobility, suboptimal cardiopulmonary function, and vision impairment (113; 70). Neurocognitive function after HSCT showed stabilization in the years following transplant, but some decline has been demonstrated decades following transplant. The decline could be related to difficulties in psychosocial functioning, mobility limitations, and isolation, as well as late-onset psychiatric symptoms like depression (70). Data on attenuated mucopolysaccharidoses I patients who received ERT-only are limited and highly variable depending on the severity of disease. Intravenous enzyme replacement therapy is not expected to treat central nervous system disease, and about 20% to 30% of patients with attenuated mucopolysaccharidoses I are cognitively impaired (172).
Long-term outcomes on systemic symptoms of mucopolysaccharidoses II are similar to those of mucopolysaccharidoses I, but results of HSCT have been equivocal and, therefore, longitudinal data are limited. As with mucopolysaccharidoses I, even some so-called attenuated mucopolysaccharidoses II patients demonstrate cognitive impairment (172), and intravenous ERT does not prevent the development of spinal cord compression (138).
The only other mucopolysaccharidoses with longer-term outcome data are mucopolysaccharidoses VI. A study of three patients who received enzyme replacement since infancy demonstrated improvements and/or stabilization in growth, hearing, cardiopulmonary function, facial dysmorphisms, and hepatomegaly. Similar to mucopolysaccharidoses I, skeletal and eye disease appeared to progress in the long-term (61).
The mucopolysaccharidoses have been demonstrated to be an etiologic cause of nonimmune hydrops fetalis. Tests for in utero diagnosis are available, and should be considered in the event of a prenatal diagnosis of nonimmune hydrops fetalis (69; 178). A successful pregnancy and breastfeeding have been described in a woman with MPS I while receiving enzyme replacement therapy (27).
General anesthesia in children with mucopolysaccharidoses constitutes a major risk (72; 168). This risk involves cervical instability, difficulty in efforts to intubate due to a narrowed airway, and acute perioperative airway problems. Airway obstruction can be caused by a narrow trachea, thickened vocal cords, adenotonsillar hypertrophy, macroglossia, and copious pulmonary and nasal secretions (72). Although maintenance of the airway during induction of anesthesia may be difficult, mask ventilation is usually effective, for example, in mucopolysaccharidosis III (31). Limitations of the temporomandibular joint also make intubation difficult. Intubation becomes increasingly difficult as these patients age. Other pathologies are common, including structural cardiac abnormalities, abnormal clotting, and bleeding diathesis (31; 168). Thus, general anesthesia in these patients should be approached with extreme caution, and should only be attempted by an experienced anesthesiologist, and only when experienced otolaryngologists are available (72).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Agnes Chen MD
Dr. Chen of Harbor-UCLA Medical Center received a research grant from Takeda.
See Profile
Andrea Gropman MD
Dr. Gropman of St. Jude Children's Research Hospital has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurobehavioral & Cognitive Disorders
Jun. 17, 2026
Neurogenetic Disorders
Jun. 01, 2026
Neurobehavioral & Cognitive Disorders
May. 20, 2026
Neurogenetic Disorders
May. 08, 2026
Infectious Disorders
May. 01, 2026
Neurogenetic Disorders
Apr. 30, 2026
General Child Neurology
Apr. 29, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026